

Abstract
Objective
Recessive mutations in the CAPN1 gene have recently been identified in spastic paraplegia 76 (SPG76), a complex hereditary spastic paraplegia (HSP) that is combined with cerebellar ataxia, resulting in an ataxia‐spasticity disease spectrum. This study aims to assess the influence of CAPN1 variants on the occurrence of SPG76 and identify factors potentially contributing to phenotypic heterogeneity.
Methods
We screened a cohort of 240 unrelated HSP families for variants in CAPN1 using high‐throughput sequencing analysis. We described in detail the clinical and genetic features of the SPG76 patients in our cohort and summarized all reported cases.
Results
Six unreported CAPN1‐associated families containing eight patients with or without cerebellar ataxia were found in our cohort of HSP cases. These patients carried three previously reported homozygous truncating mutations (p.V64Gfs*103, c.759+1G>A, and p.R285*), and three additional novel compound heterozygous missense mutations (p.R481Q, p.P498L, and p.R618W). Lower limbs spasticity, hyperreflexia, and Babinski signs developed in about 94% of patients, with ataxia developing in 63% of cases. In total, 33 pathogenic mutations were distributed along the three reported functional domains of calpain‐1 protein, encoded by CAPN1, with no hotspot region. A comparison of gender distribution between the two groups indicated that female SPG76 patients were significantly more likely to present with complicated HSP than male patients (P = 0.015).
Interpretation
Our study supports the clinically heterogeneous inter‐ and intra‐family variability of SPG76 patients, and demonstrates that gender and calpain‐1 linker structure may contribute to clinical heterogeneity in SPG76 cases.
Introduction
Hereditary spastic paraplegia (HSP) comprises a group of clinically heterogeneous disorders characterized by progressive spasticity and hyperreflexia of the legs due to corticospinal tract degeneration. 1 , 2 Historically, HSP is distinguished as pure or complicated forms on the basis of clinical grounds. 3 Pure form HSP shows predominant spasticity and weakness restricted to the lower limbs, and often presents hypertonia, hyperreflexia, and Babinski sign. In addition, bladder dysfunction and sensory disturbance are also frequently reported in the pure form. Complicated form HSP is accompanied by additional neurological or extra‐neurological signs, including ataxia, intellectual disability, peripheral neuropathy, amyotrophy, cataracts, hypopigmentation, and others.
HSP is also a genetically heterogeneous disease. To date, more than 82 known or suspected genes or genetic loci have been reported to cause HSP. 4 Notably, it has been proposed that many autosomal recessive HSP genes remain unidentified. 3 Recently, loss‐of‐function mutations in the CAPN1 gene have been described as causative for spastic paraplegia 76 (SPG76). 5 CAPN1 encodes a neutral calcium‐activated protease known as calpain‐1 protein, which is involved in processes of synaptic plasticity, neuronal migration, neuronal maintenance and necrosis, among others. 6 Calpain‐1 contains three functional domains, including a protease domain, a C2‐like (C2L) Ca2+ binding domain, and a penta‐EF‐hand (PEF) Ca2+ binding domain. 7 , 8 Notoriously, HSP patients with autosomal recessive inheritance showed higher clinical complexity, especially due to ataxia resulting in ataxia‐spasticity spectrum. 9 Even though mutations in CAPN1 were initially associated with cerebellar ataxia in dogs and humans, 6 , 10 as well as in human complicated form HSP presented with any of ataxia, dysarthria and amyotrophy, 5 reports of pure form HSP patients with CAPN1 mutations are increasing. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 Given this broad range of symptoms and contributing factors, it remains an ongoing challenge to find a specific, reliable correlation between genotype and phenotype for this disease complex.
In this study, we describe six additional CAPN1‐associated SPG76 families containing eight affected individuals with or without cerebellar signs. These patients had six homozygous or compound heterozygous variants. In addition, we provide a summary review of SPG76 literature in order to clarify the phenotypic heterogeneity associated with this disease.
Patients and Methods
Subjects recruitment
This retrospective study was carried out at the Department of Neurology of the First Affiliated Hospital of Fujian Medical University. In total, 240 families that fulfilled Fink’s diagnostic criteria for HSP symptoms were enrolled irrespective of their genetic diagnosis. 1 , 2 , 25 , 26 , 27 Clinical features, brain MRI findings, and genetic data were collected and analyzed for each available case. The ethics committees of the First Affiliated Hospital of Fujian Medical University approved the study, and written informed consent was obtained from each of the participants.
High‐throughput sequencing and data analysis
Genomic DNA was extracted from the peripheral blood of the included subjects using a QIAGEN kit. Prior to high‐throughput sequencing (HTS), multiplex ligation‐dependent probe amplification (MLPA) was applied to detect the large deletions or copy number variations (CNV) in commonly known pathogenic genes (ATL1, SPAST, NIPA1, SPG11, REEP1, and SPG7) using SALSA MLPA probe mixes (MRC‐Holland). Subsequently, point mutations and small indel (insertion/deletion) mutations (< 50 base pairs) in 82 known HSP‐causative genes (https://neuromuscular.wustl.edu) were screened by targeted next‐generation sequencing (NGS) or whole‐exome sequencing (WES) using the Illumina HiSeq 3000 platform. 25 , 26 , 27 , 28 The pathogenicity of the filtered variants was then investigated by in silico prediction tools. The variants were finally comprehensively classified according to the American College of Medical Genetics and Genomics (ACMG) criteria. 29 Specifically, each pathogenic criterion is weighted as very strong (PVS1), strong (PS1–4), moderate (PM1–6), or supporting (PP1–5). In particular, segregation of the identified pathogenic CAPN1 variants (NM_001198868.2) was investigated in their available relatives using Sanger sequencing with the variant‐specific primers (Supplemental File S1: Table S1).
Statistical analysis
Results are expressed as ratios or means with min to max values. To compute p values, Student’s t‐test and Chi‐squared followed by Bonferroni test were used, as indicated in the figure legends.
Results
Genetic spectra of CAPN1
A total of 110 families carrying the known HSP‐causative genes were diagnosed through HTS analysis (Supplemental File S2: Figure S1a). Remarkably, six unrelated families carrying CAPN1 mutations were identified, accounting for 12.2% (6/49) of the autosomal‐recessive HSP cases (Supplemental File S2: Figure S1b). Genetically, six homozygous or compound heterozygous variants of CAPN1 were detected from the aforementioned six families (Family 1 to Family 6) (Fig. 1A and Supplemental File S2: Figure S2), three of which were previously documented pathogenic homozygous mutations (c.182_183insC (p.V64Gfs*103), c.759+1G>A, and c.853C>T (p.R285*)). 6 , 11 , 21 The other three additional compound heterozygous missense variants ((c.1442G A (p.R481Q), c.1493C>T (p.P498L), and c.1852C T (p.R618W)) occurred at extremely low allele frequency (<7 × 10‐5) and have not yet been reported. The former two of these mutations were located in the C2L Ca2+‐binding domain, while the latter was found in the PEF Ca2+ binding domain of calpain‐1 (Fig. 1b). Notably, the three novel variants showed concordant segregation with the HSP phenotype, and were predicted to be deleterious. Thus, these three missense variants can be classified as likely pathogenic according to the ACMG criteria (PM1+PM2+PP1+PP3) (Supplemental File S1: Table S2). 29 Intriguingly, two separate, unrelated families (Families 1 and 4) harbored the same homozygous mutation p.V64Gfs*103, while the other two families (Families 2 and 6) carried a separate, identical heterozygous mutation p.P498L.
Figure 1.
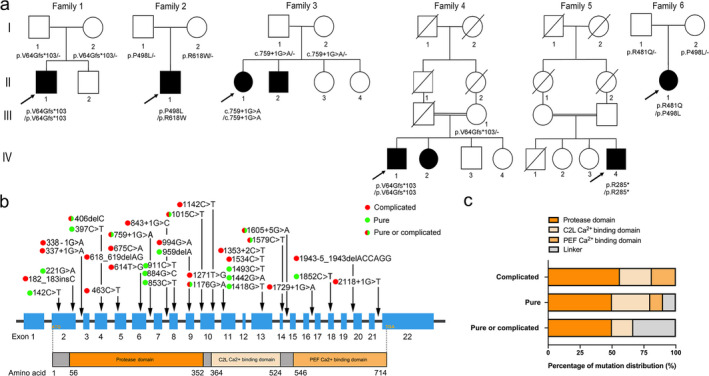
Mutational spectrum of CAPN1 in SPG76 patients. (A) The pedigrees of six SPG76 families in our cohort, with CAPN1 mutations indicated. Filled black squares and circles indicate affected individuals. The proband in each family is indicated by an arrow. The genotypes of all available family members were determined. Dash symbol indicates reference allele. (B) The mutation distribution across the CAPN1 gene and corresponding calpain‐1 protein containing a protease domain (amino acid 56‐352), a C2L Ca2+‐binding domain (amino acid 364‐524), and a PEF Ca2+‐binding domain (amino acid 546‐714). Gray bars indicate the linker structure. Filled green circles highlight the mutations causing pure form HSP. Filled red circles highlight the mutations causing complicated form HSP. The half‐red and half‐green circles highlight the mutations causing either pure or complicated form HSP. (C) Statistical analysis of the correlation between mutation distribution and HSP forms.
Clinical profiles of CAPN1‐related SPG76
The clinical profiles of eight patients from the six CAPN1‐related SPG76 families are shown in Table 1. Collectively, the mean ± SD of age at onset of the affected individuals was 27.6 ± 6.3 years, ranging from 19‐ to 38‐years‐old. The mean ± SD of disease duration was 12.4 ± 6.0 years. Clinically, bilateral lower limb spasticity, with or without weakness, was the initial symptom for all cases. Moreover the brisk deep tendon reflex observed in four extremities and extensor plantar responses were the common clinical signs among all eight patients.
Table 1.
A summary of clinical features of eight affected individuals in six families with CAPN1 mutations
| Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | Family 6 | |||
|---|---|---|---|---|---|---|---|---|
| II‐1 | II‐1 | II‐1 | II‐2 | IV‐1 | IV‐2 | IV‐4 | II‐1 | |
| Mutation | c.182_183insC | c.1493C>T and c.1852C>T | c.759+1G>A | c.759+1G>A | c.182_183insC | c.182_183insC | c.853C>T | c.1442G>A and c.1493C>T |
| Gender | Male | Male | Female | Male | Male | Female | Male | Female |
| Age at onset, year | 27 | 33 | 33 | 38 | 21 | 21 | 28 | 19 |
| Disease duration, year | 14 | 9 | 11 | 4 | 20 | 19 | 18 | 4 |
| Disability score | 3 | 2 | 2 | 2 | 3 | 3 | 2 | 2 |
| Initial symptom | Weakness and stiffness of legs | Weakness and stiffness of legs | Stiffness of legs |
Stiffness of legs; unsteadiness |
Stiffness of legs | Stiffness of legs | Weakness and stiffness of legs |
Stiffness of legs |
| Dysarthria | ‐ | ‐ | ‐ | ‐ | + | + | ‐ | ‐ |
| Cerebellar sign | Limb ataxia | ‐ | Truncal and gait ataxia | Truncal and gait ataxia | Limb ataxia | Limb ataxia | ‐ | ‐ |
| Hypermyotonia‐UL | + | + | ‐ | ‐ | + | + | ‐ | ‐ |
| Hypermyotonia‐LL | + | + | + | + | + | + | + | + |
| Hyperreflexia‐UL | + | + | + | + | + | + | + | + |
| Hyperreflexia‐LL | + | + | + | + | + | + | + | + |
| Lower limb weakness | + | + | ‐ | ‐ | ‐ | ‐ | + | ‐ |
| Extensor plantar response | + | + | + | ‐ | + | + | + | + |
| Sensory impairment | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Brain MRI | Normal | Normal | NA | Mild cerebellar atrophy | Normal | Normal | Normal | Normal |
Disability score: 1 = normal, 2 = walks but cannot run, 3 = walks with aids, 4 = wheelchair bound.
UL = upper limbs, LL = lower limbs; + = present; ‐ = absent; NA = not available.
Three probands (II‐1 in Family 2, IV‐4 in Family 5, and II‐1 in Family 6) had the core features of HSP without cerebellar signs, and were considered as prototypical pure forms. Five other affected individuals (II‐1 in Family 1, II‐1 and II‐2 in Family 3, and IV‐1 and IV‐2 in Family 4) exhibited ataxia and hypermyotonia in the lower limbs, without the dysdiadochokinesia, nystagmus, or slurred speech. Specifically, the male patient II‐2 in Family 3 first noticed symptoms of slowly progressive lower limb stiffness and unsteadiness at the age 38. He presented with truncal and gait ataxia, but was still able to walk independently without aids. His cranial MRI revealed very mild vermian atrophy. The patients IV‐1 and IV‐2 in Family 4 were born to the third‐degree consanguineous parents and started with gait difficulty at 21 years. In these two patients, dysarthria began approximately 20 years later, with their ability to walk without aid lost by age 40. Notably, none of the affected individuals complained of bladder dysfunction or any sensory abnormalities. However, the cranial MR imaging of the other available individuals showed no remarkable signs of disease.
Comprehensive analysis of phenotypic heterogeneity for CAPN1‐associated SPG76
The combination of our cohort with the other previously reported cases resulted in a total of 35 pedigrees, which included 67 SPG76‐affected individuals that carried a total of 33 homozygous or compound heterozygous CAPN1 mutations among them. 5 , 6 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 These mutations were distributed along the entire CAPN1 locus with no exon‐associated hotspot (Fig. 1B,C). Additionally, scrutiny of the integrated clinical information available for 85.1% (57/67) of the CAPN1‐associated SPG76 patients showed that 36.8% (21/57) of these patients were pure form HSP (Fig. 2A). In contrast, 63.2% (36/57) of this patient subset presented as complicated HSP with cerebellar signs, and 25% (9/36) of which exhibited cerebellar atrophy.
Figure 2.
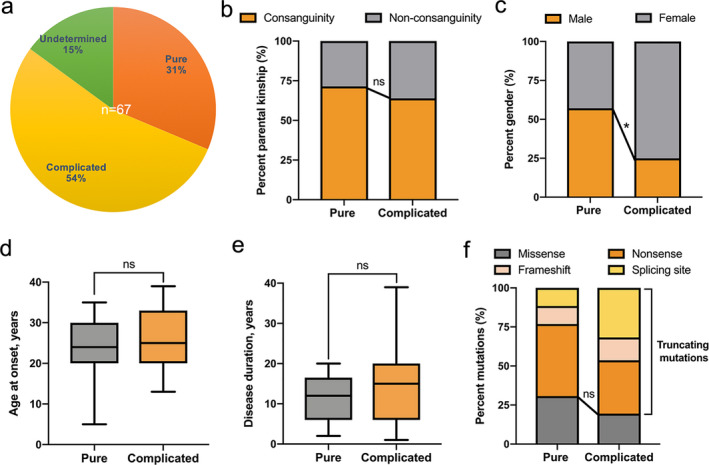
Comprehensive analysis of phenotypic heterogeneity for CAPN1‐associated SPG76. (A) Distribution of pure, complicated, and undetermined forms (no available clinical information) across all reported SPG76 patients (n = 67). (B) Statistical comparison of the percentage of parental kinship between the pure and complicated forms of SPG76. Chi‐squared followed by Bonferroni test. (C) Statistical comparison of the gender distribution between pure and complicated forms. Chi‐squared followed by Bonferroni test. * indicates P < 0.025. (D) Statistical comparison of the age at onset between pure and complicated forms. Student’s t‐test. (E) Statistical comparison of the disease duration between pure and complicated forms. Student’s t‐test. (F) Statistical comparison of the percentage of truncating or missenses mutation types between pure and complicated forms. Nonsense, frameshift, and splicing site mutations are combined into the truncating mutation category for comparison with missense mutations. In box and whisker plots, error bars indicate min to max. Chi‐squared followed by Bonferroni test. ns, not significant.
To explore the factors (i.e., gender, disease course, genetic background, etc.) potentially contributing to phenotypic heterogeneity, we first validated the parental relationship between pure form and complicated form HSP and found no significant differences in consanguinity (Fig. 2B). Subsequently, a comparison of gender distribution between the two groups indicated that female SPG76 patients were significantly more likely to present with complicated HSP than male patients (P = 0.015, Fig. 2C). Given that slowly progressive paraparesis is the prototypical characteristic of HSP, we then evaluated age at onset and disease duration. However, this analysis revealed no significant differences in the disease course between pure form and complicated form HSP (Fig. 2D,E). We investigated the relationship between genotype and phenotype. No significant differences were found in the ratio of truncating mutations (i.e., nonsense, frameshift, and splicing site mutations) versus missense mutations between the pure and complicated groups (Fig. 2F).
Discussion
In the current study, HTS analysis of 240 HSP families identified six additional autosomal‐recessive HSP families, with eight patients carrying six homozygous or compound heterozygous variants of CAPN1. Among them, the three homozygous variants (c.182_183insC (p.V64Gfs*103) in Families 1 and 4, c.759+1G>A in Family 3, and c.853C>T (p.R285*) in Family 5) were found to be known truncating mutations, 6 , 11 , 21 while the three compound heterozygous variants ((c.1442G>A (p.R481Q) in Family 6, c.1493C>T (p.P498L) in Families 2 and 6, and c.1852C> T (p.R618W) in Family 2) were defined as novel, likely‐pathogenic missense mutations. A combined total of 33 rare CAPN1 mutations were detected between our cohort and previously reported cases. 5 , 6 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 Notably, seven mutations (c.182_183insC, c.759+1G>A, c.853C>T, c.1015C>T, c.1142C>T, c.1176G>A, and c.1534C>T) were shared in ≥2 pedigrees, and in particular the nonsense mutation c.1176G>A (p.W392*) was most prevalent, recorded in four pedigrees.
In agreement with previous reports of HSP, the clinical features of the CAPN1‐associated SPG76 patients also showed inter‐ and intra‐family variability. 5 , 15 , 18 Comprehensive analysis of all available clinical information for our cohort combined with that available in previous studies showed that lower limb spasticity, manifesting as stiffness, hyperreflexia, and Babinski signs, developed in about 94% of patients, followed by ataxia, dysarthria, and cerebellar atrophy. 5 , 6 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 Meanwhile, the clinical overlap is extensive between complicated form HSP and HSP mimic diseases (e.g., adrenoleukodystrophy, spinocerebellar ataxia type 3, etc.). 30 , 31 Remarkably, the variability and fluidity of the ataxia‐spasticity disease spectrum suggest that spastic paraplegia and ataxia share not only overlapping phenotypes and causal genes, but also disease‐specific underlying mechanisms (e.g., common cellular pathways or regulatory networks, etc.). 9 This dilemma presents a highly challenging obstacle for correct clinical diagnoses of HSP. Moreover, an increasing suite of HSP‐causative genes, including PNPLA6 (SPG39), KIF1C (SPG58), CAPN1 (SPG76), ATP13A2 (SPG78), and UCHL1 (SPG79), have been associated with hereditary ataxia. 9 , 24
To clarify the causes of phenotypic heterogeneity in CAPN1‐associated SPG76 disease, we performed a comprehensive analysis of the parental kinship, gender, age at onset, disease duration, and mutation type between the pure and complicated HSP forms. Clinically, we speculated that mutations of CAPN1 are more likely to lead to complicated HSP with cerebellar ataxia, especially for female SPG76 patients, rather than pure HSP. In addition, the consanguinity and disease course potentially do not substantially contribute to differences in clinical characteristics. Genetically, all mutations have been scattered along the conserved functional domains of calpain‐1. Moreover, both truncating and missense mutations can be accompanied by either pure or complicated forms of HSP. Thus, genotype does not ostensibly share a causative relationship with specific SPG76 phenotypes. However, on further review of the calpain‐1linker structure, we speculated that mutations in this location were more likely to cause pure form HSP than mutations in the above functional domains. Additionally, this heterogeneous SPG76 phenotypic spectrum could also rationally be accounted for by as‐yet‐unknown genetic modifiers or interacting proteins of CAPN1. 27 , 32
In conclusion, our study extends the mutation spectra and clinical profiles of CAPN1‐associated SPG76 disease, stressing that CAPN1 screening is warranted in both pure and complicated forms of HSP. We demonstrated that the factors of gender and calpain‐1 linker structure may potentially contribute to SPG76 phenotype, although the extent of this contribution remains to be investigated. Further study of a large patient cohort and the use of patient‐specific models (e.g., induced pluripotent stem cells), 33 will facilitate the elucidation of mechanisms underlying this characteristic phenotypic heterogeneity and variability for CAPN1‐associated SPG76, as well as for other disease‐causing genes in both as pure or ataxic HSP.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Authors’ Contributions
X.L. and W.‐J.C. carried out the study concept and design. X.L., L.‐L.L., Y.‐J.C., and Y.‐L.L. wrote the initial manuscript and constructed the figures and tables. Y.‐J.C., Y.‐L.L., X.‐H.L., M.‐W.W., and E.‐L.D. involved in acquisition of data and analysis. X.L., W.‐J.C., and N.W. contributed to the critical revision of the manuscript, figures, and tables. All authors have read and approved the final manuscript.
Supporting information
Supplemental File S1. Tables for the primers of Sanger sequencing, and the information of novel mutations in CAPN1.
Supplemental File S2. Figures for the distribution of HSP in our cohort, and the Sanger tracing of mutations in CAPN1.
Acknowledgments
The authors thank the patients and their relatives for participation. This work was supported by grants from the National Natural Science Foundation of China (81771230, U1905210, and 81801130), the Natural Science Foundation of Fujian Province (2019J05076), the Joint Funds for the Innovation of Science and Technology of Fujian Province (2017Y9094 and 2018Y9082), the grant 2017XQ1072 and 2018QH2033 from Startup Fund for scientific research of Fujian Medical University, the Key Clinical Specialty Discipline Construction Program of Fujian.
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81771230, 81801130, and U1905210; Joint Funds for the Innovation of Science and Technology of Fujian Province grants 2017Y9094 and 2018Y9082; Key Clinical Specialty Discipline Construction Program of Fujian grant ; Natural Science Foundation of Fujian Province grant 2019J05076; Startup Fund for scientific research of Fujian Medical University grants 2017XQ1072 and 2018QH2033.
Contributor Information
Wan‐Jin Chen, Email: wanjinchen75@fjmu.edu.cn.
Xiang Lin, Email: linxiang1988@fjmu.edu.cn.
References
- 1. Fink JK, Heiman‐Patterson T, Bird T, et al. Hereditary spastic paraplegia: advances in genetic research. Hereditary Spastic Paraplegia Working group. Neurology 1996;46:1507–1514. 10.1212/wnl.46.6.1507. [DOI] [PubMed] [Google Scholar]
- 2. Fink JK. Hereditary spastic paraplegia: clinico‐pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013;126(3):307–328. 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shribman S, Reid E, Crosby AH, et al. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol 2019;18:1136–1146. 10.1016/S1474-4422(19)30235-2. [DOI] [PubMed] [Google Scholar]
- 4. Vaz FM, McDermott JH, Alders M, et al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain 2019;142:3382–3397. 10.1093/brain/awz291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gan‐Or Z, Bouslam N, Birouk N, et al. Mutations in CAPN1 cause autosomal‐recessive hereditary spastic paraplegia. Am J Hum Genet 2016;98:1038–1046. 10.1016/j.ajhg.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Y, Hersheson J, Lopez D, et al. Defects in the CAPN1 gene result in alterations in cerebellar development and cerebellar ataxia in mice and humans. Cell Rep 2016;16:79–91. 10.1016/j.celrep.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorimachi H, Suzuki K. The structure of calpain. J Biochem 2001;129:653–664. 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- 8. Cotti Piccinelli S, Bassi MT, Citterio A, et al. A Novel CAPN1 mutation causes a pure hereditary spastic paraplegia in an Italian family. Front Neurol 2019;10:580 10.3389/fneur.2019.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Synofzik M, Schüle R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov Disord 2017;32:332–345. 10.1002/mds.26944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forman OP, De Risio L, Mellersh CS. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS One 2013;8:e64627 10.1371/journal.pone.0064627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tadic V, Klein C, Hinrichs F, et al. CAPN1 mutations are associated with a syndrome of combined spasticity and ataxia. J Neurol 2017;264:1008–1010. 10.1007/s00415-017-8464-5. [DOI] [PubMed] [Google Scholar]
- 12. Travaglini L, Bellacchio E, Aiello C, et al. Expanding the clinical phenotype of CAPN1‐associated mutations: A new case with congenital‐onset pure spastic paraplegia. J Neurol Sci 2017;378:210–212. 10.1016/j.jns.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 13. Kocoglu C, Gundogdu A, Kocaman G, et al. Homozygous CAPN1 mutations causing a spastic‐ataxia phenotype in 2 families. Neurol Genet 2018;4:e218 10.1212/NXG.0000000000000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lambe J, Monaghan B, Munteanu T, Redmond J. CAPN1 mutations broadening the hereditary spastic paraplegia/spinocerebellar ataxia phenotype. Pract Neurol 2018;18:369–372. 10.1136/practneurol-2017-001842. [DOI] [PubMed] [Google Scholar]
- 15. Melo US, Freua F, Lynch DS, et al. Clinical aspects of hereditary spastic paraplegia 76 and novel CAPN1 mutations. Clin Genet 2018;94:482–483. 10.1111/cge.13428. [DOI] [PubMed] [Google Scholar]
- 16. Shetty A, Gan‐Or Z, Ashtiani S, et al. CAPN1 mutations: Expanding the CAPN1‐related phenotype: From hereditary spastic paraparesis to spastic ataxia. Eur J Med Genet 2019;62:103605 10.1016/j.ejmg.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 17. Souza PVS, Silva LHL, Badia BML, et al. SPG76: An extremely rare hereditary spastic paraplegia with a new expanding complicated phenotype. Rev Neurol (Paris) 2019;175:572–574. 10.1016/j.neurol.2019.01.397. [DOI] [PubMed] [Google Scholar]
- 18. Chen Y, Cen Z, Zheng X, et al. A novel homozygous CAPN1 pathogenic variant in a Chinese patient with pure hereditary spastic paraplegia. J Clin Neurol 2019;15:271–272. 10.3988/jcn.2019.15.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng F, Sun YM, Quan C, et al. Two novel homozygous mutations of CAPN1 in Chinese patients with hereditary spastic paraplegia and literatures review. Orphanet J Rare Dis 2019;14:83 10.1186/s13023-019-1053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim A, Kumar KR, Davis RL, et al. Increased diagnostic yield of spastic paraplegia with or without cerebellar ataxia through whole‐genome sequencing. Cerebellum 2019;18:781–790. 10.1007/s12311-019-01038-0. [DOI] [PubMed] [Google Scholar]
- 21. Wei Q, Dong HL, Pan LY, et al. Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl Neurodegener 2019;8:19 10.1186/s40035-019-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia‐Berlanga JE, Moscovich M, Palacios IJ, et al. CAPN1 variants as cause of hereditary spastic paraplegia type 76. Case Rep Neurol Med 2019;2019:7615605 10.1155/2019/7615605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia ZC, Liu ZH, Zhou XX, et al. Mutation analysis of CAPN1 in Chinese populations with spastic paraplegia and related neurodegenerative diseases. J Neurol Sci 2020;411:116691 10.1016/j.jns.2020.116691. [DOI] [PubMed] [Google Scholar]
- 24. Rahimi Bidgoli MM, Javanparast L, Rohani M, et al. CAPN1 and hereditary spastic paraplegia: a novel variant in an Iranian family and overview of the genotype‐phenotype correlation. Int J Neurosci 2020;1–13. 10.1080/00207454.2020.1763344. [DOI] [PubMed] [Google Scholar]
- 25. Dong EL, Wang C, Wu S, et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol Neurodegener 2018;13:36 10.1186/s13024-018-0269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin X, Su HZ, Dong EL, et al. Stop‐gain mutations in UBAP1 cause pure autosomal‐dominant spastic paraplegia. Brain 2019;142(8):2238–2252. 10.1093/brain/awz158. [DOI] [PubMed] [Google Scholar]
- 27. Zhao M, Chen YJ, Wang MW, et al. Genetic and clinical profile of chinese patients with autosomal dominant spastic paraplegia. Mol Diagn Ther 2019;23:781–789. 10.1007/s40291-019-00426-w. [DOI] [PubMed] [Google Scholar]
- 28. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet 2011;12:363–376. 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen YJ, Wang MW, Dong EL, et al. Chinese patients with adrenoleukodystrophy and Zellweger spectrum disorder presenting with hereditary spastic paraplegia. Parkinsonism Relat Disord 2019;65:256–260. 10.1016/j.parkreldis.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 31. Gan SR, Zhao K, Wu ZY, et al. Chinese patients with Machado‐Joseph disease presenting with complicated hereditary spastic paraplegia. Eur J Neurol 2009;16:953–956. 10.1111/j.1468-1331.2009.02639.x. [DOI] [PubMed] [Google Scholar]
- 32. Newton T, Allison R, Edgar JR, et al. Mechanistic basis of an epistatic interaction reducing age at onset in hereditary spastic paraplegia. Brain 2018;141:1286–1299. 10.1093/brain/awy034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu YQ, Dong EL, Yang WQ, et al. Generation of an integration‐free induced pluripotent stem cell line, FJMUi001‐A, from a hereditary spastic paraplegia patient carrying compound heterozygous p. P498L and p.R618W mutations in CAPN1 (SPG76). Stem Cell Res 2019;34:101354 10.1016/j.scr.2018.11.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File S1. Tables for the primers of Sanger sequencing, and the information of novel mutations in CAPN1.
Supplemental File S2. Figures for the distribution of HSP in our cohort, and the Sanger tracing of mutations in CAPN1.