

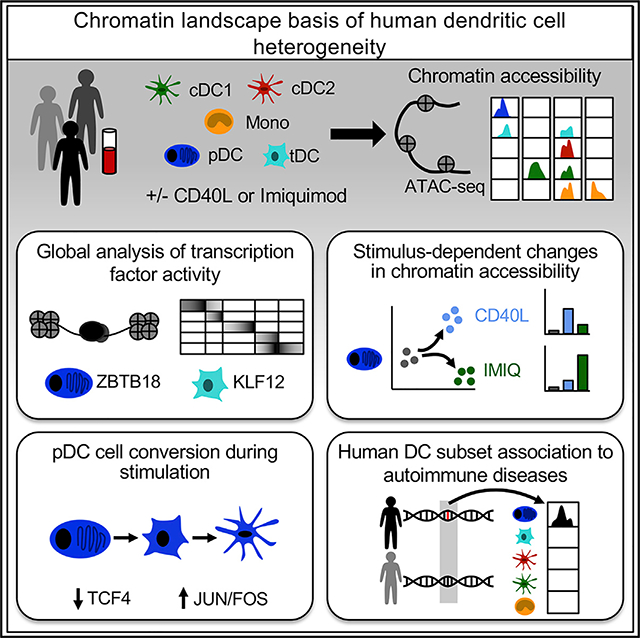
SUMMARY
Human dendritic cells (DCs) comprise subsets with distinct phenotypic and functional characteristics, but the transcriptional programs that dictate their identity remain elusive. Here, we analyze global chromatin accessibility profiles across resting and stimulated human DC subsets by means of the assay for transposase-accessible chromatin using sequencing (ATAC-seq). We uncover specific regions of chromatin accessibility for each subset and transcriptional regulators of DC function. By comparing plasmacytoid DC responses to IFN-I-producing and non-IFN-I-producing conditions, we identify genetic programs related to their function. Finally, by intersecting chromatin accessibility with genome-wide association studies, we recognize DC subset-specific enrichment of heritability in autoimmune diseases. Our results unravel the basis of human DC subset heterogeneity and provide a framework for their analysis in disease pathogenesis.
In Brief
Human dendritic cells (DCs) orchestrate immune responses by a division of labor between functionally specialized subsets; however, the transcriptional basis of this heterogeneity is poorly understood. Using ATAC-seq, Leylek et al. profile the chromatin landscape of human DC subsets, providing insight into the underlying regulatory mechanisms that modulate their function.
Graphical Abstract
INTRODUCTION
Dendritic cells (DCs) play pivotal roles in the activation of a wide range of immune responses, which are mediated through a division of labor among functionally specialized subsets. Functions within each DC subset are enabled by programs coordinated by the precise interactions of transcription factors (TFs) binding genomic sites to control gene expression. Tight regulation of these programs is essential to promote appropriate responses against infection and cancer while avoiding autoimmunity. Although several transcriptional programs dictating mouse DC subset development and function have been described, transcriptional regulation of human DC subsets remains elusive. Furthermore, we lack a comprehensive and unbiased view of the global chromatin landscape of human DCs. Revealing chromatin landscapes of primary human DCs in health is ultimately essential to pinpoint altered programs in disease.
The human DC network is composed of two major subsets, classical DCs (cDCs) and plasmacytoid DCs (pDCs) (Guilliams et al., 2014; Merad et al., 2013). cDCs can be further divided into cDC1 and cDC2, specialized in the activation of CD8+ and CD4+ T cells, respectively. pDCs are known for their capacity to produce large amounts of type I interferon (IFN-I) in response to viral infection followed by their conversion into cDC-like cells (Abbas et al., 2020; Leylek and Idoyaga, 2019; Reizis, 2019). Advances in molecular profiling allowed the characterization of human blood DCs using single-cell RNA sequencing (scRNA-seq) (See et al., 2017; Villani et al., 2017). This approach was powerful to discern previously unappreciated heterogeneity of human DCs and lead to the identification of transitional DCs (tDCs, also known as AXL+ DCs or ASDCs), an evolutionarily conserved DC population that shares transcriptomic and proteomic features with pDCs and cDCs (Alcántara-Hernández et al., 2017; Leylek et al., 2019). However, RNA-seq captures protein-coding regions that account for <3% of the genome (ENCODE Project Consortium, 2012). RNA expression is often preceded by changes in the chromatin accessibility at gene promoters and other distal regulatory elements such as enhancers. Thus, understanding human DC transcriptional regulation requires an evaluation of the chromatin landscape, which can be measured in high resolution with the assay for transposase-accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al., 2013). ATAC-seq identifies genome-wide accessible regulatory regions (cis-elements) and can infer the activity of TFs (Buenrostro et al., 2013, 2015; Schep et al., 2017). The integration of ATAC-seq data with genome-wide association studies (GWAS) of immune cell-mediated diseases also allows the identification of cell-specific enrichment of disease-causing heritability traits. ATAC-seq has been used to characterize the chromatin landscape of major mouse and human immune lineages (Calderon et al., 2019; Corces et al., 2016; Granja et al., 2019; Satpathy et al., 2019; Yoshida et al., 2019); however, a detailed study of primary human DC subsets, some of which represent <0.1% of peripheral blood mononuclear cells (PBMCs), is lacking. Furthermore, the global chromatin landscape changes that occur during DC activation are unknown. These limitations pose a barrier to dissecting the transcriptional mechanisms of DC dysfunction in disease and designing DC-based therapeutics.
Here, we applied ATAC-seq to measure chromatin accessibility in primary DC subsets in resting and activated states. We correlated the chromatin landscape with TF activity, RNA expression, protein expression, and the function of each DC subset. This approach allowed us to unmask features of the chromatin landscape associated with cell subsets or activation states and discover previously undescribed TFs that regulate DC function. By integrating these data with autoimmune disease GWAS, we found evidence for candidate single-nucleotide polymorphisms (SNPs) in a pDC-specific enhancer that explained trait heritability in systemic sclerosis. Our data constitute a comprehensive analysis of the epigenomic profiles among functionally distinct but closely related human DC subsets and provide a valuable resource for future comparisons of these cells in different tissues in health and disease.
RESULTS
Approach to Analyze the Chromatin Accessibility Landscape of Primary Human DC Subsets
We set out to generate a DC map of chromatin accessibility by performing ATAC-seq on purified primary human subsets (i.e., cDC1, cDC2 and pDCs) obtained from 7 healthy male and female donors. For comparison, we included CD14+ monocytes in our analysis (Figures 1A and S1A). cDC2s were purified as CD14− BDCA1high to exclude most CD32− inflammatory DC3 (Figure S1A; Dutertre et al., 2019; Villani et al., 2017). Our ATAC-seq data were of high quality (i.e., the samples had a median transcription start site [TSS] enrichment score of 23.7, and replicates were highly reproducible) (Figures S1B and S1C). After filtering the data, we identified 94,328 genome-wide ATAC-seq peaks (cis-elements) (p < 0.01). Analysis of these cis-elements by principal-component analysis (PCA) showed that each DC subset has a distinct chromatin signature (Figure 1B). Cell type made up >97% of the variance between samples, while sex and individual differences made minimal contributions (Figure S1D).
Figure 1. Analysis Workflow of Primary Human DC Chromatin Accessibility Profiles.

(A) Left: experimental workflow. Human myeloid populations were sorted from peripheral blood of 7 healthy adult donors and analyzed by ATAC-seq. Technical replicates were analyzed when not limited by cell number. Right: post-sort purity. The numbers indicate the frequency of parent gate. See Figure S1A for the full gating strategy.
(B) PCA based on ATAC-seq signal in all cis-elements. Each point represents 1 sample.
(C) Genome tracks from 1 representative donor showing signal near known subset-specific genes. The bottom bar represents the gene and the arrow indicates the start codon. The gray highlights indicate differentially accessible cis-elements.
(D) Top: heatmap of subset-specific cis-elements (fold change [FC] > 5 and adjusted p value [p-adj] < 0.05 in all pairwise comparisons). Color indicates Z score of ATAC-seq signal. Bottom: number of subset-specific cis-elements.
(E) Left: scatterplots comparing ATAC-seq signal (read counts) between cDC2 and other subsets. Each point represents 1 cis-element. The colored points indicate differentially accessible cis-elements (FC > 5 and p-adj < 0.05). The dark gray points indicate shared cis-elements (FC < 2 and average count > 10). Right: heatmap of cis-elements shared between cDC2 and other subsets. The color indicates ATAC-seq signal Z scores. Bottom: overlap of cDC2-specific differentially accessible cis-elements in each pairwise comparison.
(F) Genome tracks for select shared cis-elements from (E).
Open chromatin at known DC subset-specific genes validated our analysis (Figure 1C). pDCs had higher accessibility at cis-elements neighboring CLEC4C and GZMB, genes that encode the surface marker BDCA2 and granzyme B, respectively. cDC1 showed higher accessibility within CADM1 encoding CADM1/NECL2 and SNX22, a sorting nexin expressed by mouse and human cDC1 (Brähler et al., 2018; Villani et al., 2017). We found that cDC2 and monocytes shared higher accessibility at CD1C and ITGAX, which encode BDCA1 and CD11c, respectively. Monocytes had higher accessibility at cis-elements neighboring CD14 and the macrophage-inducible C-type lectin CLEC4E.
We next unbiasedly queried cis-elements accessible in each subset (Figure 1D). Of the 94,328 cis-elements, >7,000 were only accessible in pDCs. Similarly, 2,901 and 2,050 were solely accessible in cDC1 and monocytes, respectively. However, only 26 cis-elements were uniquely accessible in cDC2, suggesting that the chromatin landscape of cDC2 is shared with other DCs. The regions that were more accessible in cDC2 were also more accessible in either pDCs, cDC1, or monocytes (Figures 1E and S1E). For instance, cDC2 and pDCs shared 909 cis-elements, including regions near DENND1B and ALCAM, genes involved in endocytosis and leukocyte adhesion, respectively (Figure 1F). Similarly, cDC2 and cDC1 shared 3,463 cis-elements neighboring genes associated with the induction of T cell responses (e.g., CCR7, CD59). Finally, cDC2 had a greater degree of overlap with monocytes, sharing accessibility at 6,134 cis-elements such as CFP and CD58, 2 myeloid cell activation genes. The lack of cis-elements unique to cDC2 may reflect further heterogeneity in our purified population. This could arise from current limitations in the ability to distinguish cDC2 from DC3, given their continuum of phenotypes (Dutertre et al., 2019). Of note, the 26 cDC2-specific cis-elements surrounded genes of unknown function (e.g., the long non-coding RNA LINC007000 and lymphocyte expansion molecule LEXM) (see Table S1 for the complete list). Collectively, our approach allows analysis of chromatin accessibility in primary human DCs.
ATAC-Seq Uncovers a pDC-Specific TF, ZBTB18
We leveraged ATAC-seq to infer the activity of 870 human TFs using chromVAR, which calculates a TF activity score based on the enrichment of known binding motifs within cis-elements (Schep et al., 2017). PCA and hierarchical clustering based on TF activity showed that pDCs and monocytes were quite distinct, whereas cDC1 and cDC2 were similar to each other (Figures 2A and 2B). We next analyzed the activity of TFs known to be involved in the development and function of mouse DC subsets (Figure 2C). pDCs showed higher activity scores for TCF4 and RUNX2, as expected (Cisse et al., 2008; Sawai et al., 2013). Similarly, cDC2 showed higher activity for IRF4 and SPI1 (Anderson et al., 2000; Suzuki et al., 2004), whereas monocytes showed higher activity for FLI1, KLF4, and CEBP family members (Zhu et al., 2016). Finally, we found that the cDC1 TF BATF3 (Hildner et al., 2008) appeared more active in human monocytes than cDC1, which may be due to similarities between the binding motifs of BATF3 and other TFs of the AP-1 family (Friedman, 2007).
Figure 2. ATAC-Seq Reveals an Undescribed Transcriptional Regulator in pDCs.

(A) PCA based on TF activity scores (TF score) calculated by chromVAR.
(B) Heatmap of top 200 most variable TFs (columns) across subsets (rows). The color indicates scaled TF score.
(C) PCA as in (A) colored by scaled TF score.
(D) Chromatin accessibility around the IRF8, TCF4, CEBPA, and KLF4 loci. The tracks are from 1 representative donor. The TCF4 ChIP-seq track (Ceribelli et al., 2016) is shown for IRF8 and TCF4.
(E) pDC-specific TFs identified by chromVAR that also demonstrate higher mRNA expression in pDCs. The x axis represents the mean mRNA expression in pDCs measured by scRNA-seq (Villani et al., 2017). The bars are colored by pDC specificity compared to other DC subsets (Z score).
(F) Genome tracks of ZBTB18 locus from 1 representative donor showing transcript variant 1.
(G) ZBTB18 HINT-ATAC footprint from genome-wide binding sites. The data are pooled from all of the samples for each subset.
(H) ZBTB18 transcript variant 1 expression measured by RT-PCR, n = 3–5 in 2–4 independent experiments. The statistics are determined by 1-way ANOVA with Dunnett’s multiple comparisons test.
Bar graphs show means ± SDs. *p < 0.05 and **p < 0.01. See also Figure S2.
Although TF expression can be shared between DC subsets, mouse studies have shown that each subset can use distinct enhancers. For instance, both pDCs and cDC1 express Irf8, which regulates the function and development of these cells, respectively (Sichien et al., 2016). However, mouse pDCs use the +41-kb Irf8 enhancer, whereas cDC1 absolutely require the +32-kb Irf8 enhancer for their development (Bagadia et al., 2019; Durai et al., 2019). Similarly, human pDCs showed higher accessibility of the +58-kb enhancer and cDC1 showed higher accessibility of the +49-kb enhancer, which are equivalent to the mouse counterparts (Figure 2D; Bagadia et al., 2019; Grajales-Reyes et al., 2015). We also observed 2 cis-elements unique to pDCs located near +54 kb, which have not been reported in mice. Chromatin immunoprecipitation sequencing (ChIP-seq) data (Ceribelli et al., 2016) showed that the pDC lineage-defining TF TCF4 binds to these 2 newly described enhancers, suggesting they may drive IRF8 expression specifically in human pDCs.
We further investigated enhancers of other lineage-defining TFs (Figure 2D). We found that all DC subsets exhibited open regions at the TCF4 locus, including the TSS; however, only pDCs had accessibility at the +570-kb TCF4 enhancer, a binding site necessary for the TCF4 positive feedback loop that drives pDC development in mice (Grajkowska et al., 2017). We further observed higher accessibility of the +42-kb CEBPA enhancer in monocytes, equivalent to the +37-kb Cebpa enhancer described in mice (Cooper et al., 2015). Finally, we analyzed the KLF4 locus and found that cDC2 and monocytes shared accessibility at +25 kb, a predicted enhancer site (Fishilevich et al., 2017). Thus, our data enable identification of human DC subset-specific cis-elements around lineage-defining TFs.
We then explored TFs with high activity in pDCs to identify undescribed regulators. One limitation of ATAC-seq is its difficulty in distinguishing between TFs that share similar binding motifs. Therefore, we considered only TFs with high activity that were also specifically expressed in pDCs at the RNA level (Figures 2E and S2A). TCF4 and RUNX2 were highly expressed in pDCs, correlating with their higher activity. TGIF2 was not specific, being expressed in pDCs and other DC subsets. TCF3, which shares binding motifs with TCF4, was highly and specifically expressed in pDCs; however, mouse experiments have shown that this TF is dispensable for pDC development and function (Cisse et al., 2008). Lastly, ZBTB18 (ZNF238/RP58/ZFP238), a zinc finger TF known to inhibit ID2 expression in skeletal muscle (Yokoyama et al., 2009), was specifically expressed in pDCs.
We further evaluated ZBTB18 activity in pDCs. Analysis of the cis-elements surrounding ZBTB18 showed a pDC-specific peak at the TSS for one of the transcript variants (Figures 2F and S2B). Also, HINT (HMM-based identification of TF footprints)-ATAC, which displays the “footprint” caused by TF-mediated protection from transposition (Li et al., 2019b), showed changes surrounding ZBTB18 binding sites in pDCs but no other DC subsets (Figure 2G). Moreover, RT-PCR confirmed specific expression of ZBTB18 transcript variant 1 by pDCs (Figure 2H). Of note, ZBTB18 transcript variants 2 and 3 were not differentially expressed between the subsets, suggesting that they do not contribute to the expression levels observed by scRNA-seq (Figure S2C). These data suggest that ZBTB18 may regulate pDC gene expression. To support this hypothesis, we leveraged publicly available data of Zbtb18 silencing in mouse myoblasts and gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005). We found that genes that were downregulated upon Zbtb18 silencing were significantly enriched among human pDCs but no other DC subsets, providing additional evidence of ZBTB18 activity in pDCs (Figures S2D and S2E).
In summary, our analyses unravel TFs that regulate the chromatin landscape of human primary DC subsets. In particular, they allowed the identification of ZBTB18, a previously unrecognized TF that is active and specifically expressed by human pDCs.
Chromatin Landscape and Transcriptional Regulation of tDCs
Recently, we and others described an evolutionarily conserved DC population that shares features with both pDCs and cDCs, which we called tDCs. Given that the transcriptional regulation of tDCs remains poorly understood, we obtained high-quality ATAC-seq profiles using the optimized Omni-ATAC protocol for low cell numbers (Corces et al., 2017; Figures S3A and S3B).
To parallel our previous analyses, we divided tDCs into pDC-like CD11clo tDCs and cDC-like CD11chi tDCs (Alcántara-Hernández et al., 2017; Leylek et al., 2019). As previously observed for RNA and protein analyses (Alcántara-Hernández et al., 2017; Villani et al., 2017), TF activity scores derived from ATAC-seq profiles positioned tDCs intermediate between pDCs and cDC2 by PCA and unsupervised hierarchical clustering (Figures 3A and 3B). TFs with higher activity scores in tDCs versus pDCs tended to have higher scores in cDC2 (Figure 3C). Conversely, TFs with higher activity scores in tDCs versus cDC2 tended to have even higher scores in pDCs (Figure 3D). For example, the pDC TFs TCF4 and RUNX2 clearly showed the transition,i.e, high activity in pDCs, intermediate in tDCs, and low in cDC2 (Figure 3E). On the other hand, the cDC2 TFs CEBPA and FLI1 were low in pDCs, intermediate in tDCs, and higher in cDC2. In all of the cases, there was a directional gradient in TF activity from CD11clo tDCs to CD11chi tDCs. Finally, tDCs displayed activity for both IRF8 and IRF4, in agreement with their protein expression (Leylek et al., 2019). As expected (Alcántara-Hernández et al., 2017; Villani et al., 2017), tDCs did not show a transitional relationship between pDCs and cDC1 (Figure S3C).
Figure 3. Unique TF Profile of tDCs.

(A) PCA based on TF scores calculated by chromVAR.
(B) Heatmap of top 200 most variable TFs (columns) across subsets (rows). The color indicates scaled TF score.
(C) Left: histogram of difference in TF scores between pDCs and CD11chi tDCs. The colored points indicate significantly different TFs (ΔTF score > |0.05| and p-adj < 0.05). Right: boxplots of TF scores for differentially active TFs from indicated comparisons.
(D) Same as (C), but comparing cDC2 and CD11chi tDCs.
(E) Bar graphs of scaled TF scores for indicated TF motifs (n = 4–17 samples per subset).
(F) Left: heatmap of scaled TF scores for tDC-specific TF motifs (ΔTF score > |0.05| and p-adj < 0.05 in all pairwise comparisons; indicated by asterisk) and closely related TFs. Right: heatmap of average TF mRNA expression from scRNA-seq data (Villani et al., 2017).
(G) Genome tracks of KLF12 locus from 1 representative donor.
(H) Left: KLF12 expression in human subsets measured by RT-PCR (n = 2–3 in 2 experiments). Right: Klf12 expression in mouse subsets measured by RNA-seq (n = 2–3) (Lau et al., 2016; Leylek et al., 2019). The statistics are determined by 1-way ANOVA with Dunnett’s multiple comparisons test.
Bar graphs show means ± SDs. **p < 0.01, ***p < 0.001, and ****p < 0.0001. See also Figure S3.
We next focused on TFs that show higher activity in tDCs, i.e., BCL11A, BCL11B, KLF3 and TBX2 (Figure 3F, left, labeled with an asterisk). Due to the challenge of differentiating between TFs with similar binding motifs, we included closely related TFs in our analysis and assessed RNA expression (Figure 3F, right). We found that the TBX family was not expressed in any DC population. BCL11A and BCL11B have identical binding motifs, and both showed higher TF activity in tDCs; however, BCL11B was not expressed in any DC subset, and BCL11A was highly expressed in both pDCs and tDCs. Lastly, all 3 KLF family members were expressed in tDCs, but KLF12 expression was highest in tDCs. Analysis of cis-elements around the KLF12 locus confirmed the presence of an intronic region uniquely accessible in tDCs (Figure 3G). Furthermore, tDCs from both humans and mice expressed at least 2-fold more KLF12 mRNA than the other populations (Figure 3H). Finally, genes that were downregulated in Klf12−/− mouse natural killer (NK) cells were significantly enriched among genes expressed in tDCs, providing further evidence for KLF12 activity in tDCs (Figures S3D and S3E).
In conclusion, the transcription factor profile of tDCs is intermediate between pDCs and cDC2, corroborating their transitional features. We nevertheless identified KLF12, a TF that is uniquely active and expressed in human and mouse tDCs, suggesting it may play a role in regulating their development or function.
pDCs Undergo Large-Scale Stimulus-Dependent Chromatin Changes
We next aimed to identify regulatory elements that control functional changes during DC activation. For this, we used pDCs as an example, given their potential for multiple functional outcomes (Alculumbre et al., 2018; Swiecki and Colonna, 2015). pDCs are known for their capacity to produce IFN-I upon viral stimulation (Reizis, 2019). Also, pDCs can convert into cDC-like cells by remodeling their morphology, upregulating co-stimulatory markers, increasing antigen presentation, and decreasing IFN-I production (Leylek and Idoyaga, 2019). However, the chromatin dynamics underlying these 2 distinct functional outcomes are unclear. Thus, we performed a comprehensive analysis of chromatin changes across resting and stimulated bona fide pDCs (purified to be free of tDCs; Figure S3A). We compared a stimulus that promotes IFN-I secretion (i.e., the TLR7 agonist imiquimod [IMIQ]) and a stimulus that promotes pDC activation without IFN-I secretion (i.e., CD40L) (Figures 4A and S4A–S4C). As expected, both stimuli induced human leukocyte antigen (HLA)-DR (major histocompatibility complex II [MHC-II]) and CD80 protein upregulation, corresponding with increased chromatin accessibility around HLA-DRA, CD80, and CD83 (Figures 4B and 4C). However, only IMIQ induced IFN-I secretion, which corresponded to greater chromatin accessibility surrounding IFN-stimulated genes such as IFITM2.
Figure 4. Analysis of Chromatin Landscapes Reveals Alternative pDC Cell States following Stimulation.

(A) Experimental workflow for analysis of freshly isolated (day 0) and stimulated pDCs. Bona fide pDCs (AXL−) were sorted and analyzed immediately (day 0) or stimulated in vitro for 2 days, followed by re-sorting live cells for ATAC-seq analysis (see Figures S3A and S4A for gating strategy).
(B) Left: protein levels of HLA-DR and CD80 in freshly isolated (day 0) or 2-day stimulated bona fide pDCs measured by flow cytometry (n = 3–9 in 3–8 experiments). The statistics are determined by Kruskal-Wallis with Dunn’s multiple comparisons test. Right: IFN-α measured by ELISA in culture supernatant after 2 days (n = 5 in 5 experiments). The statistics are determined by Mann-Whitney test.
(C) Genome tracks from 1 representative donor.
(D) Left: PCA based on ATAC-seq signal in all cis-elements. Each point represents 1 sample (n = 3–4 per condition). Right: scatterplot comparing all cis-elements between CD40L- and IMIQ-stimulated pDCs. The colored points indicate significantly differentially accessible cis-elements (FC > 2 and p-adj < 0.05).
(E) Heatmap of scaled ATAC-seq signal in cis-elements identified in (D).
(F) Genome tracks from 1 representative donor.
(G) Left: PCA of sorted bona fide pDCs analyzed by CyTOF, including three time points (days 0, 2, and 6) and conditions (media alone, CD40L, IMIQ) subsampled and merged. The color indicates the branch cluster determined by Wishbone (n = 1 representative of 2 experiments). Center: percentage of pDCs in each Wishbone branch at day 6. Right: PCA colored by expression of select markers.
(H) Top Gene Ontology terms enriched in CD40L and IMIQ differentially accessible cis-elements. The bubble size represents the fold enrichment. The color indicates −log10 false discovery rate (FDR).
(I) Cytokines in culture supernatant of 2-day stimulated pDCs (n = 5 in 5 experiments). The statistics are determined by t test.
(J) Left: frequency of Ki67+ cells among fresh (day 0) or 2-day stimulated pDCs. Right: number of CellTrace Violet low (CTVlo) cells among fresh, 2-, or 6-day stimulated pDCs (n = 4–11 in 2–7 experiments). The statistics are determined by t test.
(K) MLR using fresh or 2-day stimulated pDCs (DC:T cell ratio 1:20, n = 3–4 donors in 3 experiments). The statistics are determined by 1-way ANOVA against day 0 or t test.
Bar graphs shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4 and Table S2.
Comparison of the chromatin accessibility changes between the 2 stimuli showed that the primary axis of variation distinguished resting and stimulated pDCs (Figure 4D, left). Nevertheless, we found 2,502 and 8,182 regions that were more accessible after CD40L and IMIQ stimulation, respectively (Figures 4D, right, and 4E). These regions neighbored several immune genes—for example, CADM1, CD5, and CCR7 for IMIQ and CD2 for CD40L (Figures 4E and 4F). To determine whether changes in chromatin accessibility correlated with differences in protein expression, we profiled stimulated bona fide pDCs by CyTOF (see Table S2 for the antibody cocktail). In accordance with the ATAC-seq data, IMIQ and CD40L stimulation induced different cellular phenotypes identified by 2 major differentiation arms of the Wishbone trajectory analysis (Setty et al., 2016; Figure 4G). Paralleling the ATAC-seq analysis, CD2 protein expression was higher in branch 2 corresponding to CD40L-stimulated cells, whereas CADM1 and CCR7 protein expression was higher in branch 3 corresponding to IMIQ-stimulated cells.
Next, we used the Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al., 2010) to identify significantly enriched functional pathways among IMIQ- and CD40L-remodeled chromatin landscapes (Figure 4H). IMIQ-stimulated pDCs gained more accessibility around genes involved in cytokine biosynthesis, which correlated with greater IFN-I, tumor necrosis factor α (TNF-α), and interleukin-6 (IL-6) production (Figures 4B and 4I). Also, IMIQ-stimulated pDCs had increased accessibility around proliferation genes, which functionally correlated with higher Ki67 expression and increased cell division during culture (Figure 4J). Finally, IMIQ-stimulated pDCs gained greater accessibility around genes related to lymphocyte activation. To functionally confirm this pathway, we compared the capacity of IMIQ- and CD40L-stimulated pDCs to activate T cells in a mixed leukocyte reaction (MLR) (Figures 4K, S4D, and S4E). While both stimuli increased the capacity of pDC to promote T cell proliferation, IMIQ-stimulated pDCs biased responses toward IFN-γ-producing T cells, whereas CD40L-stimulated pDCs biased responses toward regulatory T cells (Tregs) (Figure 4K).
Our analysis resolves the chromatin dynamics of pDCs stimulated under IFN-I-producing and non-IFN-I-producing conditions, providing evidence of distinct pathways of differentiation that result in alternative cell states and functionality.
CD40L-Stimulated pDCs Share Chromatin Landscape with tDCs and cDCs
We observed that stimulated bona fide pDCs cluster nearest to tDCs using unbiased global analysis (Figure S5). Using a modified GSEA, we confirmed that both CD40L and IMIQ stimulation promoted chromatin remodeling that correlated with the chromatin landscape of tDCs and cDCs (Figure 5A). This analysis also revealed a higher correlation between CD40L-stimulated pDCs and CD11chi tDCs. Thus, pDCs can undergo large-scale chromatin remodeling to primarily resemble tDCs, especially during CD40L stimulation.
Figure 5. CD40L-Stimulated pDCs Share Chromatin Accessibility Landscape with tDCs and cDCs.

(A) Modified GSEA to test for enrichment of DC subset chromatin signatures among CD40L- or IMIQ-stimulated pDCs (see Quantification and Statistical Analysis). The bubble size represents the Spearman’s rank correlation coefficient. The color indicates the normalized enrichment score (NES).
(B) Top Gene Ontology terms enriched in CD40L-stimulated pDCs compared to freshly isolated (day 0) pDCs. The terms are colored and ranked by −log10 FDR. The bubble size represents the term fold enrichment.
(C) Left: histogram of difference in TF scores between CD40L-stimulated pDCs and day 0 pDCs. The significantly different TF motifs (ΔTF score > |0.08| and p-adj < 0.05) are colored. Right: boxplots of TF scores.
(D) Heatmap of differentially active TFs from (C). The color indicates the scaled TF score for each subset.
(E) HINT-ATAC footprint plots for indicated TFs. The data are pooled from 3–4 donors per condition.
(F) Bar graphs of scaled scores for select TFs from (D) (n = 4–17 samples per cell type).
(G) Top: frequency of pDCs expressing high levels of TCF4 protein measured by flow cytometry (n = 13–17 in 10–14 experiments). Bottom: ZBTB18 transcript variant 1 expression measured by RT-PCR (n = 3–4 in 3–4 experiments). The statistics are determined by t test.
(H) Top: representative histogram of ID2 mRNA expression in 2-day CD40L-stimulated pDCs measured by PrimeFlow. The unfilled histogram represents the control. Bottom: frequency of ID2+ pDCs (n = 2 in 2 experiments).
(I) Scatterplot comparing changes between CD40L stimulation and TCF4 silencing. x axis: FC of mRNA expression between TCF4 and control small hairpin RNA (shRNA) conditions in the pDC cell line CAL-1 (microarray) (Ceribelli et al., 2016). y axis: ΔTF score between freshly isolated (day 0) and CD40L-stimulated pDCs (ATAC-seq). Shown are TFs that were significantly different in both analyses.
Bar graphs show means ± SDs. *p < 0.05 and ****p < 0.0001. See also Figure S5.
GREAT analysis of biological processes showed that, similar to tDCs and cDCs, CD40L-stimulated pDCs had greater accessibility near genes related to myeloid cell differentiation, T cell activation, and cytokine secretion (Figure 5B), which is in line with previous evidence that CD40L-stimulated bona fide pDCs can acquire cDC-like functions (Alcántara-Hernández et al., 2017). We then used chromVAR to profile the changes in TF activity between CD40L-stimulated and freshly isolated (day 0) pDCs (Figures 5C and 5D). TFs that were less active in CD40L-stimulated pDCs tended to be less active in tDCs and cDCs. Conversely, TFs that were more active in CD40L-stimulated pDCs tended to have higher activity in tDCs and cDCs. Among these, we found a marked decrease in the activity of TCF4 during CD40L stimulation, which was also less active in tDCs and cDCs (Figures 5E and 5F). However, we found a marked increase in the activity of TFs from the JUN and FOS families (i.e., TFs that can regulate myeloid cell differentiation) (Liebermann et al., 1998) in CD40L-stimulated pDCs, tDCs, and cDCs. Of note, only CD40L-stimulated pDCs showed a marked increase in the activity of TFs from the nuclear factor κB (NF-κB) family, suggesting that these have minimal activity in resting DC subsets (Figure 5D).
TCF4 is known to control the expression of several pDC-specific genes while repressing cDC hallmark genes such as ID2, thereby blocking cDC differentiation (Ghosh et al., 2010; Grajkowska et al., 2017). Accordingly, we found that CD40L-stimulated bona fide pDCs had lower TCF4 protein expression, which corresponded to higher ID2 expression (Figures 5G and 5H). ZBTB18, which has also been described as repressing ID2, was similarly less active and had a lower expression in CD40L-stimulated pDCs (Figures 5E–5G). We then asked which other stimulation-induced changes in TF activity could be attributed to the loss of TCF4. We compared changes in TF activity upon CD40L stimulation to changes in TF expression upon TCF4 silencing (Figure 5I; Ceribelli et al., 2016). We found that several members of the JUN and FOS families were upregulated upon both TCF4 silencing and CD40L stimulation, suggesting that TCF4 may repress their expression directly or indirectly.
Our chromatin landscape analysis shows that CD40L is able to promote bona fide pDC conversion into tDC- and cDC-like cells, and that this dynamic process is likely tightly regulated by TCF4, as previously suggested in the mouse (Ghosh et al., 2010). Our data further suggest that ZBTB18 may contribute to this process.
pDC Conversion into cDC-like Cells Follows a Linear Trajectory
Our bulk analysis could not dissect whether CD40L-mediated chromatin remodeling was a homogeneous process for all pDCs or whether there was heterogeneity within the pDC population. Thus, we correlated our ATAC-seq observations with phenotypic cell conversion at the single-cell level by analyzing fresh (day 0), 2-, or 6-day CD40L-stimulated bona fide pDCs using CyTOF (mass cytometry). Wanderlust analysis (Bendall et al., 2014), which predicts the time and order of phenotypic changes, showed that the trajectory of cell conversion correlated with TCF4 downregulation, as suggested by the ATAC-seq data (Figure 6A). During stimulation, pDCs downregulated classic markers such as BDCA2, and upregulated several cDC markers (e.g., CD33, HLA-DR, CD172a, CD11c) (Figures 6B and 6C).
Figure 6. Single-Cell Trajectory of pDC Cell State Conversion during Stimulation.

(A) Wanderlust trajectory of fresh (day 0), 2-, or 6-day CD40L-stimulated bona fide pDCs analyzed by CyTOF; each point represents 1 cell (1 experiment of 3–4).
(B) Normalized expression of pDC and cDC markers plotted along Wanderlust trajectory axis.
(C) As in (A), but colored by expression of key markers.
(D) Statistical Scaffold maps of CyTOF data from fresh (day 0), 2-, or 6-day CD40L-stimulated pDCs (1 representative donor).
(E) Summary graph of frequency of pDCs mapped to each landmark node (n = 2–3 donors in 3–4 experiments).
(F) Protein expression in fresh (day 0), 2-, or 6-day CD40L-stimulated bona fide pDCs analyzed by flow cytometry and CyTOF (n = 3–18 donors in 3–16 experiments). Statistics determined by Kruskal-Wallis with Dunn’s multiple comparisons test.
(G) Functional analysis of pDCs that mapped to each landmark node. Two-day CD40L-stimulated pDCs were re-sorted based on phenotype. Left: IFN-α in culture supernatant after 24 h CpG-A, measured by ELISA. Right: T cell proliferation in MLR (n = 2–3 donors in 2–3 experiments).
Bar graphs show means ± SDs. **p < 0.01, ***p < 0.001, and ****p < 0.0001. See also Figure S6 and Table S2.
Next, we aligned the phenotype of CD40L-stimulated bona fide pDCs to that of tDCs and cDCs using Scaffold (Spitzer et al., 2015). Scaffold generates a reference map that facilitates comparison across conditions by connecting single cells to landmarks based on phenotypic similarity. We used prior knowledge to denote the location of pDC (blue), tDC (cyan), cDC1 (green), and cDC2 (red) landmarks in the map (Figures S6A and S6B). Corroborating our purification strategy, ~99% of freshly isolated bona fide pDCs localized in the pDC landmark at day 0 (Figures 6D and 6E). In accordance with the ATAC-seq analysis, we found that ~15% of pDCs mapped to the tDC landmark after 2 days of culture with CD40L. These cells expressed several tDC markers, including CD5, BDCA3, CD11c, CD33, CX3CR1, and AXL (Figures 6F and S6C). The appearance of AXL+ cells in the culture was not attributable to cell proliferation (Figure S6D). By day 6, we observed that a fraction of cells positioned in the cDC2 landmark (Figures 6D and 6E). Considering that cultured pDCs mapped to the tDC landmark by day 2, but only mapped to the cDC2 landmark at day 6, our data suggest that tDCs are a transitional cell state during pDC-to-cDC conversion.
To test whether CD40L-mediated phenotypic changes correlated with functional changes, we re-sorted cells that mapped to the pDC, tDC, or cDC2 landmarks and analyzed their function (see Figure S6E for the phenotype of purified cells). Cells that mapped to the pDC landmark retained the capacity to produce IFN-α (Figure 6G). Conversely, cells that mapped to the cDC2 landmark lost IFN-α production potential and acquired strong T cell activation capacity. Finally, cells that mapped to the tDC landmark produced very little IFN-α and induced strong T cell proliferation, which corresponds to the previously described functional capabilities of tDCs (Leylek et al., 2019).
We conclude that bona fide pDCs follow a linear trajectory of cell conversion to cDC-like cells during CD40L stimulation, passing through a stage that resembles circulating tDCs. There is heterogeneity in the pDC response to stimulation at the single-cell level, such that not all pDCs proceed through the cell-conversion process simultaneously. Finally, our data are in line with a recent report showing that mouse pDCs can acquire tDC- and cDC-like gene signatures after viral infection in vivo (Abbas et al., 2020).
GWAS Associates Systemic Sclerosis Genetic Risk Variants with pDCs
Dissecting the molecular mechanisms behind autoimmune disease genetic risk variants requires pinpointing disease-relevant cell types. However, nearly 90% of these variants lie in non-coding regions (Farh et al., 2015). Our global chromatin analysis allowed us to ask whether genetic variation in coding and non-coding regions confers the risk of autoimmune diseases in a DC subset-dependent manner.
We used a publicly available database for autoimmune and non-immune (control) disorders and calculated the enrichment of disease-related SNPs in DC subsets using the Chromatin Element Enrichment Ranking by Specificity (CHEERS) algorithm (Farh et al., 2015; Soskic et al., 2019; Figures 7A and S7A). The majority of the significant autoimmune disease associations were found with cis-elements that opened upon stimulation. For instance, a SNP that confers the risk of Crohn’s disease was located in a stimulation-responsive enhancer region within STAT3, a negative regulator of DC activation (Figure S7B; Melillo et al., 2010). Thus, stimulation-responsive chromatin regions can explain significant trait heritability in DCs.
Figure 7. ATAC-Seq Identifies Regulatory Regions Overlapping Autoimmune Disease-Related SNPs.

(A) Enrichment for autoimmune disease-associated SNPs performed by CHEERS. Color indicates p value, asterisk indicates p < 0.05. See Figure S7 for the complete list.
(B) Left: genome track of the IRF8 locus showing 1 representative donor. Right: genome track of the +58-kb IRF8 enhancer showing pDCs, major immune cell lineages (Calderon et al., 2019), and TCF4 ChIP-seq data (Ceribelli et al., 2016). Bottom panel shows the schematic of SNP positions in relation to TCF4 binding sites (E-boxes).
We then focused on identifying disease-related SNPs in cis-elements for resting DCs. Resting pDCs had significant enrichment for risk variants associated with systemic sclerosis (Figure 7A). Two variants, rs12445476 and rs11642873, were located within the +58-kb pDC-specific IRF8 enhancer (Figure 7B). This enhancer was highly accessible in resting pDCs, but not in cDCs, monocytes, or stimulated pDCs. Furthermore, this enhancer was not accessible in B cells, T cells, or NK cells, demonstrating pDC specificity. Notably, rs12445476 and rs11642873 were adjacent to six E-boxes, which is in line with previous reports showing that the majority of SNPs are found near but not within TF binding sites (Bagadia et al., 2019; Farh et al., 2015). Since IRF8 regulates pDC function (Sichien et al., 2016), it is possible that these SNPs contribute to the pathogenesis of systemic sclerosis by dysregulating IRF8 expression in pDCs. Our approach shows the potential of this dataset to infer disease mechanisms that involve alterations in DC chromatin regulatory regions.
DISCUSSION
Here, we analyzed the chromatin landscape that provides the basis of primary human DC heterogeneity by unraveling the repertoire of accessible cis-elements in each subset. We inferred previously undescribed TFs that may underlie pDC and tDC development or function and exposed the dynamic activity of lineage-specific and stimulus-dependent TFs governing the outcome of stimulated pDCs. Finally, by connecting the chromatin landscape of each DC subset to disease-causing SNPs, we identified genetic variants that contribute to the risk of autoimmune diseases such as the human pDC-specific +58-kb IRF8 enhancer in systemic sclerosis.
Nearly all of the regions accessible in cDC2 were shared with ≥1 cell types. We propose that the lack of a cDC2-specific signature reflects the predominance of a generalized and shared myeloid program. Alternatively, it is possible that lack of a cDC2 signature is indicative of cDC2 heterogeneity, which has recently been highlighted in several publications (Alcántara-Hernández et al., 2017; Dutertre et al., 2019; Villani et al., 2017). Considering this heterogeneity among cDC2, further investigation of their transcriptional regulation may benefit from advances in single-cell ATAC-seq approaches (Satpathy et al., 2019).
The development, identity, and function of pDCs is dependent on the master regulator TCF4 (Cisse et al., 2008). TCF4 antagonizes ID2, and thus is critical in regulating pDC versus cDC differentiation (Grajkowska et al., 2017). Similar to TCF4, the herein-identified pDC-specific TF ZBTB18 is known to inhibit ID2 expression during muscle cell development (Yokoyama et al., 2009). Thus, it is reasonable to hypothesize that ZBTB18 cooperates with TCF4 to repress ID2 during pDC development and differentiation. A recent chromatin-accessibility study noted the specific activity of Zbtb18 in murine pDCs (Li et al., 2019b), suggesting it is conserved in both species. Future loss-of-function experiments will aim to evaluate the role of this undescribed TF on pDC function.
A feature of bona fide pDCs (AXL−) is their capacity to convert into cDC-like cells (Alcántara-Hernández et al., 2017). We showed that pDC conversion occurs efficiently during CD40L stimulation and is associated with the loss of TCF4 and ZBTB18 TF activity and the gain of ID2 expression. We also observed the increased activity of JUN and FOS families, which form AP-1 heterodimers (Shaulian and Karin, 2002). TCF4 silencing in the absence of stimulation was sufficient to induce the expression of AP-1 members, suggesting that AP-1 activity is repressed by TCF4 directly or indirectly, and may be necessary for pDC conversion into cDC-like cells. Finally, we showed that bona fide pDC conversion into cDC-like cells can pass through a stage that resembles circulating tDCs.
The recent identification of tDCs has raised questions about their origin and function. Our analysis of TF activity based on chromatin accessibility complemented previous high-dimensional protein and transcriptomic approaches demonstrating the transitional nature of these cells in relation to pDCs and cDC2 (Alcántara-Hernández et al., 2017; Leylek et al., 2019; Villani et al., 2017). Given their phenotypic and functional overlap with other DC subsets, it remains unclear whether tDCs have a distinct function within the immune system. Nevertheless, we identified KLF12 as a TF expressed in both human and mouse tDCs. Although KLF12 has not been described in myeloid development or function, other KLF family members have been described. KLF4 drives monocyte differentiation and is required for cDC2 function (Feinberg et al., 2007; Tussiwand et al., 2015). KLF12 is most closely related to KLF3 and KLF8, which act as transcriptional repressors (Ilsley et al., 2017). Within the immune compartment, Klf12 is required for NK cell proliferation (Lam et al., 2019). Further study is needed to determine the function of KLF12 in tDCs.
Our analysis enabled identification of disease-associated SNPs that lie in the regulatory regions of human DC subsets. We observed most of the significant autoimmune trait heritability within accessible regions of stimulated but not resting DCs, as observed for other immune cells (Calderon et al., 2019), suggesting an important role for DC activation in autoimmune dysregulation. Nevertheless, we did identify disease SNPs associated with resting cells, such as systemic sclerosis-associated SNPs rs12445476 and rs11642873 within the pDC-specific +58-kb IRF8 enhancer. The IRF8 locus was previously linked to systemic sclerosis (Arismendi et al., 2015; Gorlova et al., 2011; Terao et al., 2013). Similarly, pDCs have been associated with systemic sclerosis in human patients and mouse models (Ah Kioon et al., 2018). However, the molecular cause of pDC dysregulation is not fully understood. It is possible that mutations in the +58-kb IRF8 enhancer may alter IRF8 expression specifically in pDCs, and consequently, their function in systemic sclerosis.
This dataset provides insights into the transcriptional regulation that underpins the heterogeneity of primary human DCs and a resource for understanding human DC differentiation, plasticity, and function. This dataset enables connecting human DC subsets to disease pathogenesis and, consequently, provides an avenue for DC-based therapeutic design.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Juliana Idoyaga (jidoyaga@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
ATAC-seq data generated in this study are deposited in NCBI Gene Expression Omnibus (GEO: GSE146896). Previously published data analyzed in this study include human dendritic cell scRNA-seq data (Villani et al., 2017; https://singlecell.broadinstitute.org/single_cell/study/SCP43/atlas-of-human-blood-dendritic-cells-and-monocytes#study-download), mouse dendritic cell RNA-seq data (Lau et al., 2016; GEO: GSE76132), Zbtb18 silencing data (Yokoyama et al., 2009; GEO: GSE12993), Klf12−/− mouse RNA-seq data (Lam et al., 2019; GEO: GSE128962), TCF4 ChIP-seq data (Ceribelli et al., 2016; GEO: GSE76147), TCF4 silencing data (Ceribelli et al., 2016; GEO: GSE75650), and ATAC-seq of human major immune cell lineages (Calderon et al., 2019; GEO: GSE118189).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Blood was obtained with informed consent from healthy adults in accordance with the Research and Laboratory Environmental Health and Safety program of Stanford University and Institutional Review Board (IRB) protocols approved by the Stanford University Administrative Panel on Human Subjects in Medical Research. Males and females were equally represented. Our analysis revealed that sex differences did not contribute to the variation observed (see Figure S1D), thus it was not a major focus of this work. Blood donors were healthy, without acute diseases and between 20–45 years old.
METHOD DETAILS
Isolation of Human PBMCs
50 mL of blood from healthy adults were collected using EDTA-coated tubes (BD Biosciences). PBMCs were isolated by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). Cells were washed with PBS, counted, and immediately processed for sorting.
Cell Sorting
PBMCs were incubated with human gamma-globulin (Invitrogen) to block non-specific binding for 15 minutes on ice. Myeloid cells were negatively enriched using mAbs against CD3 (OKT3), CD19 (HIB19), CD335 (9E2), and CD66b (G10F5) followed by anti-mouse magnetic beads (Dynabeads, Thermo Fisher Scientific) at a concentration of 4 beads per target cell. For experiments in which tDCs were sorted, cells were negatively enriched using mAbs against CD3, CD19, CD335, and CD14 (HCD14). After enrichment, cells were stained with sorting antibody cocktail for 20 minutes at 4°C. Cells were sorted to > 98% purity using a FACSAria II or FACSAria Fusion cell sorter (BD Biosciences). See Figures S1A, S3A, and S4A for sort gating strategies. For phenotypic experiments, pDCs were enriched from PBMCs using the Human Plasmacytoid Dendritic Cell Isolation Kit II (Miltenyi Biotec), then sorted as BDCA4+AXL−.
ATAC-seq
Immediately after sorting, cells were processed as described (Buenrostro et al., 2015). Briefly, 50,000–60,000 sorted cells were spun down, washed with PBS, and lysed. Technical replicates were performed when not limited by cell numbers. The transposition reaction was performed with Tn5 transposase from the Illumina Nextera DNA Library Preparation Kit or produced as described (Picelli et al., 2014), for 30 min at 37°C. Reactions were scaled down for samples with less than 50,000 cells. Transposed DNA was purified (QIAGEN MinElute PCR Purification Kit) and stored at −20°C. Once 3–4 donors were accumulated, samples were PCR amplified. To reduce GC and size bias, the optimal number of PCR cycles was determined for each sample via qPCR according to the protocol. Samples were barcoded using published primers (Buenrostro et al., 2013). Library quality and quantification was assessed with an Agilent Bioanalyzer at the Stanford Protein and Nucleic Acid Facility.
For subsequent analysis of tDCs and stimulated pDCs, cells were processed according to the Omni-ATAC protocol as described (Corces et al., 2017). Reaction volumes for 5,000–15,000 cells were scaled down from the protocol for 50,000 cells.
Sequencing
Barcoded sample libraries were pooled for a final concentration of 4 nM. Libraries were first run on low depth MiSeq for quality assessment. Subsequent sequencing was performed on Illumina NextSeq 500 (2×75bp or 2×50bp) at the Stanford Functional Genomics Facility. NextSeq was selected to avoid problems with index swapping, which can occur in the presence of excess primers (Larsson et al., 2018). Sequencing was performed in three batches, which constituted two runs with the initial pDC, cDC1, cDC2, and CD14+ analysis, and a later analysis of pDCs, tDCs, and stimulated pDCs.
Staining cell suspensions for flow cytometry
Antibodies (Abs) for flow cytometry were purchased from Biolegend, R&D, MBL International Corp., and Thermo Fisher Scientific. Anti-TCF4 (Abcam) was labeled using the Alexa 647 Labeling Kit (Thermo Fisher Scientific) following manufacturer’s instructions or detected using anti-Rabbit-Alexa 647 (Jackson ImmunoResearch). Cells were acquired on a 5-laser LSRFortessa X-20 (BD Biosciences), and data analyzed using FlowJo software (Tree Star, Inc). Compensation was set up using compensation beads (BD Biosciences). PBMCs were incubated with human gamma-globulin to block non-specific binding for 15 minutes on ice. Cells were incubated with Ab mixes in human FACS buffer (2mM EDTA, 2% Donor equine serum in PBS) for 20 minutes at room temperature (RT). For transcription factors and cytokine detection, cells were stained with LIVE/DEAD Fixable Blue (Thermo Fisher Scientific) for 5 minutes at RT for detection of dead cells, then fixed using FoxP3 Transcription Factor Fix/Perm Buffer (Thermo Fisher Scientific) for 1 hour and stained intracellularly for 20 minutes in 1X Permwash buffer (Thermo Fisher Scientific). After intracellular stain, cells were stained for remaining markers for 20 minutes in human FACS buffer. For PrimeFlow, freshly isolated or 2-day stimulated sorted bona fide human pDCs (AXL−) were plated with 2×106 mouse splenocytes to provide a cellular bed, and stained according to manufacturer’s instructions (Thermo Fisher Scientific). Human cells were identified by gating mouse CD45− human CD123+ cells. ID2 target probes were purchased from Thermo Fisher Scientific. For a negative control, cells were processed identically and stained without target probes. To track pDC proliferation, PBMCs were resuspended at a concentration of 20×106 cells/mL and labeled with 2.5 μM CellTrace Violet (Thermo Fisher Scientific) for 10 minutes at 37°C prior to sort.
Staining cell suspensions for CyTOF
Metal-labeled Abs were obtained from Fluidigm or labeled using the MaxPar X8 labeling kit (Fluidigm) according to manufacturer’s instructions (see Table S2). For mass cytometry analysis of stimulated pDC experiments, bona fide pDCs (AXL−) were obtained by sorting as in Figure S3A. Freshly isolated and cultured pDCs were pooled with 3×106 mouse splenocytes to provide a cellular bed, stained for CyTOF, and identified as human CD45+ mouse CD45− cells. Cells were stained with 1 mL of 0.25 μM cisplatin (Fluidigm) for 5 minutes at room temperature to exclude dead cells. Cells were then washed with CyFACS buffer (2mM EDTA, 1% BSA 1% in PBS) and stained with heavy-metal-labeled Ab cocktail for 30 minutes on ice. Cells were washed twice with CyFACS then fixed with FoxP3 Transcription Factor Fix/Perm Buffer for 2 hours. Human surface Abs that were sensitive to FoxP3 buffer (i.e., CX3CR1, CD123, CD33, CD135, CD172a and CD163), were stained after fixation in 1X Permwash buffer, for 30 minutes at 4°C. After staining, samples were washed and incubated with 2% paraformaldehyde (Electron) in PBS containing 125 nM Iridium intercalator (Fluidigm) overnight. Cells were washed with water, filtered and acquired in a CyTOF2 (Fluidigm).
Cell culture for pDC activation
Prior to cell culture, pDCs were purified by sorting to be free of tDCs (AXL−), following the gating strategy in Figure S3A. 10,000 sorted bona fide pDCs were cultured in 200 μL R10 complete media consisting of RPMI (Corning) with 10% FBS, 2 mM L-glutamine (Corning), 100 IU Penicillin, 100 mg/mL Streptomycin (Corning), 25 mM HEPES (Corning), 1 mM Sodium Pyruvate (Corning), 100 mM MEM Nonessential Amino Acids (Corning) and 55 mM 2-Mercaptoethanol (GIBCO) in 96 well U-bottom plates at 37°C. All conditions included 10 ng/mL recombinant human IL-3 (R&D Systems; carrier-free) for pDC survival. Stimulation conditions were 100 ng/mL CD40L (R&D Systems; carrier-free) or 5 μg/mL Imiquimod (R837, Invitrogen). For ATAC-seq, 4–6 wells were plated per condition. After 2 days, cells were pooled and sorted as FSC-A, SSC-A, Live, Singlets, CD123+ HLA-DR+ (see Figure S4A for sorting strategy). For analysis of IFN-I production in Figure 6G, pDCs were re-sorted into “pDC-like,” “tDC-like,” and “cDC-like” after 2 days of stimulation with CD40L, then stimulated for 24h with 5 μg/mL CpG-A (ODN 2216, Invivogen).
Cytokine detection in culture supernatant
IFNα was detected with the VeriKine Human IFN Alpha Multi-Subtype ELISA Kit (PBL Assay Science). IL-6 and TNFα were detected using the Cytometric Bead Array (CBA) Enhanced Sensitivity Flex Set (BD Biosciences).
Mixed Leukocyte Reactions
PBMCs were washed with PBS, incubated with 1.7 nM CFSE (Sigma-Aldrich) at 37°C in a water bath for 10 minutes, and washed with MACS buffer (2mM EDTA, 2% BSA in PBS). After CFSE labeling, total T cells (CD4+ and CD8+ T cells) were obtained using the Pan T Cell Isolation Kit (Miltenyi Biotec) according to manufacturer’s instructions. Allogeneic T cells were co-cultured with freshly sorted (day 0) bona fide (AXL−) pDCs (1:20 ratio) or bona fide (AXL−) pDCs that were stimulated for 2 days with CD40L or IMIQ. Activated pDCs were washed, re-counted and plated with allogeneic T cells in the presence of fresh stimuli. As a control of homeostatic proliferation, T cells were cultured alone without pDCs in the presence of the corresponding stimuli. The same T cell donor was used to perform the experiments with freshly isolated versus activated pDC. Alternatively, pDCs were re-sorted into “pDC-like,” “tDC-like,” and “cDC-like” after 2 days of stimulation with CD40L, then plated with T cells as above. After 6 days of reaction, 100 ng/mL of PMA (Sigma-Aldrich) and 500 ng/mL of Ionomycin (Sigma-Aldrich) were added to the culture followed by the addition of 10 μg/mL of Brefeldin A (Sigma-Aldrich) and 1:1500 Golgi STOP (BD Biosciences) 1 hr later for a total of 6 hr. Cytokines and FoxP3 were stained after 2 hours of fixation with FoxP3 Transcription Factor Fix/Perm Buffer (ThermoFisher Scientific). Results are expressed as total number of CFSElo T cells or ratio between the numbers of cytokine-producing cells and FoxP3+ CD127low regulatory T cells (Treg).
Quantitative PCR
RNA was extracted from sorted cells with the Nucleospin RNA XS kit (Takara Bio) according to manufacturer’s instructions. Total RNA from each DC subset was reverse transcribed to cDNA using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad Laboratories). cDNA was amplified in a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories) with the iTaq Universal SYBR® Green Supermix (Bio-Rad Laboratories). Expression was calculated following a ΔΔCq method relative to RPL13A and cDC2 and shown as 2−ΔΔCq. Primers were as follows: KLF12 forward CCTTTCCATAGCCAGAGCAG; KLF12 reverse TTGCATCCCTCAAAATCACA; ZBTB18_v1 forward CAGGTTTATGTGTCCTAAAGGTTATG; ZBTB18_v1 reverse CCACCAGAACAGTGCAGTCA; ZBTB18_v2 forward AGCACAGTCAGGTAGCAAAAGT; ZBTB18_v2 reverse GTCCCACAAAACCTACAAAATAGC; ZBTB18_v3 forward GGCCGCTCCGTGTTATGAA; ZBTB18_v3 reverse CCACCAGAACAGTGCAGTCA; RPL13A forward GCCCTACGACAAGAAAAAGCG; RPL13A reverse TACTTCCAGCCAACCTCGTGA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments, including statistical tests and value of n, can be found in figure legends. Significance is depicted as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical tests were two-sided, and were performed with GraphPad Prism 6 unless otherwise indicated. All bar graphs show mean ± SD. PCA and heatmaps were performed and visualized in R with the ggplot2, prcomp, and viridis packages. gMFI indicates geometric mean fluorescence intensity. ΔMFI indicates percent change from control condition.
Initial processing of ATAC-seq data
Processing of raw sequencing data was performed with the ENCODE ATAC-seq Pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline) (ENCODE Project Consortium, 2012) on the Stanford Sherlock computing cluster. A non-overlapping union peak set was defined using a previously described method (Corces et al., 2018). Briefly, peak calling was first performed on each sample individually with MACS2. Next, 500 bp fixed width peaks were defined by identifying the peak summit and extending it by 250 bp on either side. Finally, overlapping peaks were removed through an iterative process. For combined analysis in Figure 3, Figure 5, and Figure 7, fixed width peak sets from both datasets were combined and overlaps were removed. Read counts were calculated with bedtools multicov (Quinlan and Hall, 2010).
Analysis of open chromatin regions
Differential accessibility analysis of peaks was performed with DESeq. Raw counts were normalized with conditional quantile normalization (CQN). A model was built with DESeq using the formula ~CellType + Sex + Batch. Cell-specific peaks in Figure 1 were defined as FC > 5 and p-adj < 0.05. Shared peaks in Figure 1 were defined as FC < |2| and average count > 10. Differential peaks between CD40L and IMIQ in Figure 4 were defined as FC > |2| and p-adj < 0.05. For visualization (PCA, heatmaps), counts were batch corrected using ComBat when applicable (Johnson et al., 2007). In Figure 3A, the PCA was built on one batch, and remaining two batches were overlaid using pca.predict. In Figure 3B, TF scores were averaged across samples for each subset prior to calculating Z-score.
For genome tracks, bigwig files were created from bam files with deeptools and normalized using the CPM method (Ramírez et al., 2016). Genome tracks were explored using the WashU Epigenome browser (Li et al., 2019a). Genome tracks were formatted for publication with SparK (https://www.biorxiv.org/content/10.1101/845529v1; https://github.com/harbourlab/SparK).
To connect observed chromatin accessibility at the IRF8 locus with previously described enhancers, we used UCSC BLAT to find the human hg19 genomic coordinates for the conserved sequences that overlapped the mouse +32kb Irf8 enhancer reported in Grajales-Reyes et al., 2015. These sequences were found between chr16:85981418–85981613, which overlapped the +49kb peak identified here. We also used the human hg19 genomic coordinates representing a conserved sequence that overlapped the mouse +41kb Irf8 enhancer reported in Bagadia et al. (2019). We found that this region (chr16:85991064–85991633) overlapped the +58kb peak identified here.
Enrichment of gene ontology terms was performed on differentially accessible cis-elements with GREAT (McLean et al., 2010) using the hg19 reference genome with whole genome as background and associating genomic regions by “Single nearest gene within 1000kb.” Default global controls were used (Region-based fold enrichment > 2, FDR < 0.05) except that we increased the minimum observed gene hits to 5. Redundant pathways were removed with ReVIGO (Supek et al., 2011). Remaining terms were ranked by p value and the top 8–10 were shown.
To compare chromatin accessibility signatures between stimulated pDCs and resting populations, a modified Gene Set Enrichment Analysis (GSEA) was performed with cis-elements in place of genes. The top 500 cis-elements that were differentially accessible in each DC subset or monocyte compared to Day 0 unstimulated pDCs were used to create signatures (.gmx file), representing the x axis in Figure 5A. To define the pDC signature, pDCs were compared to cDC2. Stimulation profiles (.rnk files) were created by taking the top differentially accessible cis-elements (2000 up and 2000 down) between CD40L-stimulated pDCs and Day 0 unstimulated pDCs or between IMIQ-stimulated pDCs and Day 0 unstimulated pDCs, representing the y axis in Figure 5A. The analysis was performed with GSEA software, which is available through the Broad Institute (https://www.gsea-msigdb.org/gsea/downloads.jsp) (Mootha et al., 2003; Subramanian et al., 2005).
Analysis of transcription factor activity
For transcription factor analysis, chromVAR was used to calculate deviations (TF activity scores, or TF scores) for a curated collection of 870 transcription factor motifs derived from cisBP data “human_pwms_v2” (Schep et al., 2017). t test was used for differential analysis with a Benjamini-Hochberg correction for multiple hypothesis testing. For visualization of chromVAR data (PCA, heatmaps), deviation scores were batch corrected using ComBat where applicable. Where indicated, TF scores were scaled between 0 and 1 by subtracting the minimum, then dividing by the maximum.
Transcription factor footprint plots were produced using HINT-ATAC (Li et al., 2019b). Motif position weight matrices (pwms) were acquired from CisBP, JASPAR, or chromVAR. Data was pooled from all samples for each cell type.
Analysis of autoimmune SNPs
Fine-mapped autoimmune-disease associated SNPs were downloaded from Farh et al. (Farh et al., 2015). We used the CHEERS algorithm to identify enrichment of disease variants in each cell type (Soskic et al., 2019). Briefly, peak counts were averaged for each cell type then normalized with a “reads in peak” normalization. The bottom 10th percentile of peaks was removed, and quantile normalization was performed. Euclidean normalization was performed to obtain a cell type specificity score for each peak in each cell type. Finally, the algorithm identified peaks that overlap SNPs for each disease and calculated an enrichment p value.
RNA-seq and microarray analysis
Human scRNA-seq data was downloaded from https://singlecell.broadinstitute.org/single_cell/study/SCP43/atlas-of-human-blood-dendritic-cells-and-monocytes#study-download as log transformed transcripts per million (TPM). For analysis in Figure 2E, expression from single cells was averaged for each subset. Then, Z-scores were calculated for pDCs, cDC1, cDC2, and CD14+ monocytes. pDC specificity refers to the Z-score in pDCs. Normalized counts for mouse RNA-seq bulk data were downloaded from NCBI GEO (GEO: GSE76132). For Figures S2 and S3, GSEA was performed with Zbtb18 silencing (GEO: GSE12993) and Klf12−/− mice (GEO: GSE128962) datasets, respectively, downloaded from NCBI GEO. The top 500 differentially expressed genes were used to create signatures for loss of Zbtb18 or Klf12. Differentially expressed genes (FC > 1.5 and p-adj < 0.05) between pDCs or tDCs and other DC subsets were identified with the limma package in R. Up to 2000 differentially expressed genes (1000 up and 1000 down) were ranked by log2 fold change and tested for overlap with signatures. For Figure 5I, TCF4 silencing data was downloaded from NCBI GEO (GEO: GSE75650). CAL-1 cells analyzed after 48h treatment with control shRNA or TCF4 shRNA were compared with the limma package in R to determine differentially expressed TFs (FC > |2| and p-adj < 0.05).
CyTOF data analysis
Files in FCS format were normalized using the Nolan Lab’s Normalizer (https://github.com/nolanlab/bead-normalization). For both mouse and human, live, single cells were gated using FlowJo. Human PBMCs were gated as CD3−, CD19−, CD335−, CD66b−, CD14−, CD16− and HLA-DR+. Stimulated pDCs were gated on human CD45+ live cells. Events of interest were imported into CYT and transformed using hyperbolic arcsin (asinh x/5). For Wanderlust and Wishbone analysis, 250–300 events were sampled from each condition. Trajectory analysis was performed using all of the parameters except lineage and setting freshly isolated pDCs as the starting trajectory point.
The Scaffold R package was downloaded from GitHub (https://github.com/nolanlab/scaffold). Events from the stimulated pDCs and the corresponding PBMCs were imported. DC populations were gated and exported from FlowJo. Clustering was performed using all of the parameters except lineage markers.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD11b (clone ICRF44) Nd144 | Fluidigm | Cat# 3144001B; RRID:AB_2714152 |
| Anti-human BDCA2/CD303 (clone 201A) Sm147 | Fluidigm | Cat# 3147009B; RRID:AB_2714153 |
| Anti-human CD16 (clone 3G8) Nd148 | Fluidigm | Cat# 3148004B; RRID:AB_2661791 |
| Anti-human CD127/IL-7R (clone A019D5) Sm149 | Fluidigm | Cat# 3149011B; RRID:AB_2661792 |
| Anti-human CD123/IL3R (clone 6H6) Eu151 | Fluidigm | Cat# 3151001B; RRID:AB_2661794 |
| Anti-human CD163 (clone GHI/61) Sm154 | Fluidigm | Cat# 3154007B; RRID:AB_2661797 |
| Anti-human CD45 (clone HI30) Y89 | Fluidigm | Cat# 3089003; RRID:AB_2661851 |
| Anti-human CD45 purified (clone HI30) | Biolegend | Cat# 304002; RRID:AB_314390 |
| Anti-human CCR7 (clone G043H7) Tb159 | Fluidigm | Cat# 3159003A; RRID:AB_2714155 |
| Anti-human CD14 (clone M5E2) Gd160 | Fluidigm | Cat# 3160001B; RRID:AB_2687634 |
| Anti-human CD11c (clone Bu15) Dy162 | Fluidigm | Cat# 3162005B; RRID:AB_2687635 |
| Anti-human CD117/c-kit purified (clone 104D2) | Biolegend | Cat# 313202; RRID:AB_314981 |
| Anti-human BDCA3/CD141 (clone 1A4) Yb173 | Fluidigm | Cat# 3173002B; RRID:AB_2714156 |
| Anti-human CD32 purified (clone FUN-2) | Biolegend | Cat# 303202; RRID:AB_314334 |
| Anti-human CD335/NKp46 purified (clone 9E2) | Biolegend | Cat# 331902; RRID:AB_1027637 |
| Anti-human BDCA-1/CD1c purified (clone L161) | Biolegend | Cat# 331502; RRID:AB_1088995 |
| Anti-human CD1a (clone HI149) | Biolegend | Cat# 300102; RRID:AB_314016 |
| Anti-human CD172a/b / SIRP alpha purified (clone SESA5) | Biolegend | Cat# 323802; RRID:AB_830701 |
| Anti-human HLADR purified (clone L243) | Biolegend | Cat# 307651; RRID:AB_2562826 |
| Anti-human CD34 purified (clone 561) | Biolegend | Cat# 343602; RRID:AB_1732014 |
| Anti-human CD3 purified (clone UCHT1) | Biolegend | Cat# 300443; RRID:AB_2562808 |
| Anti-human CD115/CSF1R purified (clone 9-4D2-1E4) | Biolegend | Cat# 347302; RRID:AB_2085375 |
| Anti-human CX3CR1 purified (clone K0124E1) | Biolegend | Cat# 355702; RRID:AB_2561726 |
| Anti-human CD116/GMSFR purified (clone 4H1) | Biolegend | Cat# 305902; RRID:AB_314568 |
| Anti-human CLEC9A/ DNGR1 purified (clone 8F9) | Biolegend | Cat# 353802; RRID:AB_10983070 |
| Anti-human CD135/ FLT3 purified (clone BV10A4H2) | Biolegend | Cat# 313302; RRID:AB_314987 |
| Anti-human CD45RA purified (clone HI100) | Biolegend | Cat# 304102; RRID:AB_314406 |
| Anti-human CD33 purified (clone WM53) | Biolegend | Cat# 303402; RRID:AB_314346 |
| Anti-human CD2 purified (clone RPA-2.10) | Biolegend | Cat# 300202; RRID:AB_314026 |
| Anti-human CD81 purified (clone 5A6) | Biolegend | Cat# 349502; RRID:AB_10643417 |
| Anti-human CD5 purified (clone UCHT2) | Biolegend | Cat# 300602; RRID:AB_314088 |
| Anti-human CD66b purified (clone G10F5) | Biolegend | Cat# 305102; RRID:AB_314494 |
| Anti-human CD19 purified (clone HIB19) | Biolegend | Cat# 302202; RRID:AB_314232 |
| Anti-APC (clone APC003) | Biolegend | Cat# 408005; RRID:AB_2563706 |
| Anti-human IRF4 (clone 3E4) | Biolegend | Cat# 646402; RRID:AB_2280462 |
| Anti-human IRF8 APC (clone V3GYWCH) | ThermoFisher Scientific | Cat# 17-9852-82; RRID:AB_2573318 |
| Anti-human Siglec-6/CD327 PE (clone 767329) | R&D Systems | Cat# FAB2859P; RRID:AB_2714157 |
| Anti-human CD100- APC (clone REA316) | Miltenyi Biotec | Cat# 130-104-674; RRID:AB_2654323 |
| Anti-human AXL purified (clone 108724) | R&D Systems | Cat# MAB154; RRID:AB_2062558 |
| Anti-human/mouse CADM1/ SynCAM purified (clone 3.E.1) | MBL Life Science | Cat# CM004-3; RRID:AB_592783 |
| Anti-human CD3 purified (OKT3) | Biolegend | Cat# 317302; RRID:AB_571927 |
| Anti-human CD14 purified (HCD14) | Biolegend | Cat# 325602; RRID:AB_830675 |
| Anti-human AXL Alexa Fluor 488 (clone 108724) | R&D Systems | Cat# FAB154G; RRID:AB_2714170 |
| Anti-human BDCA1/CD1c APC/Cy7 (clone L161) | Biolegend | Cat# 331520; RRID:AB_10644008 |
| Anti-human BDCA3/CD141 PE/Cy7 (clone M80) | Biolegend | Cat# 344110; RRID:AB_2561623 |
| Anti-human BDCA3/CD141 BV785 (clone M80) | Biolegend | Cat# 344116; RRID:AB_2572195 |
| Anti-human BDCA4/ CD304 APC (clone 12C2) | Biolegend | Cat# 354506; RRID:AB_11219600 |
| Anti-human BDCA4/ CD304 PE (clone 12C2) | Biolegend | Cat# 354503; RRID:AB_11219200 |
| Anti-human CD11c Alexa Fluor 700 (clone Bu15) | Biolegend | Cat# 337220; RRID:AB_2561503 |
| Anti-human CD123 FITC (clone 6H6) | Biolegend | Cat# 306014; RRID:AB_2124259 |
| Anti-human CD123 PE (clone 6H6) | Biolegend | Cat# 306006; RRID:AB_314580 |
| Anti-human CD14 APC (clone M5E2) | Biolegend | Cat# 982506; RRID:AB_2650643 |
| Anti-human CD14 BV785 (clone M5E2) | Biolegend | Cat# 301840; RRID:AB_2563425 |
| Anti-human CD14 650 (clone M5E2) | Biolegend | Cat# 301836; RRID:AB_2563799 |
| Anti-human CD16 BV650 (clone 3G8) | Biolegend | Cat# 302042; RRID:AB_2563801 |
| Anti-human CD19 PerCP/Cy5.5 (clone HIB19) | Biolegend | Cat# 302230; RRID:AB_2073119 |
| Anti-human CD20 PerCP/Cy5.5 (clone 2H7) | Biolegend | Cat# 302325; RRID:AB_893285 |
| Anti-human CD3 PerCP/Cy5.5 (clone UCHT1) | Biolegend | Cat# 300430; RRID:AB_893299 |
| Anti-human CD335 PerCP/Cy5.5 (clone 9E2) | Biolegend | Cat# 331920; RRID:AB_2561665 |
| Anti-human CD66b PerCP/Cy5.5 (clone G10F5) | Biolegend | Cat# 305108; RRID:AB_2077855 |
| Anti-human CD80 BV421 (clone 2D10) | Biolegend | Cat# 305222; RRID:AB_2564407 |
| Anti-human HLADR BV605 (clone L243) | Biolegend | Cat# 307640; RRID:AB_2561913 |
| Anti-human/mouse TCF4/E2-2 purified (clone NCI-R159-6) | Abcam | Cat# ab217668; RRID:AB_2714172 |
| Anti-human Ki67 PerCP/Cy5.5 (clone Ki-67) | Biolegend | Cat# 350520; RRID:AB_2562295 |
| Anti-human CD3 PE/Cy7 (clone UCTH1) | Biolegend | Cat# 300419; RRID:AB_439780 |
| Anti-human CD4 BV785 (clone RPA-T4) | Biolegend | Cat# 300554; RRID:AB_2564382 |
| Anti-human CD8 APC/Cy7 (clone RPA-T8) | Biolegend | Cat# 301016; RRID:AB_314134 |
| Anti-human IFNγ Alexa Fluor 700 (clone B27) | Biolegend | Cat# 506516; RRID:AB_961351 |
| Anti-human FOXP3 APC (clone PCH101) | ThermoFisher Scientific | Cat# 17-4776-41; RRID:AB_1603281 |
| Anti-human CD33 BV650 (clone WM53) | Biolegend | Cat# 303430; RRID:AB_2650934 |
| Anti-human CD5 BUV737 (clone UCHT2) | BD Biosciences | Cat# 564452; RRID:AB_2714177 |
| Biological Samples | ||
| Whole blood from healthy donors | Obtained from donors with informed consent. IRB approved by Stanford University Research Compliance Office. | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ficoll-Paque PLUS | GE Healthcare | Cat# 300-25 |
| 1M Tris-HCl, pH 7.4 | VWR | Cat# 100216-458 |
| NaCl | ThermoFisher Scientific | Cat# AM9760G |
| MgCl2 | ThermoFisher Scientific | Cat# AM9530G |
| Tween-20 | Millipore-Sigma | Cat# 11332465001 |
| Digitonin | VWR | Cat# PAG9441 |
| Nonidet P40 Substitute | Millipore-Sigma | Cat# 11332473001 |
| IGEPAL CA-630 | Millipore-Sigma | Cat# 18896 |
| Tn5 transposase | Produced as described in Picelli et al., 2014 | N/A |
| SYBR Green I Nucleic Acid Gel | ThermoFisher Scientific | Cat# S7563 |
| Dulbecco’s Phosphate Buffered Saline | Corning | Cat# 21-031-CV |
| Fetal Bovine Serum, qualified, US origin | GIBCO | Cat# 26140079 |
| RPMI 1640 with L- Glutamine | Corning | Cat# 10040CV |
| L-glutamine Solution | Corning | Cat# 25005CI |
| Sodium Pyruvate Solution | Corning | Cat# 25000CI |
| Penicillin-Streptomycin | Corning | Cat# 30002CI |
| HEPES solution | Corning | Cat# 25060CI |
| MEM Nonessential Amino Acid Solution | Corning | Cat# 25025CI |
| Recombinant Human IL-3 | R&D Systems | Cat# 203IL010CF |
| EDTA 0.5M pH 8.0 | Corning | Cat# 46034CI |
| ACK Lysis Buffer | Lonza | Cat# 10-548E |
| Benzonase Nuclease | Millipore-Sigma | Cat# E1014-25KU |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192A |
| Cell-ID Cisplatin | Fluidigm | Cat# 201064 |
| 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester | Millipore-Sigma | Cat# 21888-25MG-F |
| CellTrace Violet Cell Proliferation Kit | ThermoFisher Scientific | Cat# C34557 |
| Bovine Serum Albumin solution 30% ± 2% in 0.85% sodium chloride, aseptically filled | Millipore-Sigma | Cat# A7284-50ML |
| Dimethyl sulfoxide > 95% | Millipore-Sigma | Cat# D4540 |
| Paraformaldehyde 16% aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| Indium 113 metal chloride | Trace Sciences International | In-113 |
| Indium 115 metal chloride | Trace Sciences International | In-115 |
| CpG-A ODN 2216 | Invivogen | Cat# tlrl-2216-1 |
| Imiquimod | Invivogen | Cat# tlrl-imqs |
| Recombinant Human CD40 Ligand | R&D Systems | Cat# 6420CL025CF |
| Critical Commercial Assays | ||
| Foxp3 / Transcription Factor Fixation/Permeabilization Concentrate and Diluent | ThermoFisher Scientific | Cat# 00-5521-00 |
| Permeabilization Buffer (10X) | ThermoFisher Scientific | Cat# 00-8333-56 |
| Maxpar X8 Multimetal Labeling Kit Fluidigm | Fluidigm | Cat# 201300 |
| LIVE/DEAD Fixable Dead Cell Stain Sampler Kit | ThermoFisher Scientific | Cat# L34960 |
| Dynabeads Pan Mouse IgG | ThermoFisher Scientific | Cat# 11042 |
| Plasmacytoid Dendritic Cell Isolation Kit II, human | MACS, Miltenyi Biotec | Cat# 130-097-415 |
| Pan T cell Isolation Kit, human | MACS, Miltenyi Biotec | Cat# 130-096-535 |
| Human IFN Alpha Multi-Subtype ELISA Kit (TCM) | PBL Assay Science | Cat# 41105-1 |
| BD Cytometric Bead Array Human Enhanced Sensitivity Master Buffer Kit | BD Biosciences | Cat# 561521 |
| BD Cytometric Bead Array Human IL-6 Enhanced Sensitivity Flex Set | BD Biosciences | Cat# 561512 |
| BD Cytometric Bead Array Human TNF Enhanced Sensitivity Flex Set | BD Biosciences | Cat# 561516 |
| BD CompBead Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD Biosciences | Cat# 552843 |
| Alexa Fluor 647 Antibody Labeling Kit | ThermoFisher Scientific | Cat# A20186 |
| NucleoSpin RNA XS kit | Takara Bio | Cat# 740902.10 |
| NEBNext High Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541S |
| iScript Reverse Transcription Supermix | Bio-Rad Laboratories | Cat# 1708840 |
| iTaq Universal SYBRGreen Supermix | Bio-Rad Laboratories | Cat# 1725120 |
| QIAGEN MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| Nextera DNA Library Preparation Kit | Illumina | Cat# FC-121-1030 |
| Zymo DNA Clean and Concentrator-5 Kit | Zymo Research Corporation | Cat# D4013 |
| PrimeFlow RNA Assay Kit | ThermoFisher Scientific | Cat# 88-18005-204 |
| ID2 PrimeFlow Probe Set (A488) | ThermoFisher Scientific | Cat# PF210; Assay ID: VA4-3086868-PF |
| Deposited Data | ||
| ATAC-seq data (generated here) | NCBI GEO | GEO: GSE146896 |
| Human DC scRNA-seq | https://singlecell.broadinstitute.org/single_cell/study/atlas-of-human-blood-dendritic-cells-and-monocytes | Broad Single Cell Portal study “Atlas of human blood dendritic cells and monocytes” |
| Mouse DC RNA-seq | NCBI GEO | GEO: GSE76132 |
| Other Immune Lineage ATAC-seq | NCBI GEO | GEO: GSE118189 |
| TCF4 ChIP-seq | NCBI GEO | GEO: GSE76147 |
| TCF4 silencing microarray | NCBI GEO | GEO: GSE75650 |
| Zbtb18 silencing microarray | NCBI GEO | GEO: GSE12993 |
| Klf12−/− mouse RNA-seq | NCBI GEO | GEO: GSE128962 |
| Software and Algorithms | ||
| GraphPad Prism 6 | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| MATLAB | N/A | https://www.mathworks.com/products/matlab.html |
| Cytofkit | N/A | https://bioconductor.riken.jp/packages/3.7/bioc/html/cytofkit.html |
| FlowJo Software v10.0.8 | TreeStar, Inc | https://www.flowjo.com/solutions/flowjo |
| R | N/A | https://www.R-project.org/ |
| ggplot2 v2.2.1 | N/A | https://github.com/tidyverse/ggplot2 |
| viridis v0.3.0 | N/A | https://github.com/sjmgarnier/viridis |
| limma v3.44.3 | N/A | https://bioconductor.org/packages/release/bioc/html/limma.html |
| DESeq2 v1.28.1 | N/A | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| cqn v1.34.0 | N/A | https://www.bioconductor.org/packages/release/bioc/html/cqn.html |
| PVCA v1.28.0 | N/A | https://www.bioconductor.org/packages/release/bioc/html/pvca.html |
| chromVAR v1.10.0 | Schep et al., 2017 | http://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| SparK | N/A | https://github.com/harbourlab/SparK/blob/master/README.md |
| HINT-ATAC | Li et al., 2019b | http://www.regulatory-genomics.org/hint/introduction/ |
| deeptools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| ENCODE-DCC atac-seq pipeline | N/A | https://github.com/ENCODE-DCC/atac-seq-pipeline |
| ENCODE-DCC chip-seq pipeline | N/A | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| bedtools | Quinlan and Hall., 2010 | https://github.com/arq5x/bedtools2 |
| ComBat (sva package) | Johnson et al., 2007 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| CHEERS | Soskic et al., 2019 | https://github.com/trynkaLab/CHEERS |
| GSEA | Mootha et al., 2003; Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/downloads.jsp |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/index.php |
| ReVIGO | Supek et al., 2011 | http://revigo.irb.hr/ |
| Wanderlust | Bendall et al., 2014 | https://dpeerlab.github.io/dpeerlab-website/cyt-download.html |
| Wishbone | Setty et al., 2016 | https://github.com/ManuSetty/wishbone |
| Scaffold | Spitzer et al., 2015 | https://github.com/nolanlab/scaffold |
Highlights.
Mapping the chromatin landscape of primary human DC subsets using ATAC-seq
The transcription factors ZBTB18 and KLF12 are active in pDCs and tDCs, respectively
The chromatin landscape of stimulated pDCs converges toward tDCs and cDC2
DC cis-elements, including the pDC IRF8 enhancer, harbor autoimmune disease SNPs
ACKNOWLEDGMENTS
R. Leylek was the recipient of an NSF GRFP fellowship (no. DGE-1147470). This work was supported by NIH grant 1DP2AR069953, the Baxter and Freidenrich Foundations, and a Stanford WHSDM seed grant awarded to J.I.; RM1-HG007735 and a Scleroderma Research Foundation grant to H.Y.C.; and the Parker Institute for Cancer Immunotherapy and NIH grant K08CA230188 to A.T.S. CyTOF and sorting were performed on Stanford Shared FACS Facility instruments funded by S10OD016318-01, S10RR025518-01, and the Parker Institute for Cancer Immunotherapy. The computational analysis was performed through Stanford Sherlock. We thank the blood donors for their participation, the Idoyaga lab members for technical support and discussions, and Dr. Gottfried for critical reading of the manuscript.
DECLARATION OF INTERESTS
H.Y.C. is affiliated with Accent Therapeutics, Boundless Bio, 10x Genomics, Arsenal Biosciences, and Spring Discovery. A.T.S. is a scientific co-founder of Immunai and receives research funding from Arsenal Biosciences.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108180.
REFERENCES
- Abbas A, Vu Manh T-P, Valente M, Collinet N, Attaf N, Dong C, Naciri K, Chelbi R, Brelurut G, Cervera-Marzal I, et al. (2020). The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection. Nat. Immunol 21, 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, Gordon JK, and Barrat FJ (2018). Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med 10, eaam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara-Hernández M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, Davis MM, Nolan GP, and Idoyaga J (2017). High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 47, 1037–1050.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alculumbre SG, Saint-André V, Di Domizio J, Vargas P, Sirven P, Bost P, Maurin M, Maiuri P, Wery M, Roman MS, et al. (2018). Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat. Immunol 19, 63–75. [DOI] [PubMed] [Google Scholar]
- Anderson KL, Perkin H, Surh CD, Venturini S, Maki RA, and Torbett BE (2000). Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor-derived dendritic cells. J. Immunol 164, 1855–1861. [DOI] [PubMed] [Google Scholar]
- Arismendi M, Giraud M, Ruzehaji N, Dieudé P, Koumakis E, Ruiz B, Airo P, Cusi D, Matucci-Cerinic M, Salvi E, et al. (2015). Identification of NF-κB and PLCL2 as new susceptibility genes and highlights on a potential role of IRF8 through interferon signature modulation in systemic sclerosis. Arthritis Res. Ther 17, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagadia P, Huang X, Liu T-T, Durai V, Grajales-Reyes GE, Nitschké M, Modrusan Z, Granja JM, Satpathy AT, Briseño CG, et al. (2019). An Nfil3-Zeb2-Id2 pathway imposes Irf8 enhancer switching during cDC1 development. Nat. Immunol 20, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brähler S, Zinselmeyer BH, Raju S, Nitschké M, Suleiman H, Saunders BT, Johnson MW, Böhner AMC, Viehmann SF, Theisen DJ, et al. (2018). Opposing Roles of Dendritic Cell Subsets in Experimental GN. J. Am. Soc. Nephrol 29, 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D, Nguyen MLT, Mezger A, Kathiria A, Müller F, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV, et al. (2019). Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet 51, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceribelli M, Hou ZE, Kelly PN, Huang DW, Wright G, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL, Marcucci G, et al. (2016). A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell 30, 764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. (2008). Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Guo H, and Friedman AD (2015). The +37 kb Cebpa Enhancer Is Critical for Cebpa Myeloid Gene Expression and Contains Functional Sites that Bind SCL, GATA2, C/EBPα, PU.1, and Additional Ets Factors. PLOS ONE 10, e0126385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai V, Bagadia P, Granja JM, Satpathy AT, Kulkarni DH, Davidson JT 4th, Wu R, Patel SJ, Iwata A, Liu T-T, et al. (2019). Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol. 20, 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre C-A, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, et al. (2019). Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 51, 573–589.e8. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, et al. (2015). Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg MW, Wara AK, Cao Z, Lebedeva MA, Rosenbauer F, Iwasaki H, Hirai H, Katz JP, Haspel RL, Gray S, et al. (2007). The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J 26, 4138–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AD (2007). Transcriptional control of granulocyte and monocyte development. Oncogene 26, 6816–6828. [DOI] [PubMed] [Google Scholar]
- Ghosh HS, Cisse B, Bunin A, Lewis KL, and Reizis B (2010). Continuous expression of the transcription factor e2–2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 33, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlova O, Martin J-E, Rueda B, Koeleman BPC, Ying J, Teruel M, Diaz-Gallo L-M, Broen JC, Vonk MC, Simeon CP, et al. ; Spanish Scleroderma Group (2011). Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLOS Genet. 7, e1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseño CG, Durai V, Bagadia P, et al. (2015). Batf3 maintains autoactivation of Irf8 for commitment of a CD8α(+) conventional DC clonogenic progenitor. Nat. Immunol 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajkowska LT, Ceribelli M, Lau CM, Warren ME, Tiniakou I, Nakandakari Higa S, Bunin A, Haecker H, Mirny LA, Staudt LM, and Reizis B (2017). Isoform-Specific Expression and Feedback Regulation of E Protein TCF4 Control Dendritic Cell Lineage Specification. Immunity 46, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, et al. (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilsley MD, Gillinder KR, Magor GW, Huang S, Bailey TL, Crossley M, and Perkins AC (2017). Krüppel-like factors compete for promoters and enhancers to fine-tune transcription. Nucleic Acids Res 45, 6572–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Lam VC, Folkersen L, Aguilar OA, and Lanier LL (2019). KLF12 Regulates Mouse NK Cell Proliferation. J. Immunol 203, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson AJM, Stanley G, Sinha R, Weissman IL, and Sandberg R (2018). Computational correction of index switching in multiplexed sequencing libraries. Nat. Methods 15, 305–307. [DOI] [PubMed] [Google Scholar]
- Lau CM, Nish SA, Yogev N, Waisman A, Reiner SL, and Reizis B (2016). Leukemia-associated activating mutation of Flt3 expands dendritic cells and alters T cell responses. J. Exp. Med 213, 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leylek R, and Idoyaga J (2019). The versatile plasmacytoid dendritic cell: function, heterogeneity, and plasticity. Int. Rev. Cell Mol. Biol 349, 177–211. [DOI] [PubMed] [Google Scholar]
- Leylek R, Alcántara-Hernández M, Lanzar Z, Lüdtke A, Perez OA, Reizis B, and Idoyaga J (2019). Integrated Cross-Species Analysis Identifies a Conserved Transitional Dendritic Cell Population. Cell Rep 29, 3736–3750.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Hsu S, Purushotham D, Sears RL, and Wang T (2019a). WashU Epigenome Browser update 2019. Nucleic Acids Res 47 (W1), W158–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019b). Identification of transcription factor binding sites using ATAC-seq. Genome Biol 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebermann DA, Gregory B, and Hoffman B (1998). AP-1 (Fos/Jun) transcription factors in hematopoietic differentiation and apoptosis. Int. J. Oncol 12, 685–700. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, and Schindler C (2010). Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol 184, 2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, and Mortha A (2013). The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B (2019). Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 50, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai CM, Sisirak V, Ghosh HS, Hou EZ, Ceribelli M, Staudt LM, and Reizis B (2013). Transcription factor Runx2 controls the development and migration of plasmacytoid dendritic cells. J. Exp. Med 210, 2151–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See P, Dutertre C-A, Chen J, Günther P, McGovern N, Irac SE, Gunawan M, Beyer M, Händler K, Duan K, et al. (2017). Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356, eaag3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, Choi K, Bendall S, Friedman N, and Pe’er D (2016). Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 34, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol 4, E131–E136. [DOI] [PubMed] [Google Scholar]
- Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, et al. (2016). IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640. [DOI] [PubMed] [Google Scholar]
- Soskic B, Cano-Gamez E, Smyth DJ, Rowan WC, Nakic N, Esparza-Gordillo J, Bossini-Castillo L, Tough DF, Larminie CGC, Bronson PG, et al. (2019). Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nat. Genet 51, 1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Gherardini PF, Fragiadakis GK, Bhattacharya N, Yuan RT, Hotson AN, Finck R, Carmi Y, Zunder ER, Fantl WJ, et al. (2015). IMMUNOLOGY. An interactive reference framework for modeling a dynamic immune system. Science 349, 1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bo_snjak M, _Skunca N, and _Smuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, Yamamoto K, Suematsu T, Nakamura M, Yui K, and Kumatori A (2004). Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha-dendritic cell development. Proc. Natl. Acad. Sci. USA 101, 8981–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, and Colonna M (2015). The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol 15, 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao C, Ohmura K, Kawaguchi Y, Nishimoto T, Kawasaki A, Takehara K, Furukawa H, Kochi Y, Ota Y, Ikari K, et al. (2013). PLD4 as a novel susceptibility gene for systemic sclerosis in a Japanese population. Arthritis Rheum 65, 472–480. [DOI] [PubMed] [Google Scholar]
- Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, et al. (2015). Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42, 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Ito Y, Ueno-Kudoh H, Shimizu H, Uchibe K, Albini S, Mitsuoka K, Miyaki S, Kiso M, Nagai A, et al. (2009). A systems approach reveals that the myogenesis genome network is regulated by the transcriptional repressor RP58. Dev. Cell 17, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. ; Immunological Genome Project (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YP, Thomas GD, and Hedrick CC (2016). 2014 Jeffrey M. Hoeg Award Lecture: Transcriptional Control of Monocyte Development. Arterioscler. Thromb. Vasc. Biol 36, 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAC-seq data generated in this study are deposited in NCBI Gene Expression Omnibus (GEO: GSE146896). Previously published data analyzed in this study include human dendritic cell scRNA-seq data (Villani et al., 2017; https://singlecell.broadinstitute.org/single_cell/study/SCP43/atlas-of-human-blood-dendritic-cells-and-monocytes#study-download), mouse dendritic cell RNA-seq data (Lau et al., 2016; GEO: GSE76132), Zbtb18 silencing data (Yokoyama et al., 2009; GEO: GSE12993), Klf12−/− mouse RNA-seq data (Lam et al., 2019; GEO: GSE128962), TCF4 ChIP-seq data (Ceribelli et al., 2016; GEO: GSE76147), TCF4 silencing data (Ceribelli et al., 2016; GEO: GSE75650), and ATAC-seq of human major immune cell lineages (Calderon et al., 2019; GEO: GSE118189).