

Polymeric nanogel with on-demand release of hirudin prevents and inhibits clot formation.
Abstract
Pathological coagulation, a disorder of blood clotting regulation, induces a number of cardiovascular diseases. A safe and efficient system for the delivery of anticoagulants to mimic the physiological negative feedback mechanism by responding to the coagulation signal changes holds the promise and potential for anticoagulant therapy. Here, we exploit a “closed-loop” controlled release strategy for the delivery of recombinant hirudin, an anticoagulant agent that uses a self-regulated nanoscale polymeric gel. The cross-linked nanogel network increases the stability and bioavailability of hirudin and reduces its clearance in vivo. Equipped with the clot-targeted ligand, the engineered nanogels promote the accumulation of hirudin in the fibrous clots and adaptively release the encapsulated hirudin upon the thrombin variation during the pathological proceeding of thrombus for potentiating anticoagulant activity and alleviating adverse effects. We show that this formulation efficiently prevents and inhibits the clot formation on the mouse models of pulmonary embolism and thrombosis.
INTRODUCTION
Unintended deposition of clots in arteries or veins induces many life-threatening diseases, such as venous thromboembolism (1, 2), acute myocardial infarction (3), and stroke (4, 5). The clot formation is induced by the damage of vascular endothelial cells, abnormality of the coagulation system, and change of hemorheology (6). The main composition of the blood clot includes activated platelets, red blood cells (RBCs), and fibrins (7, 8). Multiple coagulation factors participate in the coagulation process (9). Among them, thrombin is a pivotal effector enzyme in coagulation cascade (10), which is responsible for transferring soluble fibrinogen into insoluble fibrin to stabilize the aggregated platelets and RBCs for the clot formation (11). Once the clot occludes the major blood vessels, ischemia and hypoxia occur in the downstream tissues, leading to their necrosis (12). For example, carotid arterial embolism causes ischemia and hypoxia of the brain tissue, resulting in tissue necrosis and neurological dysfunction (5).
A number of anticoagulants have been used to prevent or inhibit thrombus and clot formation in clinic (13, 14), including heparin, warfarin, and rivaroxaban. Heparin as a widely used anticoagulant acts by potentiating the activity of antithrombin III to neutralize the enzymatic activity of thrombin for blood clotting control (15). However, the clinical application of heparin leads to bleeding risk and thrombocytopenia (16). The heparin-induced thrombocytopenia (HIT) is a severe side effect in an incidence of 0.1 to 5%, which is mediated by the interactions between immunoglobulin G antibodies and complexes of heparin-platelet factor 4 (17, 18). Great efforts have been made to develop alternative anticoagulant drugs (19). Hirudin is one of these potent thrombin inhibitors, which is extracted from the peripharyngeal glands of leeches. Unlike heparin, hirudin can form a stable complex with thrombin to suppress its coagulative function directly (20). This thrombin-inhibiting process is independent upon antithrombin III, which brings about a definite dose-response relationship in the applications of hirudin for relatively more accurate dose control. Moreover, hirudin does not cause any thrombocytopenia, which makes it a preferable anticoagulant alternative to heparin for the control of blood clotting (21).
To address the drawbacks of natural hirudin including insufficient sources and poor quality, recombinant hirudins have been exploited and approved by the U.S. Food and Drug Administration for anticoagulant therapy (13), such as lepirudin, desirudin, and bivalirudin. Lepirudin is the first approved recombinant hirudin for the treatment of the patients with HIT (21). The indications of the recombinant hirudins have been further expanded to percutaneous coronary intervention and angioplasty (22, 23). However, the recombinant hirudins suffer from poor serum stability and susceptibility to protease degradation, leading to rapid clearance, short half-life, and, therefore, low bioavailability (24, 25). In addition, a strict dose control of the recombinant hirudins is also required because of their unexpected bleeding risk (26, 27).
Here, we propose a closed-loop hirudin delivery platform consisting of self-regulated nanogels for anticoagulant therapy, which can precisely tailor the release profile of hirudin in response to the pathological proceeding of thrombus (Fig. 1). The nanogel formulation is composed of two components, a chemically cross-linked thrombin-responsive polymeric matrix and recombinant hirudin variant 3 (HV) as a model anticoagulant agent (Fig. 1A). The HV-loaded clot-targeted thrombin-responsive nanogels (designated as HV/ctNGs) are prepared using a “one-pot” synthesis method, single emulsion polymerization (28–31). In addition to acrylamide (AAm), an allyl-modified peptide monomer [allyl-GGCR(NMe)EKA] and a thrombin-cleavable peptide (TCP) cross-linker (allyl-GGGLVPRGSGGG-allyl) are incorporated in the polymerization. The CREKA peptide that was found by an in vivo phage-displayed screening technique has a superior binding ability to the clotted plasma proteins including fibrin and fibronectin in the tumor vasculatures (32, 33). Further methyl modification of the amino group (NMe) of glutamic acid (E) makes the oligopeptide more stable without impairment of its clot-targeting potential (34). The LVPRGS peptide in the TCP cross-linker can be degraded at the cleavage site of the arginine (R) residue by thrombin, which is derived from the cleaved peptide sequence in bovine coagulation factor XIII in response to thrombin within the activation process of coagulation (35). The thrombin-mediated cleavage of the TCP cross-linker causes the dissociation of the polymeric matrix (36), further accelerating the release of the encapsulated HV. After intravascular administration, HV/ctNGs protect HV against plasma proteolysis and prolong blood circulation of HV. The clot-targeted CR(NMe)EKA peptide ligands modified on the nanogel surface facilitate the accumulation of HV/ctNGs in the formed clots. Thrombin that is highly concentrated in the clots (37) triggers a high intraclot release of HV (Fig. 1B). The released HV stably binds to thrombin to inhibit its coagulative activity. The release of HV is self-regulated. The complex formation of thrombin and HV also hinders the hydrolytic activity of thrombin to the cross-linker and, therefore, slows the release rate of HV to basal release. We show that this HV/ctNG-mediated closed-loop anticoagulant regulation offers a safe and efficient therapeutic strategy on the mouse models of pulmonary embolism and thrombosis.
Fig. 1. Schematic of self-regulated hirudin delivery for anticoagulant therapy.
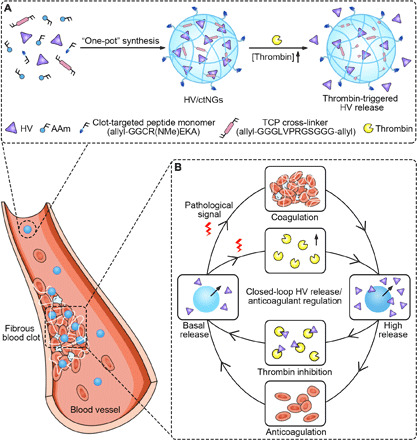
(A) Schematic of synthesis of HV/ctNGs and adaptive release of HV in response to thrombin. (B) Schematic of mechanism of HV/ctNGs for closed-loop HV delivery and anticoagulant regulation.
RESULTS
Self-regulated hirudin release
HV/ctNGs prepared by single emulsion method had a particle size of ~77 nm and a neutral surface charge (table S1). As examined using transmission electron microscope (TEM), HV/ctNGs showed uniform and spherical morphology (Fig. 2A). The HV-loaded nontargeted thrombin-cleavable nanogels (HV/tNGs) and clot-targeted nondegradable nanogels (HV/cNGs) as control were also prepared and had particle sizes of ~57 and ~65 nm, respectively (table S1). The stability of HV/ctNGs in the presence of serum protein was investigated by monitoring changes in particle size and surface charge. No notable size and charge variations were observed after HV/ctNGs were incubated with bovine serum albumin (BSA) over time (fig. S1A). In addition, the protein leakage was also evaluated when the nanogels were exposed to serum in vitro. The rhodamine-labeled HV (Rho-HV) was synthesized and encapsulated in ctNGs. The release profile of Rho-HV was determined after Rho-HV/ctNGs were incubated with fetal bovine serum (FBS) over time. The presence of FBS did not induce any marked leakage of Rho-HV from Rho-HV/ctNGs (fig. S1B). These results suggest that HV/ctNGs are highly stable to serum proteins.
Fig. 2. Thrombin-responsive self-regulated HV release.
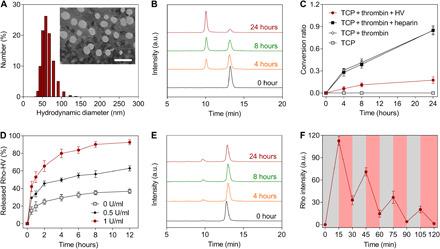
(A) Representative histogram of the hydrodynamic diameter distribution and TEM image of HV/ctNGs. Scale bar, 100 nm. (B) Representative HPLC profiles of the TCP cross-linker after incubation with thrombin for different time. a.u., arbitrary unit. (C) Change in the conversion ratio of the TCP cross-linker after incubation under different conditions over time. Data are shown as means ± SD (n = 3). (D) Release profiles of Rho-HV from Rho-HV/ctNGs after incubation with different concentrations of thrombin. Data are shown as means ± SD (n = 3). (E) Representative HPLC profiles of the TCP cross-linker after incubation with thrombin in the presence of HV for different time. (F) Release profiles of Rho-HV from Rho-HV/ctNGs after repeated incubation with and without thrombin. Data are shown as means ± SD (n = 3).
To evaluate the self-regulated HV release property of HV/ctNGs, we first investigated the degradability of the TCP cross-linker in response to thrombin using high-performance liquid chromatography (HPLC). As shown in the HPLC profiles, the peak area of the TCP cross-linker at the retention time of ~13 min reduced as the incubation time increased (Fig. 2B). The chromatographic peak at the retention time of ~10 min was further confirmed to be that of the allyl-GGGLVPR (allyl-GR7) peptide, one of the degradation products from the TCP cross-linker by thrombin (fig. S2). The conversion ratio of the TCP cross-linker was calculated to be ~0.85 at 24 hours after incubation with thrombin (Fig. 2C) (38), suggesting that TCP incorporated in the construction of HV/ctNGs is stable but to be responsively cleaved in the presence of thrombin. Subsequently, we assessed the thrombin-promoted release of HV from HV/ctNGs. The release profiles of Rho-HV were determined after Rho-HV/ctNGs were exposed to different concentrations of thrombin over time (Fig. 2D). Increased concentration of thrombin resulted in increased release rate of Rho-HV from Rho-HV/ctNGs. In contrast, no notable increase in the release rate of Rho-HV was observed when Rho-HV/cNGs containing the nondegradable linkers were incubated with thrombin (fig. S3). These results indicate that HV/ctNGs can controllably release HV under the pathological environment of thrombus with an increasing level of thrombin. Furthermore, the closed-loop regulation of HV/ctNGs on the release of HV was assessed. To demonstrate whether the released HV could inhibit the degradation of thrombin on TCP, we used the HPLC analysis on the TCP cross-linker incubated with both HV and thrombin. A slight decrease in the chromatographic peak area of the TCP cross-linker was observed after treatment with thrombin in the presence of HV (Fig. 2E). The 24-hour conversion ratio was extremely lower than that in the absence of HV (Fig. 2C). These data suggest that HV is able to efficiently suppress the thrombin-mediated TCP degradation. We also estimated the effect of heparin on the TCP cleavage. As expected, heparin could not inhibit the activity of thrombin on degrading the responsive TCP linkage (Fig. 2C and fig. S4). Furthermore, the release profile of Rho-HV displayed a pulsatile manner (36, 39), when Rho-HV/ctNGs were cyclically incubated with and without thrombin every 15 min (Fig. 2F). HV/ctNGs could respond to thrombin concentration changes. The accelerated release phase was attributed to the degradation of the TCP linker followed by the disruption of the nanogels. Collectively, HV/ctNGs can promptly release the encapsulated HV in response to elevation of thrombin at the blood clot region. The released HV directly inhibits the activity of thrombin on aggravating thrombosis on one hand and degrading the TCP linker and disintegrating the nanogels, which turn to preventing the excessive release of HV, leading to adverse effects on the other. Like an automatic control valve system, the HV release from HV/ctNGs is expedited at a high thrombin level but impeded at a low thrombin level adaptively.
In vitro and in vivo clot-targeting studies
The in vitro blood clot was obtained by incubating platelet-rich plasma with thrombin and calcium chloride (CaCl2) for 1 hour (40) and subsequently incubated with the Cy7-labeled BSA-loaded ctNGs (Cy7-BSA/ctNGs) for 5 min. After repeatedly washing with saline, the blood clots were detected using in vivo imaging system (IVIS) (Fig. 3A). The fluorescent images displayed that higher intensity of the Cy7 fluorescent signal was observed in the clots treated with Cy7-BSA/ctNGs than that with Cy7-BSA/tNGs, suggesting that the clot-targeted CR(NMe)EKA peptide supports an increased intraclot accumulation of the nanogels by effectively binding to the fibrin-fibronectin complexes. Preincubation with a high concentration of the free CR(NMe)EKA peptide led to a noticeable decrease of the Cy7 fluorescent intensity of the Cy7-BSA/ctNG–treated group, which is ascribed to the competitive inhibition effect of the free peptide on the clot-targeting efficiency of ctNGs. Quantitative results using region-of-interest (ROI) analysis substantiated that the Cy7-BSA/ctNG–treated group had significantly higher intraclot fluorescent intensity than the counterparts (Fig. 3B). These data indicate that ctNGs have a favorable targeting ability to the ex vivo formed blood clots.
Fig. 3. In vitro and in vivo clot-targeting evaluation.
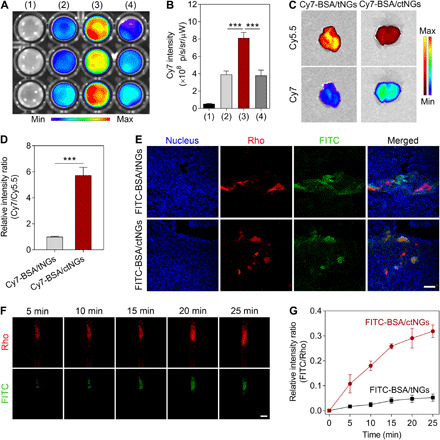
(A) Fluorescent images of the blood clots after different treatments: (1) saline, (2) Cy7-BSA/tNGs, (3) Cy7-BSA/ctNGs, and (4) the clot-targeted peptide + Cy7-BSA/ctNGs. (B) Fluorescent intensities of Cy7 in the blood clots after different treatments. Data are shown as means ± SD (n = 3). ***P < 0.001. (C) Representative fluorescent images of the lung tissue harvested from the thromboplastin-induced pulmonary embolism mouse models after intravenous injection of Cy7-BSA/ctNGs and Cy7-BSA/tNGs at 15 min after induction. The pulmonary emboli were labeled with Cy5.5. (D) Relative fluorescent intensity ratio of Cy7 to Cy5.5 in the lung tissues. Data are shown as means ± SD (n = 3). ***P < 0.001. (E) Representative fluorescent images of the lung tissues harvested from the thromboplastin-induced pulmonary embolism mouse models after intravenous injection of FITC-BSA/ctNGs and FITC-BSA/tNGs at 15 min after induction. Scale bar, 100 μm. (F) Representative fluorescent images of the carotid arteries of the FeCl3-induced thrombosis mouse models after intravenous injection of FITC-BSA/ctNGs for different time. Scale bar, 500 μm. (G) Changes in the relative fluorescent intensity ratio of FITC to Rho in the carotid arteries over time after intravenous injection of FITC-BSA/ctNGs and FITC-BSA/tNGs. Data are shown as means ± SD (n = 3).
Three different mouse models were used to appraise the in vivo clot-targeted capacity of ctNGs, including the thromboplastin-induced pulmonary embolism model (41) as well as the ferric chloride (FeCl3)–induced carotid arterial and mesenteric thrombosis models (42, 43). Thromboplastin is one of the tissue factors responsible for inducing coagulation cascade, causing the generation of thrombin and the formation of clot. The formed clots are inclined to deposit in the lung tissue with rich capillaries. In this study, the mice were intravenously injected with the Cy5.5-labeled fibrinogen (Cy5.5-fibrinogen), nanogels (Cy7-BSA/ctNGs or Cy7-BSA/tNGs), and thromboplastin in order (fig. S5A). The lung tissues were sampled and imaged using IVIS at 15 min after injection of thromboplastin. As presented in the fluorescent tissue images, the formed emboli with the fibrin-fibronectin complexes, which were labeled by Cy5.5 through a thromboplastin-induced fibrinogen-to-fibrin conversion, were observed to accumulate in the lung (Fig. 3C). Encouragingly, Cy7-BSA/ctNGs showed preferable lung-targeting ability than Cy7-BSA/tNGs, as evidenced by higher Cy7 fluorescent intensity in the lung tissue. The targeting ratio of Cy7-BSA/ctNGs to the Cy5.5-labeled clots was quantified to be 5.7-fold that of Cy7-BSA/tNGs (Fig. 3D). To further confirm whether ctNGs were specifically targeted to the pulmonary emboli, the intrapulmonary distribution was monitored using a confocal microscope (Fig. 3E). The rhodamine-labeled fibrinogen (Rho-fibrinogen) and fluoroscein isothiocyanate (FITC)–labeled BSA-loaded ctNGs (FITC-BSA/ctNGs) were used instead. As displayed in the confocal images of the lung section, most of the FITC signals were colocalized with the Rho signals in the FITC-BSA/ctNG group, in contrast to lower colocalization observed in the FITC-BSA/tNG group. The results indicate that the surface modification of the CR(NMe)EKA peptide facilitates the targeting of the nanogels to the fibrinous pulmonary emboli in vivo.
We further assessed the in vivo clot-targeted capability of ctNGs on both FeCl3-induced carotid arterial and mesenteric thrombosis mouse models. Rhodamine 6G (Rho 6G) was intravenously injected into the mice to label the platelets (44), and the left carotid artery was exposed and treated with FeCl3 (fig. S5B). The FeCl3-treated mice were intravenously injected with FITC-BSA/ctNGs. The clot formation and targeting were visualized in real time using a stereo zoom microscope (Fig. 3F). High Rho fluorescent signal was detected in the carotid artery of the mice at the FeCl3-treated site, which is indicative of the in vivo blood clot formation. The intravenously injected FITC-BSA/ctNGs could efficiently target the formed thrombus, as validated by most of the FITC fluorescent signals colocalized with the Rho fluorescent signals, while low FITC fluorescent signal was observed at the blood clot region of the mice receiving injection of FITC-BSA/tNGs (fig. S6). The relative intensity ratio of FITC-BSA/ctNGs to the Rho-labeled thrombi was increased markedly over time (Fig. 3G), indicating that more and more circulating ctNGs accumulate in the blood clots constantly. In relative quantification, FITC-BSA/ctNGs showed more than a fivefold increase in the clot-targeting ratio compared with FITC-BSA/tNGs. In addition, the superior thrombus-targeted potential of ctNGs was also validated on the FeCl3-induced mesenteric thrombosis mouse model compared with tNGs (fig. S7).
In vivo anticoagulant efficacy studies
The in vivo anticoagulant activity of HV/ctNGs was first estimated on the thromboplastin-induced pulmonary embolism model (45). The mice were intravenously injected with Cy5.5-fibronogen, HV formulations (free HV, HV/tNGs, HV/cNGs, or HV/ctNGs), and thromboplastin in sequence. The anticoagulant effect was evaluated by monitoring the Cy5.5 fluorescent signals in the lung using IVIS. Treatment with different HV formulations all suppressed the formation of the pulmonary embolism, as indicated by lower Cy5.5 fluorescent intensity detected in the lung compared with that treated with saline (Fig. 4, A and B). Among them, the HV/ctNG treatment exhibited the strongest inhibitory effect on the pulmonary embolism. The anticoagulant efficacy of either HV/tNGs or HV/cNGs was greatly limited by the poor clot-targeted capacity or inefficient drug release property. After treatment, hematoxylin and eosin (H&E) and Masson’s trichrome staining were used for histological examination of the lung tissues, respectively (Fig. 4C). A large area of embolization was observed in the lung of the mice treated with saline. Treatment with HV/ctNGs resulted in the lowest degree of fibrin deposition and the smallest area of pulmonary embolism, as expected, which markedly reduced the percentage of clot area to 7% that of treatment with saline (Fig. 4D). We also monitored the survival of the mice with the acute pulmonary embolism after different treatments. All the mice treated with HV/ctNGs survived, further confirming the efficient anticoagulant activity of HV/ctNGs (Fig. 4E).
Fig. 4. In vivo therapeutic efficacy study on the thromboplastin-induced pulmonary embolism mouse model.
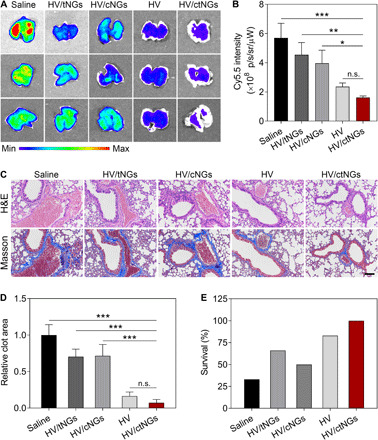
(A) Fluorescent images of the lung tissues harvested from the mice after different treatments at 15 min after induction. The pulmonary emboli were labeled with Cy5.5. (B) Fluorescent intensity of Cy5.5 in the lung tissues. Data are shown as means ± SD (n = 3). P > 0.05 (no significance, n.s.), *P < 0.05, **P < 0.01, and ***P < 0.001. (C) Representative H&E- and Masson’s trichrome–stained images of the lung sections after different treatments at 15 min after induction. Scale bar, 100 μm. (D) Relative clot areas in the lung tissues after different treatments at 15 min after induction. Data are shown as means ± SD (n = 3). P > 0.05 (no significance, n.s.) and ***P < 0.001. (E) Survival rates of the mice after different treatments within 2 hours (n = 6).
Next, we assessed the in vivo clot-inhibiting effect of HV/ctNGs on the FeCl3-induced carotid arterial thrombosis mouse model. The carotid artery of the FeCl3-treated mice before injected with Rho 6G was imaged, and the changes in the clot size reflected by the Rho fluorescent intensity were monitored after intravenous injection of different HV formulations (Fig. 5A). The Rho fluorescent intensity markedly increased in the carotid artery of the mice treated with saline, suggesting that the thrombi form and aggravate owing to the FeCl3-induced injury of the blood vessels. Treatment with the free HV efficiently inhibited the exacerbation of thrombosis, as evidenced by significant reduction in the Rho fluorescent intensity. By comparison, neither HV/tNGs nor HV/cNGs brought about efficient clot-inhibiting activity in vivo, although the nanogel encapsulation can protect HV against plasma proteolysis, which resulted from inferior thrombus-targeted capacity of HV/tNGs and inefficient drug release property of HV/cNGs. Of note, HV/ctNGs showed a more potent effect on suppressing the aggravation of thromboembolism than other control groups, including HV, HV/tNGs, and HV/cNGs. At 30 min after the injection of different formulations, the carotid arteries were sampled, and their transverse sections were histologically examined (Fig. 5B). Severe embolism was observed in the injured blood vessels treated with saline. A large area of the vessel was examined to be still clotted after treatment with either HV/tNGs or HV/cNGs. Both HV and HV/ctNGs efficiently inhibited the thrombus formation, as shown by a few clots observable in the injured carotid arterial vessels after treatment. Quantitative data showed that treatment with HV/ctNGs resulted in the lowest clot level (Fig. 5C) and embolization rate (Fig. 5D). In addition, we used the acoustic imaging technique to detect the changes in the blood flow velocity in the FeCl3-induced injured carotid artery before and after treatment with HV/ctNGs. As presented in the pulse wave Doppler images, the vibration pattern disappeared after treatment with FeCl3 (fig. S8), indicating that the FeCl3-induced clot formation blocked the vessels and, therefore, reduced the blood flow velocity. The carotid arterial flow velocity time integral (VTI) (46) was quantified to markedly drop after the FeCl3 treatment (Fig. 5E). As expected, the FeCl3-treated mice receiving treatment of HV/ctNGs recovered the carotid arterial flow velocity, and the VTI value concomitantly increased to be comparable to that of the mice without any treatment. In parallel, the anticoagulant efficacy of HV/ctNGs was explored on the FeCl3-induced mesenteric thrombosis mouse model. The obtained results were consistent with the findings of the in vivo studies on the carotid arterial thrombosis mouse model. HV/ctNGs could efficiently control the formation and aggravation of thrombi in the mesenteric blood vessels (figs. S9 and S10). These data indicate that self-regulated HV/ctNGs with the capabilities of enhanced clot targeting and closed-loop drug release potentiate the in vivo anticoagulant efficacy of HV.
Fig. 5. In vivo therapeutic efficacy study on the FeCl3-induced carotid artery thrombosis mouse model.
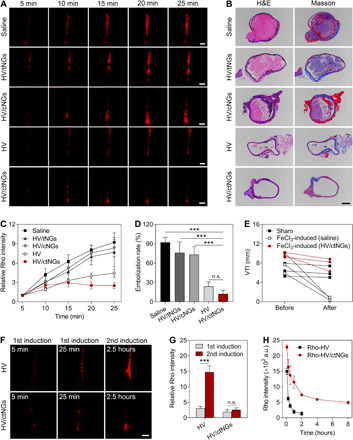
(A) Representative fluorescent images of the carotid arteries of the mice after intravenous injection of different HV formulations for different time. The blood clots were labeled with Rho 6G. Scale bars, 500 μm. (B) Representative H&E- and Masson’s trichrome–stained images of the carotid arterial transverse sections after intravenous injection of different HV formulations for 25 min. Scale bar, 100 μm. (C) Changes in the relative fluorescent intensity of Rho after different HV formulations over time. Data are shown as means ± SD (n = 3). (D) Embolization rates in the H&E-stained carotid arterial transverse sections after different treatments. Data are shown as means ± SD (n = 3). P > 0.05 (no significance, n.s.) and ***P < 0.001. (E) VTI of the carotid arteries of the mice before and after intravenous injection of HV/ctNGs for 25 min. (F) Representative fluorescent images of the carotid arteries of the mice after intravenous injection of HV and HV/ctNGs for different time upon sequential induction. The blood clots were labeled with Rho 6G. Scale bar, 500 μm. (G) Relative fluorescent intensity ratios of Rho after intravenous injection of HV and HV/ctNGs upon sequential induction. Data are shown as means ± SD (n = 3). P > 0.05 (no significance, n.s.) and ***P < 0.001. (H) Pharmacokinetic profiles of Rho-HV and Rho-HV/ctNGs after intravenous injection into the rats. Data are shown as means ± SD (n = 3).
We further performed comparative assessment of therapeutic efficacies between HV and HV/ctNGs using the sequential FeCl3-induced thrombosis mouse model with secondary injury (fig. S5C). The variations in the size of the Rho-labeled thrombi were determined by monitoring the Rho fluorescent intensity after secondary FeCl3 induction (Fig 5, F and G, and fig. S11). In the HV treatment group, a significantly higher Rho fluorescent intensity in the blood vessel was observed in the thrombosis mouse model by inducing twice using FeCl3, while there was no significant difference in the Rho fluorescent intensity between single and sequential FeCl3-induced thrombosis mouse models in the HV/ctNG treatment group. Moreover, treatment with HV/ctNGs resulted in notably lower Rho fluorescent intensity than treatment with HV in the mouse model using sequential FeCl3 induction. These findings indicate that HV/ctNGs have superior effects on preventing the reformation of thrombi than HV. We speculated that such enhancement resulted from the prolonged circulation of HV in the blood by HV/ctNGs. To validate our assumption, the pharmacokinetic profiles of Rho-HV/ctNGs and Rho-HV were determined after intravenous injection into the rats (Fig. 5H). As expected, the Rho fluorescent intensity of Rho-HV rapidly dropped in the blood over time because of quick renal clearance and protein degradation. Rho-HV/ctNGs noticeably increased the circulation time and bioavailability of Rho-HV, which supported the enhanced activity of HV to prevent and inhibit the thrombus formation.
Safety evaluation
With the confirmation on the in vivo anticoagulant effects of HV/ctNGs, the safety of HV/ctNGs was evaluated both in vitro and in vivo. We first investigated whether HV/ctNGs caused hemolysis. The RBCs were collected from the mice and incubated with HV/ctNGs over time. After centrifugation, the absorbance of the supernatant was quantified. Triton X-100, a surfactant for cell lysis, could disrupt RBCs, as evidenced by high absorbance of the supernatant containing a large amount of hemoglobins (fig. S12). No significant difference in the absorbance was observed and determined between saline and HV/ctNGs, suggesting that HV/ctNGs does not cause any hemolytic reaction. Moreover, the bleeding time test was performed to explore whether HV/ctNGs affected the ability of the coagulation system to stop bleeding. The mice were intravenously injected with HV/ctNGs. After 24 hours, a small cut was made on the tail, and the time was recorded until the bleeding terminated. Treatment with HV/ctNGs did not result in any marked change in the bleeding time (fig. S13). In addition, at 24 hours after injection of HV/ctNGs, the blood and major tissues were collected. The total amounts of RBCs and platelets were counted using a hematology analyzer, and the serum levels of different biochemical indices for liver and kidney functions were quantified, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine. The tissues were stained by H&E and observed using a microscope. The HV/ctNG treatment did not induce any variation in the total amounts of RBCs and platelets (fig. S14) or the serum levels of the biochemical indices (fig. S15) as well as any obvious pathological changes in the major tissues (fig. S16). These results indicate that HV/ctNGs are highly safe nanocarriers for the delivery of HV within the studied period of time.
DISCUSSION
On the basis of the pathological characteristics of thrombosis and the demand for precise delivery of anticoagulants, we proposed the concept of exploiting a self-regulated polymeric nanogel integrated with thrombin-sensing constituents, which could enable the laden HV to adaptively release in a closed-loop manner by responding to the changes of thrombin concentration. This nanogel formulation was obtained using a facile one-pot synthetic approach, which composed of thrombin-degradable polymeric matrices with embellishment of clot-targeted peptide ligands. After formulation optimization, HV/ctNGs displayed robust thrombin-responsive HV release behaviors. As validated, the HV release rate of HV/ctNGs sped up under a high level of thrombin but slowed down in the absence of thrombin. Moreover, HV/ctNGs showed a pulsating HV release pattern in response to cyclical variations in thrombin concentration, which mimicked the physiological negative feedback control mechanism, potentiating anticoagulant activity and alleviating adverse effects.
ctNGs stabilized the encapsulated HV and protected it from degradation by plasma enzymes in the blood after intravenous injection, which enhanced the blood persistence and reduced clearance rate of HV. Meanwhile, ctNGs promoted the accumulation of HV in the fibrous clots by high binding affinity between the surface-modified ligands and the intraclot fibrins/fibronectins. The half-life of HV delivered by HV/ctNGs was demonstrated to be substantially prolonged compared with that of free HV. The clot-targeting studies confirmed that ctNGs could efficiently bind to the ex vivo formed clots and preferentially accumulated in the intrapulmonary emboli and intravascular thrombi of the mouse models in vivo. We further showed that treatment with HV/ctNGs was able to effectively prevent and inhibit the clot formation in the mouse models of pulmonary embolism and thrombosis. While the enhanced anticoagulant activity of HV/ctNGs was attained, the safety evaluation results suggested that HV/ctNGs did not cause any hematolysis, thrombocytopenia, bleeding, liver and kidney function impairment, and pathological tissue changes within the studied period of time. Further long-term toxicity studies are required to be carried out for potential translation. Together, the self-regulated system with a closed-loop delivery of anticoagulants can be a promising strategy to treat thrombosis and embolism.
MATERIALS AND METHODS
Materials
Recombinant HV was provided by B. He from Nanjing Tech University. AAm and N,N′-methylene-bisacrylamide (MBA) were purchased from Aladdin. The TCP cross-linker and the clot-targeted GGCR(NMe)EKA peptide were purchased from GL Biochem. Fibrinogen and thromboplastin were purchased from Sigma-Aldrich.
Nanogel preparation and characterization
HV (200 U), AAm (45 mg), the TCP cross-linker (23 mg), the clot-targeted peptide (8 mg), and ammonium persulfate (6 mg) were dissolved in phosphate-buffered saline (PBS) (500 μl) and added dropwise into hexane (5 ml) containing dioctyl sulfosuccinate sodium salt (356 mg) and polyoxyethylene lauryl ether (Brij L4) (688 mg) under stirring at 4°C. After addition of N,N,N′,N′-tetramethylethylenediamine (20 μl), the solution was stirred at 4°C for 2 hours, followed by rotary evaporation and repeated ethanol washing. Last, HV/ctNGs were obtained after vacuum drying. HV/tNGs were prepared without the clot-targeted peptide, while HV/cNGs were prepared by replacing the TCP cross-linker with the nondegradable MBA cross-linker. The particle size and zeta potential of nanogels were measured using Zetasizer (Malvern). The morphology was visualized using TEM (Hitachi HT7700).
Fluorescent labeling
Proteins (HV, fibrinogen, or BSA) dissolved in sodium bicarbonate buffer (100 mM, pH 8.5) were incubated with a 10-fold molar equivalent of Rho-NHS (N-hydroxysuccinimide) or Cy5.5-NHS at room temperature for 1 hour or FITC at 4°C for 8 hours under stirring. After dialysis against PBS (pH 7.4) for 24 hours to remove excess fluorescent dyes, the fluorescent-labeled proteins were obtained. To prepare Cy7-BSA/ctNGs, BSA/ctNGs dissolved in PBS (pH 7.4) were incubated with a 10-fold molar equivalent of Cy7-Mal under stirring at 37°C for 2 hours. After dialysis against PBS (pH 7.4) for 24 hours to remove excess fluorescent dyes, Cy7-BSA/ctNGs were obtained.
Thrombin-responsive degradation of the TCP cross-linker
The TCP cross-linker (0.2 mg/ml) was incubated with thrombin (1 U/ml) in the absence and presence of HV (2 U/ml) or heparin (2 U/ml) at 37°C over time, respectively, and subsequently analyzed using HPLC (Shimadzu). The mobile phase was the mixture of acetonitrile and water (16:84, v/v) containing acetic acid (0.1%, w/w). The detection wavelength was set to 220 nm. The column temperature was 40°C. The flow rate was 1 ml/min.
In vitro release studies
To assess the thrombin responsiveness, Rho-HV/ctNGs that were put into the dialysis bag were incubated in PBS (pH 7.4) containing different concentrations of thrombin (0, 0.5, and 1 U/ml) or FBS (50%, v/v) at 37°C. At specified time intervals, the fluorescent intensity of Rho in the medium was determined at 585 nm (excitation at 552 nm) using a microplate reader (Tecan Infinite M1000 Pro). To evaluate the self-regulated release property, Rho-HV/ctNGs that were put into the dialysis bag were first incubated in PBS (pH 7.4) containing thrombin (1 U/ml) at 37°C for 15 min and subsequently incubated without thrombin at 37°C for 15 min. This cycle was repeated four times, and similarly, the fluorescent intensity of Rho in the medium was determined using a microplate reader.
In vitro clot-targeting study
The mouse plasma was incubated with thrombin (10 U/ml) and CaCl2 (40 mM) at 37°C for 1 hour to form clots. The clots were subsequently incubated with Cy7-BSA/ctNGs and Cy7-BSA/tNGs for another 5 min, respectively. For competitive inhibition assay, the formed clots were preincubated with the clot-targeted peptide (0.5 mg/ml) for 5 min. After repeated saline washing, the clots were observed using IVIS (PerkinElmer IVIS Spectrum). The fluorescent intensity of Cy7 was quantified by ROI analysis using Living Image software.
Animals
All animals were treated according to the Guide for Care and Use of Laboratory Animals, approved by the Animal Experimentation Ethics Committee of China Pharmaceutical University. The mice (C57BL/6, female, 18 to 22 g) and the rats (Wistar, male, 200 to 220 g) were offered by the Comparative Medicine Centre of Yangzhou University. The mice and rats were housed with a 12-hour light/dark cycle and free access to food and water.
In vivo clot-targeting studies
In the thromboplastin-induced pulmonary embolism model, the mice were intravenously injected with Cy5.5-fibrinogen (70 nmol/kg), Cy7-BSA/ctNGs (5-min interval), and thromboplastin (4 ml/kg) (5-min interval) successively. The mice were euthanized at 15 min after injection of thromboplastin. The lung tissues were harvested and imaged using IVIS. The fluorescent intensities of Cy5.5 and Cy7 were quantified by ROI analysis using Living Image software. Cy7-BSA/tNGs were taken as control. To monitor the intrapulmonary distribution, Rho-fibrinogen and FITC-BSA/ctNGs were used instead. The lung tissues harvested from the mice were treated by freezing microtomy. The frozen lung tissue sections were stained with Hoechst 33342 (5 μg/ml; Invitrogen) and observed using a confocal microscope (Olympus FV3000). FITC-BSA/tNGs were taken as control.
In both FeCl3-induced carotid arterial and mesenteric thrombosis models, the mice were first intravenously injected with Rho 6G (0.5 mg/kg). After 10 min, the carotid artery and mesenteric vessels were exposed and adhered by a filter paper moistened with FeCl3 at concentrations of 10 and 6% for 1 min, respectively. After 5 min, the mice were intravenously injected with FITC-BSA/ctNGs. At specified time intervals, the blood vessels were imaged using a stereo zoom microscope (ZEISS Axio Zoom.V16). The fluorescent intensity of Rho and FITC was quantified using ImageJ software. FITC-BSA/tNGs were taken as a control.
In vivo therapeutic efficacy studies
In the thromboplastin-induced pulmonary embolism model, the mice were intravenously injected with Cy5.5-fibrinogen (70 nmol/kg), different HV formulations (5-min interval), and thromboplastin (4 ml/kg) (5-min interval) successively. After 15 min, the mice were euthanized. The lung tissues were harvested and imaged using IVIS. The fluorescent intensity of Cy5.5 was quantified by ROI analysis using Living Image software. For histological examination, the lung tissues harvested from the mice were examined by H&E and Masson’s trichrome staining, respectively. The stained lung tissue sections were observed using a microscope (Nikon Ts2R). The survival rates of the mice receiving different HV formulations were determined within 2 hours after intravenous injection of thromboplastin.
In both FeCl3-induced carotid arterial and mesenteric thrombosis models, the mice were first intravenously injected with Rho 6G (0.5 mg/kg). After 10 min, the carotid artery and mesenteric vessels were exposed and treated with FeCl3 for 1 min. After 5 min, the mice were intravenously injected with different HV formulations. At specified time intervals, the blood vessels were imaged using a stereo zoom microscope. The fluorescent intensity of Rho was quantified using ImageJ software. The blood vessels were also monitored at 25 min after injection of HV/ctNGs using a micro-ultrasound imaging system (FUJIFILM VisualSonics Vevo 3100 LT). For histological examination, the blood vessels were harvested from the mice at 25 min after injection of different HV formations and examined by H&E and Masson’s trichrome staining. The stained blood vessel sections were observed using a microscope. In addition, the mice were treated with FeCl3 for 1 min once again at 2 hours after injection of HV and HV/ctNGs. After 30 min, the blood vessels were imaged using a stereo zoom microscope. The fluorescent intensity of Rho was quantified using ImageJ software.
Pharmacokinetics
The rats were intravenously injected with Rho-HV and Rho-HV/ctNGs. At specified time intervals, the blood was collected and centrifuged. The fluorescent intensity of Rho in the plasma supernatant was measured using a microplate reader.
Safety evaluation
For in vitro hemolysis assay, the mouse RBCs (2%) were incubated with HV/ctNGs and Triton X-100 (0.1%) for 4 hours. After centrifugation, the absorbance of the supernatant was measured at 570 nm using a microplate reader. Triton X-100 was taken as a control. For bleeding time assay, the mice were intravenously injected with HV/ctNGs and saline. After 24 hours, a small cut was made on the tail, and the time was recorded until the bleeding stopped. In addition, the blood and major tissues were collected. The total amounts of RBCs and platelets were counted using a hematology analyzer (Mindray BC-2800Vet). The serum levels of ALT, AST, BUN, and creatinine were assayed by the corresponding ELISA (enzyme-linked immunosorbent assay) kits (Nanjing Jiancheng) according to the manufacturers’ protocols. The tissues were stained by H&E. The stained tissue sections were observed using a microscope.
Statistical analysis
The data are indicated as means ± SD. Two-tailed Student’s t test and one-way analysis of variance (ANOVA) were used for test of statistical difference in two and multiple groups, respectively.
Supplementary Material
Acknowledgments
We acknowledge the Public platform of State Key Laboratory of Natural Medicines for the use of analytical instrumentation facilities, and also thank P. Zhou, X. Ma, and L. Guo for their help with taking images. We thank B. He at Nanjing Tech University for providing the recombinant HV. Funding: This work was supported by the National Natural Science Foundation of China (81971730), the National Ten Thousand Talents Program for Young Top-Notch Talents, the Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (171028), the Funding of Double First-Rate Discipline Innovation Team (CPU2018GF05), and the Project of State Key Laboratory of Natural Medicines of China Pharmaceutical University (SKLNMZZ202024). Author contributions: R.M. conceived and supervised the project. X.X. and R.M. designed the experiments, analyzed the data, and wrote the paper. X.X., X.H., Y.Z., Z.F., S.S., and H.D. performed the experiments. C.Z. contributed to discussion and provided relevant advice. Competing interests: R.M., X.X., and X.H. are inventors on a patent application related to this work filed by the China Pharmaceutical University (application no. 202010869769.2, filed on 26 August 2020). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/41/eabc0382/DC1
REFERENCES AND NOTES
- 1.Tritschler T., Kraaijpoel N., Le Gal G., Wells P. S., Venous thromboembolism: Advances in diagnosis and treatment. JAMA 320, 1583–1594 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Di Nisio M., van Es N., Buller H. R., Deep vein thrombosis and pulmonary embolism. Lancet 388, 3060–3073 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Crea F., Libby P., Acute coronary syndromes: The way forward from mechanisms to precision treatment. Circulation 136, 1155–1166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamel H., Healey J. S., Cardioembolic stroke. Circ. Res. 120, 514–526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hankey G. J., Stroke. Lancet 389, 641–654 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Versteeg H. H., Heemskerk J. W. M., Levi M., Reitsma P. H., New fundamentals in hemostasis. Physiol. Rev. 93, 327–358 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Furie B., Furie B. C., Mechanisms of disease: Mechanisms of thrombus formation. N. Engl. J. Med. 359, 938–949 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Fleissner F., Bonn M., Parekh S. H., Microscale spatial heterogeneity of protein structural transitions in fibrin matrices. Sci. Adv. 2, e1501778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triplett D. A., Coagulation and bleeding disorders: Review and update. Clin. Chem. 46, 1260–1269 (2000). [PubMed] [Google Scholar]
- 10.Dahlback B., Blood coagulation. Lancet 355, 1627–1632 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Mann K. G., Thrombin generation in hemorrhage control and vascular occlusion. Circulation 124, 225–235 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolberg A. S., Rosendaal F. R., Weitz J. I., Jaffer I. H., Agnelli G., Baglin T., Mackman N., Venous thrombosis. Nat. Rev. Dis. Primers 1, 15006 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Mackman N., Bergmeier W., Stouffer G. A., Weitz J. I., Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 19, 333–352 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Fredenburgh J. C., Gross P. L., Weitz J. I., Emerging anticoagulant strategies. Blood 129, 147–154 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Mulloy B., Hogwood J., Gray E., Lever R., Page C. P., Pharmacology of heparin and related drugs. Pharmacol. Rev. 68, 76–141 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Greinacher A., Heparin-induced thrombocytopenia. N. Engl. J. Med. 373, 1883–1884 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Salter B. S., Weiner M. M., Trinh M. A., Heller J., Evans A. S., Adams D. H., Fischer G. W., Heparin-induced thrombocytopenia: A comprehensive clinical review. J. Am. Coll. Cardiol. 67, 2519–2532 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Arepally G. M., Heparin-induced thrombocytopenia. Blood 129, 2864–2872 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppens M., Eikelboom J. W., Gustafsson D., Weitz J. I., Hirsh J., Translational success stories: Development of direct thrombin inhibitors. Circ. Res. 111, 920–929 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Rydel T. J., Ravichandran K. G., Tulinsky A., Bode W., Huber R., Roitsch C., Fenton J. W. II, The structure of a complex of recombinant hirudin and human alpha-thrombin. Science 249, 277–280 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Greinacher A., Volpel H., Janssens U., Hach-Wunderle V., Kemkes-Matthes B., Eichler P., Mueller-Velten H. G., Potzsch B., Recombinant hirudin (lepirudin) provides safe and effective anticoagulation in patients with heparin-induced thrombocytopenia: A prospective study. Circulation 99, 73–80 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Rassen J. A., Mittleman M. A., Glynn R. J., Brookhart M. A., Schneeweiss S., Safety and effectiveness of bivalirudin in routine care of patients undergoing percutaneous coronary intervention. Eur. Heart J. 31, 561–572 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone G. W., Witzenbichler B., Guagliumi G., Peruga J. Z., Brodie B. R., Dudek D., Kornowski R., Hartmann F., Gersh B. J., Pocock S. J., Dangas G., Wong S. C., Kirtane A. J., Parise H., Mehran R.; HORIZONS-AMI Trial Investigators , Bivalirudin during primary PCI in acute myocardial infarction. N. Engl. J. Med. 358, 2218–2230 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Tamez H., Pinto D. S., Kirtane A. J., Litherland C., Yeh R. W., Dangas G. D., Mehran R., Deliargyris E. N., Ortiz G., Gibson C. M., Stone G. W., Effect of short procedural duration with bivalirudin on increased risk of acute stent thrombosis in patients with STEMI: A secondary analysis of the HORIZONS-AMI randomized clinical trial. JAMA Cardiol. 2, 673–677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laine M., Frere C., Cuisset T., Paganelli F., Morange P. E., Dignat-George F., Berbis J., Camoin-Jau L., Bonello L., Potential mechanism of acute stent thrombosis with bivalirudin following percutaneous coronary intervention in acute coronary syndromes. Int. J. Cardiol. 220, 496–500 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Iijima R., Ndrepepa G., Mehilli J., Byrne R. A., Schulz S., Neumann F. J., Richardt G., Berger P. B., Schomig A., Kastrati A., Profile of bleeding and ischaemic complications with bivalirudin and unfractionated heparin after percutaneous coronary intervention. Eur. Heart J. 30, 290–296 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Tardy B., Lecompte T., Boelhen F., Tardy-Poncet B., Elalamy I., Morange P., Gruel Y., Wolf M., Francois D., Racadot E., Camarasa P., Blouch M. T., Nguyen F., Doubine S., Dutrillaux F., Alhenc-Gelas M., Martin-Toutain I., Bauters A., Ffrench P., de Maistre E., Grunebaum L., Mouton C., Huisse M. G., Gouault-Heilmann M., Lucke V.; GEHT-HIT study group , Predictive factors for thrombosis and major bleeding in an observational study in 181 patients with heparin-induced thrombocytopenia treated with lepirudin. Blood 108, 1492–1496 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Liu M., Shen S., Wen D., Li M., Li T., Chen X., Gu Z., Mo R., Hierarchical nanoassemblies-assisted combinational delivery of cytotoxic protein and antibiotic for cancer treatment. Nano Lett. 18, 2294–2303 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Clegg J. R., Irani A. S., Ander E. W., Ludolph C. M., Venkataraman A. K., Zhong J. X., Peppas N. A., Synthetic networks with tunable responsiveness, biodegradation, and molecular recognition for precision medicine applications. Sci. Adv. 5, eaax7946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen J. A., Beaudette T. T., Tseng W. W., Bachelder E. M., Mende I., Engleman E. G., Frechet J. M., T-cell activation by antigen-loaded pH-sensitive hydrogel particles in vivo: The effect of particle size. Bioconjug. Chem. 20, 111–119 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong J. X., Clegg J. R., Ander E. W., Peppas N. A., Tunable poly(methacrylic acid-co-acrylamide) nanoparticles through inverse emulsion polymerization. J. Biomed. Mater. Res. A 106, 1677–1686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasqualini R., Ruoslahti E., Organ targeting in vivo using phage display peptide libraries. Nature 380, 364–366 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Simberg D., Duza T., Park J. H., Essler M., Pilch J., Zhang L. L., Derfus A. M., Yang M., Hoffman R. M., Bhatia S., Sailor M. J., Ruoslahti E., Biomimetic amplification of nanoparticle homing to tumors. Proc. Natl. Acad. Sci. U.S.A. 104, 932–936 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agemy L., Sugahara K. N., Kotamraju V. R., Gujraty K., Girard O. M., Kono Y., Mattrey R. F., Park J. H., Sailor M. J., Jimenez A. I., Cativiela C., Zanuy D., Sayago F. J., Aleman C., Nussinov R., Ruoslahti E., Nanoparticle-induced vascular blockade in human prostate cancer. Blood 116, 2847–2856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takagi T., Doolittle R. F., Amino acid sequence studies on factor XIII and the peptide released during its activation by thrombin. Biochemistry 13, 750–756 (1974). [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y., Yu J., Wang J., Hanne N. J., Cui Z., Qian C., Wang C., Xin H., Cole J. H., Gallippi C. M., Zhu Y., Gu Z., Thrombin-responsive transcutaneous patch for auto-anticoagulant regulation. Adv. Mater. 29, 1604043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolberg A. S., Thrombin generation and fibrin clot structure. Blood Rev. 21, 131–142 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Jiang T., Shen S., Wang T., Li M., He B., Mo R., A substrate-selective enzyme-catalysis assembly strategy for oligopeptide hydrogel-assisted combinatorial protein delivery. Nano Lett. 17, 7447–7454 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Wang J., Yu J., Zhang Y., Zhang X., Kahkoska A. R., Chen G., Wang Z., Sun W., Cai L., Chen Z., Qian C., Shen Q., Khademhosseini A., Buse J. B., Gu Z., Charge-switchable polymeric complex for glucose-responsive insulin delivery in mice and pigs. Sci. Adv. 5, eaaw4357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung E., Kang C., Lee J., Yoo D., Hwang D. W., Kim D., Park S. C., Lim S. K., Song C., Lee D., Molecularly engineered theranostic nanoparticles for thrombosed vessels: H2O2-activatable contrast-enhanced photoacoustic imaging and antithrombotic therapy. ACS Nano 12, 392–401 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Weiss E. J., Hamilton J. R., Lease K. E., Coughlin S. R., Protection against thrombosis in mice lacking PAR3. Blood 100, 3240–3244 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Musumeci L., Kuijpers M. J., Gilio K., Hego A., Theatre E., Maurissen L., Vandereyken M., Diogo C. V., Lecut C., Guilmain W., Bobkova E. V., Eble J. A., Dahl R., Drion P., Rascon J., Mostofi Y., Yuan H., Sergienko E., Chung T. D., Thiry M., Senis Y., Moutschen M., Mustelin T., Lancellotti P., Heemskerk J. W., Tautz L., Oury C., Rahmouni S., Dual-specificity phosphatase 3 deficiency or inhibition limits platelet activation and arterial thrombosis. Circulation 131, 656–668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue M., Luo D., Yu S., Liu P., Zhou Q., Hu M., Liu Y., Wang S., Huang Q., Niu Y., Lu L., Hu H., Misshapen/NIK-related kinase (MINK1) is involved in platelet function, hemostasis, and thrombus formation. Blood 127, 927–937 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Cooley B. C., Murine arterial thrombus induction mechanism influences subsequent thrombodynamics. Thromb. Res. 135, 939–943 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Hu Q., Qian C., Sun W., Wang J., Chen Z., Bomba H. N., Xin H., Shen Q., Gu Z., Engineered nanoplatelets for enhanced treatment of multiple myeloma and thrombus. Adv. Mater. 28, 9573–9580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez de Lizarrondo S., Gakuba C., Herbig B. A., Repesse Y., Ali C., Denis C. V., Lenting P. J., Touze E., Diamond S. L., Vivien D., Gauberti M., Potent thrombolytic effect of N-acetylcysteine on arterial thrombi. Circulation 136, 646–660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/41/eabc0382/DC1