

Lnc-UC, an oscillating and NF-κB–driven lncRNA, functions as a Rev-erbα modulator and links circadian clock to colitis.
Abstract
We uncover a cycling and NF-κB–driven lncRNA (named Lnc-UC) that epigenetically modifies transcription of circadian clock gene Rev-erbα, thereby linking circadian clock to colitis. Cycling expression of Lnc-UC is generated by the central clock protein Bmal1 via an E-box element. NF-κB activation in experimental colitis transcriptionally drives Lnc-UC through direct binding to two κB sites. Lnc-UC ablation disrupts colonic expressions of clock genes in mice; particularly, Rev-erbα is down-regulated and its diurnal rhythm is blunted. Consistently, Lnc-UC promotes expression of Rev-erbα (a known dual NF-κB/Nlrp3 repressor) to inactivate NF-κB signaling and Nlrp3 inflammasome in macrophages. Furthermore, Lnc-UC ablation sensitizes mice to experimental colitis and abolishes the diurnal rhythmicity in disease severity. Mechanistically, Lnc-UC physically interacts with Cbx1 protein to reduce its gene silencing activity via H3K9me3, thereby enhancing Rev-erbα transcription and expression. In addition, we identify a human Lnc-UC that has potential to promote Rev-erbα expression and restrain inflammations.
INTRODUCTION
Ulcerative colitis (UC) and Crohn’s disease, collectively known as inflammatory bowel diseases (IBDs), are global disorders with a high incidence in developed countries (>0.3% prevalence) and an accelerating incidence in developing countries, resulting in severe morbidity, substantial health care costs, and loss of productivity at work (1, 2). IBDs are characterized by repeated relapses and remissions over long periods (3). Unfortunately, the pathogenesis of IBDs is poorly understood and the diseases are incurable so far, although some genetic and environmental factors are shown to make a contribution (4). The current goal of IBD treatment is to maintain stable remission without clinical symptoms and to decrease the incidence and duration of relapse (5). It is therefore of clinical importance to study IBD pathogenesis and to develop new therapeutic strategies (6).
Life on earth is subjected to circadian rhythms (a ~24-hour oscillation) due to daily changes in the environment (e.g., sunlight and temperature) (7). In mammals, circadian rhythms are driven by the circadian timing system consisting of a master clock (located in the suprachiasmatic nucleus of hypothalamus) and peripheral clocks (present in peripheral tissues) (8). At the molecular level, all clocks are composed of dozens of genes and proteins, forming interlocked autoregulatory feedback loops (9, 10). In the main loop, Bmal1 heterodimerizes with Clock or Npas2 to activate transcription of circadian genes including Per and Cry, whose protein products, in turn, inhibit the activators’ activity to repress their expressions (9, 10). Additional loops involve Rev-erbs, Rors, Dbp, and E4bp4 (9, 10). Their contributions to circadian rhythms are attained by regulating Bmal1 or Per2 expression (9, 10).
Circadian clock has been implicated in the regulation of inflammatory diseases (immune responses), accounting for diurnal rhythmicity in symptom severity (11, 12). UC is no exception because of a regulatory action from the clock component Rev-erbα (13). Rev-erbα regulates colitis development through transcriptional repression of nuclear factor κB (NF-κB) signaling and Nlrp3 inflammasome pathways (13). On the other hand, patients or mice with inflammatory diseases (e.g., IBDs) show disturbed circadian rhythms, highlighting potential mutual interactions between circadian and immune systems (14–16). This bidirectional regulation may be involved in the pathogenesis of IBDs (10). Although how circadian clock affects inflammations is becoming clearer, the molecular mechanisms by which inflammations regulate circadian clock remain elusive.
LncRNAs (long noncoding RNAs), defined as having ≥200 nucleotides, are a class of noncoding RNAs (ncRNAs). LncRNAs regulate gene transcription by interacting physically with DNA, other RNA, and protein via nucleotide base pairing or formation of structural domains generated by RNA folding (17). Several lncRNAs (e.g., lincRNA-EPS, Mirt2, and Arid2-IR) have been implicated in immune responses, playing a role in th3e development of inflammatory diseases such as endotoxemia and renal inflammation (18–20). In addition, lncRNAs have potential impact on circadian biology, and, in turn, they may be under the control of circadian clock (21, 22). However, the role of lncRNAs in the cross-talk of colitis with circadian clock is largely unknown. In this study, we uncover a cycling and colitis-related lncRNA (named Lnc-UC) in mice and in humans. Lnc-UC, driven by NF-κB signaling in colitis, promotes Rev-erbα transcription to alter circadian gene expression. Mechanistically, Lnc-UC interacts with Cbx1 to reduce H3K9me3 at Rev-erbα promoter and to induce Rev-erbα transcription and expression. We therefore propose that Lnc-UC functions as a Rev-erbα modulator, linking circadian clock to colitis.
RESULTS
Lnc-UC is a cycling and colitis-related lncRNA
Diurnal expression of lncRNAs was assessed by sampling mouse colon every 4-hour over a 24-hour light-dark cycle. RNA sequencing (RNA-seq) revealed overall disruption of colonic cycling lncRNAs (i.e., diurnally expressed lncRNAs detected by the JTK_CYCLE algorithm) in mice with dextran sulfate sodium (DSS)–induced colitis as compared to normal mice (Fig. 1A and fig. S1A). We also observed disrupted rhythmicity in cycling mRNAs (n = 3504) in colitis mice consistent with a previous study (fig. S1, B and C) (13). The 197 differentially expressed lncRNAs were identified between colitis and normal mice, and 28 of them were cycling lncRNAs (Fig. 1, B and C) including Lnc-UC (ENSMUST00000124503.1) that is highly conserved across various species such as mice, rats, and humans (fig. S2A). Coding potential analyses indicated Lnc-UC as a “real” noncoding transcript (i.e., does not code a protein) (fig. S2B). Lnc-UC did not affect expression of its neighboring gene Slit3 (fig. S2C). Colonic Lnc-UC with a peak expression at ZT10 was markedly up-regulated in colitis mice (Fig. 1D). Chromogenic in situ hybridization (CISH) confirmed the presence of Lnc-UC in mouse colon (Fig. 1E). In addition to the colon, Lnc-UC was expressed in many other tissues including the lung, heart, brain, kidney, and peritoneal macrophages (PMs) (Fig. 1F). Moreover, Lnc-UC was predominantly localized in the nuclei of colon cells and bone marrow–derived macrophages (BMDMs) (Fig. 1, G and H).
Fig. 1. Lnc-UC is a cycling and colitis-related lncRNA.
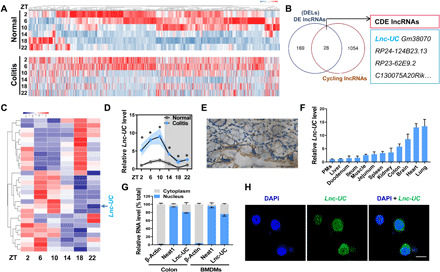
(A) Heat map for cycling lncRNAs in colons of colitis and normal mice at six circadian time points. Red indicates high expression, and blue indicates low expression of lncRNAs as shown in the scale bar. (B) Venn diagram showing numbers of cycling, DE (differentially expressed), and CDE (cycling and differentially expressed) lncRNAs. (C) Heat map for CDE lncRNAs in colons of normal mice at six circadian time points. (D) Quantitative polymerase chain reaction measurements of colonic Lnc-UC at six circadian time points. Data are mean ± SD (n = 3). *P < 0.05 at individual time points as determined by two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. (E) CISH analysis of Lnc-UC expression in colon of WT mice. Scale bar, 50 μm. (F) Lnc-UC levels in PMs and various tissues of WT mice. Data are mean ± SD (n = 5). (G) Cytoplasmic and nuclear levels of Lnc-UC in colon and BMDMs derived from WT mice. Data are mean ± SD (n = 5). (H) FISH analysis of subcellular Lnc-UC (green) location in BMDMs. Scale bar, 10 μm.
Circadian clock gene Bmal1 regulates rhythmic expression of Lnc-UC
Circadian gene expression is generally produced by a transcriptional mechanism in which core clock genes act on three cis-elements (E-box, D-box, and RevRE or RORE) in target gene promoter (23). We thus first investigated the roles of these three cis-elements in generation of rhythmic Lnc-UC using Bmal1−/−, E4bp4−/−, and Rev-erbα−/− mice. Bmal1, E4bp4, and Rev-erbα are respectively representative cis-acting proteins for E-box, D-box, and RevRE. Lnc-UC expression was down-regulated, and its rhythm was blunted in Bmal1−/− mice (fig. S3A). By contrast, Lnc-UC remained unaffected in E4bp4−/− or Rev-erbα−/− mice (fig. S3A). Supporting an activation effect, Bmal1 overexpression in BMDMs led to increased Lnc-UC and Bmal1 knockdown suppressed Lnc-UC expression (fig. S3B). Consistently, Bmal1 ablation down-regulated Lnc-UC and Dbp (a known Bmal1 target gene) and dampened their rhythms in serum-shocked (synchronized) BMDMs (fig. S3C). Serum shock is a well-recognized method that can synchronize circadian cycles and induce circadian gene expression in cultured cells. Furthermore, Bmal1 dose dependently induced the activity of the luciferase reporter driven by a 2.1-kb Lnc-UC promoter (−2.0/+0.1 kb) (fig. S3D). A canonical E-box (−1390/−1385 bp) in Lnc-UC promoter was identified to be critical for Bmal1 action, by testing Bmal1 effects on different promoter regions with E-box or without E-box site (fig. S3E). Chromatin immunoprecipitation (ChIP) assays showed that colonic Bmal1 protein was recruited to the E-box of Lnc-UC in wild-type (WT) mice in a circadian time-dependent manner (fig. S3F). Bmal1 recruitment was more extensive at ZT6 than at ZT18 (fig. S3F). However, Bmal1 recruitment was reduced and its time dependency was abolished in Bmal1−/− mice (fig. S3F). Together, Bmal1 transcriptionally regulates Lnc-UC and generates its diurnal rhythmicity (fig. S3G).
NF-κB drives Lnc-UC expression in colitis mice
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated that DSS-induced colitis was associated with activation of NF-κB and NF-κB–related signaling pathways [e.g., tumor necrosis factor (TNF), Rap1, and phosphatidylinositol 3-kinase (PI3K)–Akt signaling] (Fig. 2A and fig. S4A). This was supported by elevated expressions of NF-κB target genes such as Ptgs2, IL-1β, and Cxcl2 (Fig. 2B). Because Lnc-UC was induced in colitis mice (Fig. 1, C and D), it was of interest to test whether NF-κB played a role in regulation of Lnc-UC. Lipopolysaccharide (LPS; an NF-κB activator) dose and time dependently increased Lnc-UC expression in BMDMs (Fig. 2, C and D). Similar induction effects of LPS on Lnc-UC were observed in serum-shocked BMDMs (Fig. 2E) and in PMs (Fig. 2F). Fluorescence in situ hybridization (FISH) assays confirmed an increased nuclear Lnc-UC expression in LPS-stimulated BMDMs (Fig. 2G). Also, both recombinant Tnfα protein (an NF-κB activator) and p65 (an NF-κB subunit) overexpression plasmid induced Lnc-UC expression in BMDMs (Fig. 2H). Furthermore, JSH-23 (a specific NF-κB inhibitor) inhibited LPS activation of Lnc-UC in BMDMs (Fig. 2I). Moreover, Lnc-UC expression was correlated with NF-κB activity (reflected by IL-1β and Nlrp3 levels) during colitis development (fig. S4B). On the basis of luciferase reporter analyses, we identified two κB sites (i.e., −137/−127 bp and − 108/−98 bp) in Lnc-UC promoter for transcriptional action of p65 (Fig. 2J). Recruitment of p65 to these two κB sites was confirmed by ChIP assays (Fig. 2K). p65 recruitment was enhanced by recombinant Tnfα protein (Fig. 2K). Collectively, NF-κB drives expression of Lnc-UC via a transcriptional activation mechanism.
Fig. 2. NF-κB drives expression of Lnc-UC.
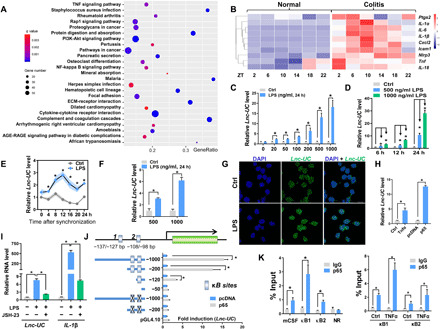
(A) KEGG pathway analysis of differentially expressed mRNAs associated with colitis at six circadian time points. (B) Heat map for colonic transcripts of NF-κB target genes in colitis and normal mice at six circadian time points. (C) Lnc-UC levels in BMDMs treated with LPS or vehicle for 24 hours. (D) Lnc-UC levels in BMDMs treated with LPS or vehicle for 6, 12, or 24 hours. (E) Lnc-UC levels in serum-shocked BMDMs treated with LPS or vehicle. (F) Lnc-UC levels in PMs treated with LPS or vehicle for 24 hours. (G) FISH analysis of Lnc-UC (green) in BMDMs treated with LPS or vehicle. Scale bar, 10 μm. (H) Lnc-UC levels in BMDMs treated with Tnfα or p65. (I) Lnc-UC and IL-1β levels in BMDMs treated with LPS or JSH-23. (J) Effects of p65 on different versions of Lnc-UC promoters. Data are mean ± SD (n = 6). “X” mark denotes a mutation. (K) ChIP assays showing enrichment of p65 to two κB sites of Lnc-UC promoter (left) and enhanced enrichment by Tnfα (right). A κB site of mCSF (macrophage colony-stimulating factor) promoter and a nonspecific region (NR) of Lnc-UC promoter were used as a positive and negative control, respectively. In (C) to (E), (F), (H), (I), and (K), data are mean ± SD (n = 5). In (C), (F), (H), (J), and (K), *P < 0.05 as determined by Student’s t test; in (D) and (I), *P < 0.05 as determined by one-way ANOVA followed by Bonferroni post hoc test; in (E), *P < 0.05 at individual time points as determined by two-way ANOVA followed by Bonferroni post hoc test.
Lnc-UC acts as an inflammatory regulator in vitro
Lnc-UC–overexpressed and control BMDMs were subjected to RNA-seq analyses. Lnc-UC induced notable expression changes in genes involved in immune responses (NF-κB and related signaling pathways) (Fig. 3, A and B, and fig. S5A). In line with RNA-seq data, Lnc-UC was a negative regulator of NF-κB target genes (including Nlrp3, IL-6, Tnfα, IL-1β, and IL-18) in BMDMs and in PMs based on gain- and loss-of-function experiments (Fig. 3, C and D, and fig. S5, B to D). Lnc-UC knockdown–induced changes in expressions of NF-κB target genes can be restored by Lnc-UC overexpression, confirming a specific effect of Lnc-UC on NF-κB (Fig. 3E). Supporting this, Lnc-UC inhibited the transcriptional activity of NF-κB according to luciferase reporter assay and electrophoretic mobility shift assay (Fig. 3, F and G). In addition, Lnc-UC inhibited p65 phosphorylation and the translocation of p65 from cytoplasm to nucleus (fig. S6, A to D). Moreover, Lnc-UC inactivated Nlrp3 inflammasome (whose activity depends on NF-κB signaling) as evidenced by reduced maturation of and release of proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 (fig. S6, E and F). Overall, these data indicated a repressive role of Lnc-UC in regulation of NF-κB signaling and inflammations.
Fig. 3. Lnc-UC acts as an inflammatory regulator.
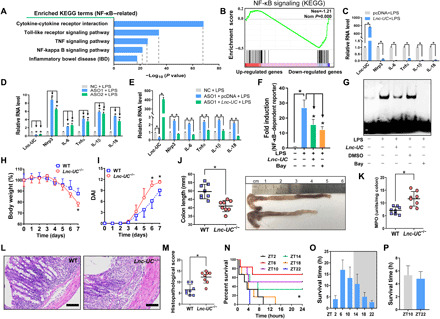
(A) KEGG analysis of Lnc-UC–induced differentially expressed genes in BMDMs (NF-κB–related pathways are shown). (B) Gene set enrichment analysis (GSEA) analysis showing activation of NF-κB signaling pathway by Lnc-UC. (C) Lnc-UC overexpression down-regulates NF-κB target genes in LPS-treated BMDMs. (D) Lnc-UC knockdown by ASOs up-regulates NF-κB target genes in LPS-treated BMDMs. (E) Rescue experiments showing that Lnc-UC knockdown–induced changes in expressions of NF-κB target genes can be restored by Lnc-UC overexpression. (F) Effects of Lnc-UC and Bay 11-7082 (Bay) on NF-κB–dependent reporter activity. (G) Effects of Lnc-UC and Bay on the binding of NF-κB DNA probe to nuclear proteins derived from BMDMs. (H) Weight loss measurements of Lnc-UC−/− and WT mice treated with DSS. (I) DAI scores of Lnc-UC−/− and WT mice. (J) Colon lengths of Lnc-UC−/− and WT mice. Photo credit: Y. Lin, College of Pharmacy, Jinan University. (K) MPO activities of Lnc-UC−/− and WT mice. (L) Representative images of hematoxylin and eosin staining in the colon. Scale bars, 100 μm. (M) Histopathological scores of Lnc-UC−/− and WT mice. Survival rate (N) and survival time (O) of WT mice after oxazolone treatment. (P) Survival time of Lnc-UC−/− mice after oxazolone treatment. In (C) to (F), data are mean ± SD (n = 5). In (H) to (K), (M), (O), and (P), data are mean ± SD (n = 8). In (C), (J), (K), and (M), *P < 0.05 as determined by Student’s t test; in (D) to (F), *P < 0.05 as determined by one-way ANOVA followed by Bonferroni post hoc test; in (H) and (I), *P < 0.05 at individual days as determined by two-way ANOVA followed by Bonferroni post hoc test; in (N), *P < 0.05 as determined by log-rank test.
Lnc-UC ablation sensitizes mice to experimental colitis
We established Lnc-UC–deficient (Lnc-UC−/−) mice by deleting ENSMUSG00000086311 sequence (fig. S7, A and B). Both Lnc-UC−/− and WT mice were subjected to colitis inducement with DSS. Lnc-UC−/− mice showed an increased sensitivity to colitis development, as evidenced by more extensive weight loss, higher disease activity index (DAI), shorter colons, and higher myeloperoxidase (MPO) activity (Fig. 3, H to K). More severe colitis in Lnc-UC−/− mice was confirmed by histological examinations (Fig. 3, L and M). Lnc-UC deficiency was also associated with an increased number of macrophages, aggravated cell apoptosis, and reduced cell proliferation (fig. S7, C to E). In addition, colonic protein levels of phosphorylated p65 and mature caspase-1 as well as proinflammatory cytokines (i.e., IL-1β and IL-6) were higher in Lnc-UC−/− mice than in WT mice, indicating exacerbated activation of NF-κB signaling and Nlrp3 inflammasome in the genetically modified mice (fig. S7, F and G). Overall, these data supported that Lnc-UC regulated the development of experimental colitis in mice. Because Lnc-UC is diurnally rhythmic, it was of interest to test circadian time-varying severity in colitis. Mice were treated with oxazolone to induce colitis at six different circadian points. Colitis severity (reflected by the mortality rate or survival time) displayed a significant diurnal rhythm (most severe at ZT22 and least severe at ZT6) (Fig. 3, N and O). However, the time difference in disease severity was lost in Lnc-UC−/− mice (Fig. 3P), supporting circadian regulation of colitis by Lnc-UC.
Lnc-UC regulates circadian clock gene Rev-erbα and inflammations
Lnc-UC ablation led to disrupted expressions of circadian clock genes (Fig. 4A and fig. S7H). In particular, Rev-erbα was markedly down-regulated (Fig. 4, A and B). Rev-erbα has been established as an integrator of circadian clock and colonic inflammation via inactivation of NF-κB signaling and Nlrp3 inflammasome (19, 20, 22). We thus investigated a potential role of Rev-erbα in Lnc-UC regulation of inflammation. As expected, overexpression of Rev-erbα in BMDMs induced expression changes in genes involved in immune responses (NF-κB and related signaling pathways) (Fig. 4C). Rev-erbα ablation led to increased levels of NF-κB–related inflammatory factors, confirming Rev-erbα as a negative regulator of inflammatory responses (Fig. 4D). We observed a high overlap (~50%) between Lnc-UC– and Rev-erbα–associated enriched pathways (including inflammatory responses based on KEGG analysis) in BMDMs (Fig. 4E). In addition, Lnc-UC reduced the mRNA level of Nlrp3 (a direct Rev-erbα target gene), and this inhibitory effect was lost in Rev-erbα−/− mice (Fig. 4F). All these data indicated a mediating role for Rev-erbα in Lnc-UC regulation of inflammation.
Fig. 4. Lnc-UC regulates circadian clock gene Rev-erbα and inflammations.
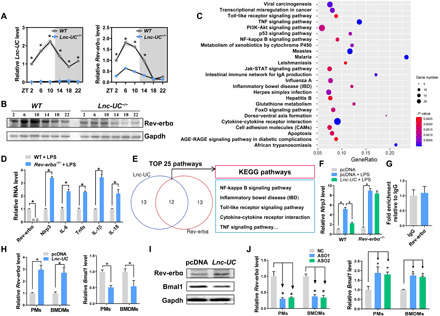
(A) Expression of Lnc-UC and Rev-erbα mRNA in the colons of Lnc-UC−/− and WT mice at six circadian time points. (B) Protein expression of Rev-erbα in the colons of Lnc-UC−/− and WT mice at six circadian time points. (C) KEGG analysis of Rev-erbα–induced differentially expressed genes in BMDMs. (D) Rev-erbα ablation up-regulates NF-κB target genes in BMDMs. (E) Venn diagram showing an extensive overlap of Lnc-UC– and Rev-erbα–associated pathways. (F) mRNA levels of Nlrp3 in BMDMs derived from Rev-erbα−/− and WT mice. (G) RIP assays showing no interaction between Rev-erbα protein and Lnc-UC. (H) Effects of Lnc-UC overexpression on mRNA levels of Rev-erbα and Bmal1 in PMs and BMDMs. (I) Effects of Lnc-UC overexpression on protein levels of Rev-erbα and Bmal1 in PMs and BMDMs. (J) Effects of Lnc-UC knockdown by ASO1 and ASO2 on mRNA levels of Rev-erbα and Bmal1 in PMs and BMDMs. In (H) to (J), cells were transfected with overexpression plasmid or ASO for 24 hours before harvest. Data are mean ± SD (n = 5). In (A), *P < 0.05 at individual time points as determined by two-way ANOVA followed by Bonferroni post hoc test; in (D) and (H), *P < 0.05 as determined by Student’s t test; in (F) and (J), *P < 0.05 as determined by one-way ANOVA followed by Bonferroni post hoc test.
We next explored the mechanisms by which Lnc-UC regulated Rev-erbα. Because there were no interactions between Lnc-UC and Rev-erbα according to RNA immunoprecipitation (RIP) assay (Fig. 4G), we tested whether Lnc-UC regulated the expression of Rev-erbα. Overexpression of Lnc-UC led to an increase in cellular Rev-erbα expression and therefore to a decrease in Bmal1 (a Rev-erbα target gene) expression (Fig. 4, H and I). Consistently, Lnc-UC knockdown resulted in reduced Rev-erbα expression and in increased Bmal1 expression (Fig. 4J). The data suggested that the anti-inflammatory effect of Lnc-UC was attained probably by regulating Rev-erbα expression.
Lnc-UC epigenetically regulates Rev-erbα by interacting with Cbx1
Lnc-UC contains four stem loop structures [i.e., 1–74, 123–193, 213–267, and 326–432 nucleotides (nt)] that may bind to target proteins to affect their functions (Fig. 5A). RNA pull-down assays followed by silver staining, mass spectrometric analyses, and Western blotting identified Cbx1 (about 25 kDa, also known as HP1β) as a nuclear protein interacting with Lnc-UC (Fig. 5, B and C). A direct interaction of Lnc-UC with Cbx1 was further confirmed by RIP assays (Fig. 5D). Moreover, Lnc-UC and Cbx1 colocalized in the nuclei of BMDMs (Fig. 5E). Truncation analyses suggested that the fragments of 1–74 nt and 326–432 nt were responsible for Cbx1 binding (Fig. 5C). Supporting this, Lnc-UC carrying mutations of both 1–74 nt and 326–432 nt fragments failed to induce Rev-erbα expression or to suppress inflammatory responses (Fig. 5F and fig. S8, A and B).
Fig. 5. Lnc-UC interacts with Cbx1 and modulates Rev-erbα transcription by reducing trimethylation of H3K9.
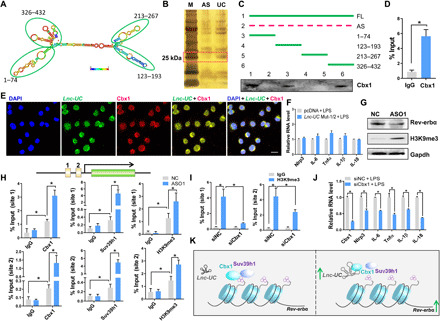
(A) Secondary structure of Lnc-UC with minimum free energy predicted by Vienna RNA web server. (B) Silver staining of protein mixtures following an RNA pull-down assay. (C) Western blotting analysis of interactions between Cbx1 protein and different versions of Lnc-UC probes following an RNA pull-down assay. Full-length Lnc-UC (FL), antisense (AS), and truncated fragments are labeled in the top panel. (D) RIP assay showing an interaction between Cbx1 and Lnc-UC. (E) FISH-immunofluorescence assays showing colocalization of Lnc-UC with Cbx1 protein in BMDMs. Nuclei are stained with DAPI (blue). Scale bar, 10 μm. (F) Effects of Lnc-UC mutation on mRNA levels of inflammatory factors in BMDMs. Lnc-UC mut-1/2 [Lnc-UC with mutated sites of (1–74 nt) and (326–432 nt)]. (G) Effects of Lnc-UC knockdown by ASO1 on protein levels of H3K9me3, Rev-erbα, and Gapdh in BMDMs. (H) Lnc-UC knockdown by ASO1 increases enrichments of Cbx1, H3K9me3, and Suv39h1 onto Rev-erbα promoter in BMDMs. (I) Cbx1 knockdown by small interfering RNA (siRNA) decreases enrichment of H3K9me3 onto Rev-erbα promoter in BMDMs. (J) Effects of Cbx1 knockdown on mRNA levels of inflammatory factors in BMDMs. (K) Schematic diagram showing the molecular mechanism for Lnc-UC regulation of Rev-erbα expression. Data are mean ± SD (n = 5). In (D) and (J), *P < 0.05 as determined by Student’s t test; in (H) and (I), *P < 0.05 as determined by two-way ANOVA followed by Bonferroni post hoc test.
Functionally, Cbx1 binds to sites of histone H3 lysine 9 (H3K9) methylation and recruits histone methyltransferases such as Suv39h1 (a histone-lysine N-methyltransferase), playing an important role in gene silencing and formation of heterochromatin (24–26). Lnc-UC knockdown enhanced the trimethylation of H3K9 in BMDMs, raising a possibility for epigenetic regulation of Rev-erbα by the lncRNA (Fig. 5G). ChIP assays showed significant enrichments of Cbx1, Suv39h1, and H3K9me3 to Rev-erbα promoter (Fig. 5H). Enrichments of these three proteins were enhanced when Lnc-UC was knocked down (Fig. 5H). In addition, knockdown of Cbx1 reduced enrichment of H3K9me3 to Rev-erbα promoter, promoting Rev-erbα transcription and expression (Fig. 5I and fig. S8C). Supporting this, knockdown of Cbx1 suppressed the expression of inflammatory factors in LPS-stimulated BMDMs (Fig. 5J). Lnc-UC failed to increase Rev-erbα expression in Cbx1-silenced BMDMs (fig. S8D). Collectively, Lnc-UC regulates Rev-erbα transcription through attenuation of Cbx1-mediated trimethylation of H3K9 (Fig. 5K). To be specific, Lnc-UC may act as a decoy and titrate away Cbx1 and its partners such as Suv39h1 from the Rev-erbα promoter (Fig. 5K). Thereby, trimethylation of H3K9 is diminished and Rev-erbα transcription is activated (Fig. 5K).
Human Lnc-UC (hLnc-UC) is a cycling and anti-inflammatory lncRNA
It is of interest to test whether function of Lnc-UC in mice can be translated to humans due to high species conservation (fig. S2A). Full length of human Lnc-UC (hLnc-UC) was identified by performing rapid amplification of complementary DNA (cDNA) ends (RACE) and mapped to the region of Chr5:169,052,253-169,052,777 (Fig. 6A and fig. S9, A and B). CISH assays confirmed the presence of hLnc-UC in human colons (Fig. 6B). Patients with colitis showed increased hLnc-UC as well as NLRP3 and IL-1β in colonic mucosae as compared to healthy individuals (Fig. 6C). This lncRNA was rhythmically expressed in serum-shocked THP-1 cells with a pattern similar to BMAL1 (Fig. 6D). Moreover, overexpression of hLnc-UC in THP-1 cells resulted in increased REV-ERBα expression and in reduced levels of REV-ERBα–dependent inflammatory factors (Fig. 6, E to G). Similar observations were noted in monocyte-derived macrophages (MDMs) (Fig. 6, H and I). Together, these data suggest that colitis-related hLnc-UC may promote REV-ERBα expression to restrain inflammations as its counterpart does in mice.
Fig. 6. Human Lnc-UC (hLnc-UC) is a cycling and anti-inflammatory lncRNA.
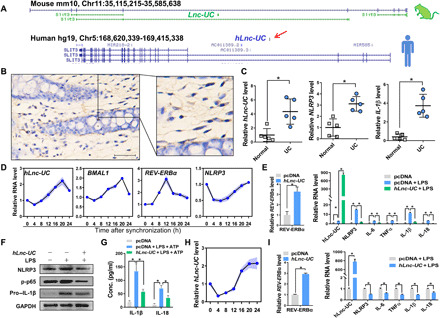
(A) Genomic locations of Lnc-UC and hLnc-UC. (B) CISH analysis of hLnc-UC in human colons. Scale bar, 50 μm. (C) hLnc-UC, Nlrp3, and IL-1β levels in the mucosae of patients with colitis and healthy individuals. (D) Rhythmic expressions of hLnc-UC, BMAL1, REV-ERBα, and NLRP3 in serum-shocked THP-1 cells. (E) hLnc-UC overexpression induces mRNA expression of REV-ERBα and represses inflammatory factors in THP-1 cells. (F) hLnc-UC overexpression increases REV-ERBα protein and reduces NLRP3, phosphorylated p65 (p-p65), and pro–IL-1β in THP-1 cells. (G) hLnc-UC reduces IL-1β and IL-18 in BMDM incubation medium. (H) Rhythmic expression of hLnc-UC in serum-shocked MDMs. (I) hLnc-UC overexpression induces mRNA expression of REV-ERBα and represses inflammatory factors in MDMs. Data are mean ± SD (n = 5). In (C), (E, left) and (I), *P < 0.05 as determined by Student’s t test; in (E, right) and (G), *P < 0.05 as determined by one-way ANOVA followed by Bonferroni post hoc test.
DISCUSSION
We have uncovered an NF-κB–driven lncRNA (named Lnc-UC) that epigenetically modifies transcription of circadian clock genes (Rev-erbα as a representative), thereby linking circadian clock to colitis. Defining this mediating role for Lnc-UC enhances a deeper understanding of the cross-talk between colitis and circadian clock (particularly on the side of regulation of circadian clock by colitis) and may promote new lncRNA-related strategies for management of colitis. LncRNA-based therapy has been shown to be effective against other diseases such as osteoporosis and cancers (27, 28). In an attempt to identify potential lncRNAs associated with both colitis and circadian clock, we applied two screening criteria, namely, rhythmically expressed and colitis-regulated (Fig. 1). Lnc-UC was a cycling lncRNA that up-regulated the most in colitis mice. There is a possibility that other cycling lncRNAs (e.g., Gm38070) may also serve as molecular linkers of colitis with circadian clock. However, this was not addressed in the current study.
Our findings may suggest a self-protective mechanism against colitis because NF-κB–driven Lnc-UC promotes Rev-erbα transcription to restrain colonic inflammation (Fig. 4). Mechanistically, Lnc-UC physically interacts with Cbx1 protein to reduce its gene-silencing activity via H3K9me3 and to enhance Rev-erbα expression. This conforms to a general RNA-protein interaction mechanism for lncRNA actions (29, 30). Rev-erbα enhancement down-regulates inflammatory responses via inactivation of NF-κB signaling and Nlrp3 inflammasome consistent with previous studies (13, 31, 32). This self-protective mechanism may support the notion that the body has a certain ability to self-heal diseases (33, 34).
We observed a dependence of colitis severity on the time of induction in mice (Fig. 3). Similar observations have been noted for fulminant hepatitis and rheumatoid arthritis (12, 32). Patients with colitis show diurnal (and seasonal) rhythmicity in flares of symptoms (35). The diurnal rhythmicity in colitis severity was antiphase to circadian Rev-erbα expression, supporting a central role of Rev-erbα as an inflammatory repressor in generating rhythmicity of colitis (36). Because Lnc-UC is an activator of Rev-erbα, it is postulated that Lnc-UC may act as a modulator of diurnal rhythmicity in inflammatory symptoms. This is supported by the fact that Lnc-UC knockout blunted the rhythm of colitis severity in mice (Fig. 3).
Our study highlighted a critical role of NF-κB signaling in bridging colitis with circadian clock via Lnc-UC. This helps to explain why inflammatory cytokines such as Tnfα and IL-1β can modulate the expression of clock genes (e.g., Per2 and Dbp) because both Tnfα and IL-1β are activators of NF-κB signaling pathway (37, 38). Although NF-κB signaling and related pathways (TNF, Toll-like receptor, and RIG-I–like receptor signaling) are primarily activated during colitis development, other pathways such as the mitogen-activated protein kinase (MAPK) signaling may be also involved (39). However, whether MAPK signaling contributes to mutual interactions of colitis with circadian clock remains unknown.
We demonstrated suppressive effects of Lnc-UC on inflammations in macrophages and in colon of mice with colitis. In addition, Lnc-UC repressed LPS-induced inflammatory cytokines in colonic epithelial cells (CECs; fig. S5E). Collectively, these data define Lnc-UC as an anti-colitis factor. The anti-inflammatory function of Lnc-UC may not be restricted to the colon tissue because Lnc-UC and its target Rev-erbα are ubiquitously distributed in the body with high expressions in other noncolon tissues such as lung, heart, and brain (Fig. 1F). However, whether Lnc-UC has anti-inflammatory effects in other tissues awaits further investigations.
Previous studies show down-regulation of Rev-erbα in colitis and pulmonary inflammation (13, 40). We confirmed a reduction in colonic Rev-erbα expression in WT mice during the induction of colitis (fig. S4C). This seems to be in conflict with an elevation in Lnc-UC, a Rev-erbα activator, in colitis development. Colitis-associated down-regulation of Rev-erbα was much more extensive in Lnc-UC−/− mice (fig. S4C), supporting a role for Lnc-UC in resisting down-regulation of Rev-erbα by colitis (i.e., a Rev-erbα–promoting effect). It is therefore speculated that a down-regulation mechanism should be involved in the colitis-caused reduction of Rev-erbα; however, it was unaddressed in the current study. This down-regulation mechanism, probably involving NF-κB–mediated transcriptional repression (41, 42), overwhelms the activation effect of Lnc-UC, resulting in an “apparent” reduction in Rev-erbα expression.
In summary, NF-κB–driven Lnc-UC functions as an epigenetic regulator of Rev-erbα, thereby orchestrating circadian clock and colitis. Lnc-UC might be targeted to intervene in communications between inflammation and circadian clock and to manage colitis.
MATERIALS AND METHODS
Materials
Human and murine macrophage colony-stimulating factor (CSFs) were purchased from PeproTech (Rocky Hill, NJ). LPS, oxazolone, phorbol 12-myristate 13-acetate (PMA), adenosine triphosphate (ATP), and collagenase I were purchased from Sigma-Aldrich (St. Louis, MO). JSH-23 was purchased from Selleck (Houston, TX). DSS (molecular weight of 36 to 50 kDa) was obtained from MP Biomedicals (Irvine, CA). Murine Tnfα, BAY 11-7082, and NF-κB-Luc were purchased from Beyotime (Shanghai, China). Cytoplasmic/nuclear RNA purification kit was purchased from Norgen Biotek (Belmont, CA). FISH kit was purchased from GenePharma (Shanghai, China). RNAiso Plus reagent and PrimeScript RT Master Mix were purchased from Takara (Shiga, Japan). ChamQ Universal SYBR qPCR Master Mix was purchased from Vazyme (Nanjing, China). Dual-Luciferase Reporter Assay System was purchased from Promega (Madison, WI). Pierce magnetic RNA-protein pull-down kit was purchased from Thermo Fisher Scientific (San Jose, CA). Murine IL-1β and IL-18 enzyme-linked immunosorbent assay (ELISA) kits were purchased from Neobioscience (Shenzhen, China). JetPrime transfection kit was purchased from POLYPLUS Transfection (Ill kirch, France). ChIP kit was purchased from Cell Signaling Technology (Beverly, MA).
Probes, plasmids, ASOs (antisense oligonucleotides), and siRNAs (small interfering RNAs): Lnc-UC probes for RNA pull-down were provided by GenePharma (Shanghai, China). Probes for FISH and CISH were obtained from BersinBio (Guangzhou, China). Luc-UC luciferase reporters (−2000/+100 bp, −1000/+100 bp, −200/+100 bp, −120/+100 bp, −50/+100 bp, and mutated versions), pcDNA3.1, pcDNA3.1-p65, pcDNA3.1-Bmal1, pcDNA3.1-Lnc-UC, siBmal1, siCbx1, ASO-NC, Lnc-UC-ASO, pGL4.10, and pRL-TK were obtained from Transheep Technologies (Shanghai, China).
Antibodies: Anti-Nlrp3 and anti–IL-1β were purchased from AdipoGen (San Diego, CA). Anti-Bmal1, anti-Cbx1, anti-Suv39h1, anti-H3K9me3, and anti-Gapdh were purchased from Abcam (Cambridge, UK). Anti-p65, anti–Rev-erbα, anti–p-p65, anti– p-IKBα, and anti-rabbit immunoglobulin G (IgG) were obtained from Cell Signaling Technology (Danvers, MA). Anti-mouse IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Histone H3 was purchased from Proteintech (Wuhan, China).
Mice
WT C57BL/6 mice were obtained from HFK Biotechnology (Beijing, China). E4bp4−/− (a C57BL/6 background) was obtained from M. Kubo at RIKEN Institute in Japan (43). Bmal1−/− and Rev-erbα−/− mice (a C57BL/6 background) have been established and validated in our laboratory (13, 44). Lnc-UC−/− mice were generated by deleting ENSMUSG00000086311 gene with the aid of Cyagen Biosciences Inc. (Guangzhou, China). All mice were maintained under a 12-hour light/12-hour dark cycle [light on 7:00 a.m. (= ZT0) to 7:00 p.m. (= ZT12)] with ad libitum access to food and water. Mice with age of 6 to 12 weeks were used for experiments. All experiments were performed using protocols approved by the Institutional Animal Care and Use Committees of Jinan University.
Colitis models
DSS-induced colitis was established by feeding mice with 2.5% (w/v) DSS in drinking water for 7 days. Mice were sacrificed on day 7, and colons were collected for biochemical analyses (colonic IL-1β, IL-18, IL-6, Tnfα levels, and MPO activity). DAI was scored based on body weight loss, occult blood, and stool consistency as described previously (13). In addition, colon tissues were fixed in 4% paraformaldehyde and embedded in paraffin, followed by hematoxylin and eosin staining. Histological damage was scored based on goblet cells loss, mucosa thickening, inflammatory cells infiltration, submucosa cell infiltration, ulcers, and crypt abscesses (13). A score of 1 to 3 or 1 to 4 was given for each parameter with a maximal total score of 20.
Oxazolone-induced colitis was established with mice as previously described (45). In brief, a small area (approximately 2 × 2 cm) of dorsal skin was shaved, and 200 μl of 3% oxazolone was applied (presensitization). Seven days later, mice were challenged intrarectally with 180 μl of 2% oxazolone in 50% ethanol under light anesthesia. Animal survival was recorded in the following 24 hours.
Human specimens
Colonic specimens were obtained from the First Affiliated Hospital of Jinan University. Biopsies were obtained from inflamed areas of colonic tissues of patients with UC. Colonic mucosae were collected from patients with UC and healthy individuals. Research protocol was approved by the Medical Ethical Committee of the First Affiliated Hospital of Jinan University.
Isolation of macrophages
Primary PMs and BMDMs were isolated from mice as previously described (13). In brief, peritoneal fluid was collected and cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). Two hours later, adherent cells (PMs) were obtained and used for further experiments. Tibias were separated from the femurs, and the bones were washed using 75% ethanol. Bone marrow cavities were rinsed with RPMI 1640 medium using a syringe. The rinsing solutions were collected and centrifuged at 1000 rpm for 5 min. The pellet cells (BMDMs) were collected and cultured in RPMI 1640 medium containing 10% FBS, 1% penicillin-streptomycin, and murine CSF (20 ng/ml). MDMs were prepared as previously described (32). Human peripheral blood mononuclear cells were first isolated from the blood of healthy donors by Ficoll density gradient centrifugation. After 7 days of culture in RPMI 1640 with 10% FBS, 1% penicillin-streptomycin, and human CSF (20 ng/ml), monocytes were differentiated into macrophages.
Isolation of CECs
Mouse colons were dissected and flushed with phosphate-buffered solution containing 1 mM dithiothreitol (DTT) to remove fecal contents. Colons were digested by incubation with a solution containing 0.1% collagenase I at 37°C for 30 min. CECs were recovered by centrifugation at 1000 rpm for 5 min and suspended in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium containing 10% FBS.
Serum shock
BMDMs, MDMs, and THP-1 cells were cultured in RPMI medium containing 10% FBS and 1% penicillin-streptomycin. On the next day, the culture medium was replaced with serum-free RPMI medium. Twelve hours later, 50% FBS was added for 2 hours and the medium was changed back to serum-free RPMI medium. Cells were harvested for RNA extraction at 0, 4, 8, 12, 16, 20, and 24 hours after serum shock. In particular, BMDMs were primed with LPS (500 ng/ml) for 3 hours before harvest.
Quantitative polymerase chain reaction
Total RNA from colon samples or cells was isolated and reversely transcribed to cDNA. Polymerase chain reaction (PCR) amplification procedures were as follows: initial denaturation at 95°C for 5 min, 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Mouse Hmbs or human GAPDH were used as internal controls. Relative mRNA or lncRNA levels were determined using the 2−ΔΔCT method. Primers are provided in table S1.
Luciferase reporter assay
Cells were cotransfected with luciferase reporter, pRL-TK, and overexpression plasmid (Lnc-UC, p65, or Bmal1 plasmid). After 24-hour transfection, cells were lysed and luciferase activities were determined using Dual-Luciferase Reporter Assay System and GloMax 20/20 luminometer (Promega). Firefly luciferase activity was normalized to renilla luciferase activity and expressed as relative luciferase unit. For assessment of Lnc-UC effect on NF-κB transcription, BAY 11-7082 was added for 1 hour and LPS for 12 hours after cotransfection. Cells were then collected for measurements of luciferase activities.
Chromatin immunoprecipitation
ChIP assays were performed using BMDMs or colon tissues from mice with the Enzymatic Chromatin IP kit as previously described (13). In brief, cells or tissues were cross-linked in 37% formaldehyde for 10 min and the reaction was quenched by glycine. Cells were lysed, and nuclei were digested with micrococcal nuclease. Sheared chromatin was immunoprecipitated with antibodies against p65, Bmal1, Cbx1, H3K9me3, Suv39h1, or normal IgG. Immunoprecipitated chromatin was decross-linked at 65°C for 4 hours and purified with spin columns. Purified DNAs were analyzed by quantitative PCR (qPCR) with specific primers.
Electrophoretic mobility shift assay
Nuclear proteins were prepared from BMDMs using a cytoplasmic/nuclear protein extraction kit (Beyotime, Shanghai, China). Nuclear proteins were incubated with biotin-labeled oligonucleotide probes containing NF-κB binding site in a binding buffer containing poly(dI-dC), MgCl2, NaCl, EDTA, DTT, and glycerol. The mixture was loaded onto a 4% nondenaturing polyacrylamide gel. After electrophoresis, the products were transferred onto a Hybond-N+ membrane and visualized by using the enhanced chemiluminescence reagent and an Omega Lum G imaging system (Aplegen, Pleasanton, CA).
Western blotting
Protein samples were subjected to SDS–polyacrylamide gel electrophoresis (10% acrylamide gels). Products were transferred onto a polyvinylidene difluoride membrane, followed by incubation with primary antibody and with horseradish peroxidase–conjugated secondary antibody. The blots were visualized by using enhanced chemiluminescence and Omega Lum G imaging system (Aplegen, Pleasanton, CA).
RNA sequencing
Eighteen colitis mice and 18 normal mice were used for RNA-seq experiments. Three colitis mice and three normal mice were sacrificed at each circadian time point (ZT2, ZT6, ZT10, ZT14, ZT18, and ZT22), and the colons were collected, snap-frozen, and stored at −80°C. RNA was extracted using RNAiso Plus reagent and analyzed for quality using Agilent 2100 BioAnalyzer Expert (Agilent Technologies, Santa Clara, CA). Ribosomal RNA (rRNA) was depleted using a Ribo-zero Gold rRNA Removal kit (Illumina, San Diego, CA, USA). Strand-specific libraries were prepared using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (Ipswich, MA, USA). The libraries were sequenced on Illumina HiSeq X Ten to generate 2 × 150–base pair (bp) paired-end reads. Reads were aligned to mouse GRC38/mm10 genome with HISAT2 (v2.0.4). FPKMs (fragments per kilobase of transcript per million mapped reads) were calculated using the StringTie-eB software. Ensembl (GENCODE) transcripts were used for annotation. Differential gene expression analysis was performed using Ballgown software. Genes were defined as differentially expressed when P < 0.05 and fold change >1.5. Cycling mRNAs and lncRNAs were determined by the JTK-cycle algorithm with adjusted P < 0.05. In addition, RNA-seq was performed with BMDMs (Lnc-UC–transfected and LPS-stimulated BMDMs, Rev-erbα–transfected and LPS-stimulated BMDMs, and control BMDMs). Poly(A) RNA was extracted using poly-T oligo-attached magnetic beads. Poly(A) RNA was used for construction of libraries using NEBNext Ultra RNA Library Prep Kit for Illumina (Ipswich, MA, USA). Gene set enrichment analysis was performed using the GSEA software (Broad Institute). KEGG pathways fulfilling the criterion of a hypergeometric P < 0.05 were defined as significantly enriched in differentially expressed genes. In bubble plots (Figs. 2A and 4C and fig. S5A), GeneRatio is the number of observed divided by the number of expected genes from each KEGG term.
Fluorescence in situ hybridization
Subcellular localization of Lnc-UC was determined using a FISH kit according to the manufacturer’s protocol (GenePharma, Shanghai, China). In brief, BMDMs were fixed in 4% paraformaldehyde and hybridized with Lnc-UC probe (10 nM) in a hybridization buffer. After washing with saline sodium citrate, cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and the images were captured by using a laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde, permeated in 0.1% Triton X-100, and blocked with 5% bovine serum albumin. Thereafter, cells were incubated with primary antibody against p65 or Cbx1 overnight at 4°C, followed by incubation with an Alexa Fluor 488–conjugated anti-rabbit antibody or Alexa Fluor 594–conjugated anti-rabbit antibody for 1 hour. Cells were then stained with DAPI, and the images were captured by using a laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Chromogenic in situ hybridization
CISH assay was performed using a CISH kit according to the manufacturer’s protocol (BersinBio, Guangzhou, China). In brief, mouse and human colon samples were fixed in 4% paraformaldehyde. Digoxin-labeled probe targeting Lnc-UC was obtained from BersinBio. Samples were incubated with proteinase K at 37°C for 10 min, followed by prehybridization in hybridization buffer for 3 hours, and hybridization with denatured probes overnight at 42°C. The images were acquired using the streptavidin-biotin complex method with a Nikon Eclipse Ci-S optical microscope (Tokyo, Japan).
RNA pull-down
Nuclear proteins were prepared from BMDMs using a nuclear protein extraction kit (Beyotime, Shanghai, China). Full-length Lnc-UC and four truncated versions of Lnc-UC were cloned into T7 promoter–based vector and transcribed using TranscriptAid T7 High Yield Transcription kit (Thermo Fisher Scientific, Madison, WI). Purified RNA was biotinylated using Magnetic RNA-Protein Pull-down kit (Thermo Fisher Scientific, Madison, WI). Biotinylated RNAs (50 pmol) were incubated with 30 μl of streptavidin magnetic beads at room temperature for 30 min and then incubated with 300 μg of nuclear proteins at 4°C for 1 hour. Proteins were eluted from the beads with elution buffer. An aliquot of proteins was subjected to mass spectrometric analysis, while remaining proteins were separated using SDS-PAGE (polyacrylamide gel electrophoresis) and silver staining was subsequently performed using a fast silver stain kit (Beyotime, Shanghai, China) and Western blotting.
RNA immunoprecipitation
RIP assays were performed using a Magna RIP (RNA binding protein immunoprecipitation) kit according to the manufacturer’s protocol (Millipore, Bedford, MA). Colons were homogenized and lysed in RIP lysis buffer supplemented with a ribonuclease (RNase) inhibitor and protease inhibitor cocktail. The lysate was centrifuged at 14,000 rpm for 10 min, and supernatant was incubated overnight with magnetic beads conjugated with anti-Cbx1 (5 μg) or IgG in immunoprecipitation buffer at 4°C. Beads were then incubated with RIP washing buffer containing proteinase K, and immunoprecipitates were collected. RNA was extracted from the immunoprecipitates. Purified RNA was quantified by qPCR analysis.
Rapid amplification of cDNA ends
Total RNA was extracted from human colon. RNA with poly(A) was isolated from total RNA using Library Preparation VAHTS mRNA Capture Beads and reversely transcribed using SMARTer RACE 5′/3′ Kit. PCR amplification was performed using Phusion Hot Start II DNA Polymerase with 5′ or 3′ RACE specific primers. Products of PCR were recovered from the gel using gel extraction kit and subjected to sequencing. The full length of hLnc-UC was obtained by integrating sequencing data of 5′ and 3′ RACE.
Statistical analyses
Data are presented as mean ± SD. Statistical significance was determined using Student’s t test or analysis of variance (ANOVA) (one- or two-way) with Bonferroni post hoc test. Statistical analysis for survival curves was performed with the log-rank test. The level of significance was set at *P < 0.05.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (no. 81722049) and China Postdoctoral Science Foundation (no. 2019TQ0120). Author contributions: B.W. and S.W. designed the study; S.W., Y.L., F.L., Z.Z., L.G., and Z.Y. performed experiments; S.W. and Y.L. collected and analyzed data; Z.Q. and Z.W. collected clinical samples; B.W. and S.W. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The data from the Illumina sequencing were deposited in the NCBI Sequence Read Archive (submission ID: SUB7301301). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabb5202/DC1
REFERENCES AND NOTES
- 1.Ng S. C., Shi H. Y., Hamidi N., Underwood F. E., Tang W., Benchimol E. I., Panaccione R., Ghosh S., Wu J. C. Y., Chan F. K. L., Sung J. J. Y., Kaplan G. G., Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 390, 2769–2778 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Khalili H., Chan S. S. M., Lochhead P., Ananthakrishnan A. N., Hart A. R., Chan A. T., The role of diet in the aetiopathogenesis of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 15, 525–535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diamanti A., Colistro F., Basso M. S., Papadatou B., Francalanci P., Bracci F., Muraca M., Knafelz D., De Angelis P., Castro M., Clinical role of calprotectin assay in determining histological relapses in children affected by inflammatory bowel diseases. Inflamm. Bowel Dis. 14, 1229–1235 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Khor B., Gardet A., Xavier R. J., Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Däbritz J., Bonkowski E., Chalk C., Trapnell B. C., Langhorst J., Denson L. A., Foell D., Granulocyte macrophage colony-stimulating factor auto-antibodies and disease relapse in inflammatory bowel disease. Am. J. Gastroenterol. 108, 1901–1910 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Loddo I., Romano C., Inflammatory bowel disease: Genetics, epigenetics, and pathogenesis. Front. Immunol. 6, 551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roenneberg T., Merrow M., The circadian clock and human health. Curr. Biol. 26, R432–R443 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Reppert S. M., Weaver D. R., Coordination of circadian timing in mammals. Nature 418, 935–941 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Curtis A. M., Bellet M. M., Sassone-Corsi P., O’Neill L. A. J., Circadian clock proteins and immunity. Immunity 40, 178–186 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Zhang E. E., Kay S. A., Clocks not winding down: Unravelling circadian networks. Nat. Rev. Mol. Cell Biol. 11, 764–776 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Gibbs J., Ince L., Matthews L., Mei J., Bell T., Yang N., Saer B., Begley N., Poolman T., Pariollaud M., Farrow S., De Mayo F., Hussell T., Worthen G. S., Ray D., Loudon A., An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat. Med. 20, 919–926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hand L. E., Hopwood T. W., Dickson S. H., Walker A. L., Loudon A. S. I., Ray D. W., Bechtold D. A., Gibbs J. E., The circadian clock regulates inflammatory arthritis. FASEB J. 30, 3759–3770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S., Lin Y., Yuan X., Li F., Guo L., Wu B., REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat. Commun. 9, 4246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bechtold D. A., Gibbs J. E., Loudon A. S. I., Circadian dysfunction in disease. Trends Pharmacol. Sci. 31, 191–198 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Kouri V.-P., Olkkonen J., Kaivosoja E., Ainola M., Juhila J., Hovatta I., Konttinen Y. T., Mandelin J., Circadian timekeeping is disturbed in rheumatoid arthritis at molecular level. PLOS ONE 8, e54049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X., Yu R., Zhu L., Hou X., Zou K., Bidirectional regulation of circadian disturbance and inflammation in inflammatory bowel disease. Inflamm. Bowel Dis. 23, 1741–1751 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Vance K. W., Ponting C. P., Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 30, 348–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atianand M. K., Hu W., Satpathy A. T., Shen Y., Ricci E. P., Alvarez-Dominguez J. R., Bhatta A., Schattgen S. A., McGowan J. D., Blin J., Braun J. E., Gandhi P., Moore M. J., Chang H. Y., Lodish H. F., Caffrey D. R., Fitzgerald K. A., A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 165, 1672–1685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du M., Yuan L., Tan X., Huang D., Wang X., Zheng Z., Mao X., Li X., Yang L., Huang K., Zhang F., Wang Y., Luo X., Huang D., Huang K., The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun. 8, 2049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Q., Huang X. R., Yu J., Yu X., Lan H. Y., Long noncoding RNA Arid2-IR is a novel therapeutic target for renal inflammation. Mol. Ther. 23, 1034–1043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coon S. L., Munson P. J., Cherukuri P. F., Sugden D., Rath M. F., Møller M., Clokie S. J. H., Fu C., Olanich M. E., Rangel Z., Werner T.; NISC Comparative Sequencing Program, Mullikin J. C., Klein D. C., Circadian changes in long noncoding RNAs in the pineal gland. Proc. Natl. Acad. Sci. U.S.A. 109, 13319–13324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan Z. H., Zhao M., Joshi P. D., Li P., Zhang Y., Guo W., Xu Y., Wang H., Zhao Z., Yan J., A class of circadian long non-coding RNAs mark enhancers modulating long-range circadian gene regulation. Nucleic Acids Res. 45, 5720–5738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi J. S., Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allshire R. C., Madhani H. D., Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 19, 229–244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skvortsova K., Iovino N., Bogdanović O., Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 19, 774–790 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Mosch K., Franz H., Soeroes S., Singh P. B., Fischle W., HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLOS ONE 6, e15894 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arun G., Diermeier S., Akerman M., Chang K.-C., Wilkinson J. E., Hearn S., Kim Y., MacLeod A. R., Krainer A. R., Norton L., Brogi E., Egeblad M., Spector D. L., Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 30, 34–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M.-C., Hung T., Argani P., Rinn J. L., Wang Y., Brzoska P., Kong B., Li R., West R. B., van de Vijver M. J., Sukumar S., Chang H. Y., Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long Y., Wang X., Youmans D. T., Cech T. R., How do lncRNAs regulate transcription? Sci. Adv. 3, eaao2110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopp F., Mendell J. T., Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffin P., Dimitry J. M., Sheehan P. W., Lananna B. V., Guo C., Robinette M. L., Hayes M. E., Cedeño M. R., Nadarajah C. J., Ezerskiy L. A., Colonna M., Zhang J., Bauer A. Q., Burris T. P., Musiek E. S., Circadian clock protein Rev-erbalpha regulates neuroinflammation. Proc. Natl. Acad. Sci. U.S.A. 116, 5102–5107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pourcet B., Zecchin M., Ferri L., Beauchamp J., Sitaula S., Billon C., Delhaye S., Vanhoutte J., Mayeuf-Louchart A., Thorel Q., Haas J. T., Eeckhoute J., Dombrowicz D., Duhem C., Boulinguiez A., Lancel S., Sebti Y., Burris T. P., Staels B., Duez H. M., Nuclear receptor subfamily 1 group D member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology 154, 1449–1464.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goudie D. R., D’Alessandro M., Merriman B., Lee H., Szeverényi I., Avery S., O’Connor B. D., Nelson S. F., Coats S. E., Stewart A., Christie L., Pichert G., Friedel J., Hayes I., Burrows N., Whittaker S., Gerdes A.-M., Broesby-Olsen S., Ferguson-Smith M. A., Verma C., Lunny D. P., Reversade B., Lane E. B., Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat. Genet. 43, 365–369 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Delić D., Warskulat U., Borsch E., Al-Qahtani S., Al-Quraishi S., Häussinger D., Wunderlich F., Loss of ability to self-heal malaria upon taurine transporter deletion. Infect. Immun. 78, 1642–1649 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai A., Guo Y., Shen Y., Xie Y., Zhu X., Lu N., Seasonality in flares and months of births of patients with ulcerative colitis in a Chinese population. Dig. Dis. Sci. 54, 1094–1098 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Zhou Z., Lin Y., Gao L., Yang Z., Wang S., Wu B., Circadian pharmacological effects of berberine on chronic colitis in mice: Role of the clock component Rev-erbα. Biochem. Pharmacol. , 113773 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Cavadini G., Petrzilka S., Kohler P., Jud C., Tobler I., Birchler T., Fontana A., TNF-α suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. U.S.A. 104, 12843–12848 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo B., Yang N., Borysiewicz E., Dudek M., Williams J. L., Li J., Maywood E. S., Adamson A., Hastings M. H., Bateman J. F., White M. R. H., Boot-Handford R. P., Meng Q. J., Catabolic cytokines disrupt the circadian clock and the expression of clock-controlled genes in cartilage via an NFкB-dependent pathway. Osteoarthr. Cartil. 23, 1981–1988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ten Hove T., van den Blink B., Pronk I., Drillenburg P., Peppelenbosch M. P., van Deventer S. J. H., Dichotomal role of inhibition of p38 MAPK with SB 203580 in experimental colitis. Gut 50, 507–512 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pariollaud M., Gibbs J. E., Hopwood T. W., Brown S., Begley N., Vonslow R., Poolman T., Guo B., Saer B., Jones D. H., Tellam J. P., Bresciani S., Tomkinson N. C., Wojno-Picon J., Cooper A. W., Daniels D. A., Trump R. P., Grant D., Zuercher W., Willson T. M., MacDonald A. S., Bolognese B., Podolin P. L., Sanchez Y., Loudon A. S., Ray D. W., Circadian clock component REV-ERBα controls homeostatic regulation of pulmonary inflammation. J. Clin. Invest. 128, 2281–2296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong H.-K., Maury E., Ramsey K. M., Perelis M., Marcheva B., Omura C., Kobayashi Y., Guttridge D. C., Barish G. D., Bass J., Requirement for NF-κ B in maintenance of molecular and behavioral circadian rhythms in mice. Genes Dev. 32, 1367–1379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang G., Wright C. J., Hinson M. D., Fernando A. P., Sengupta S., Biswas C., La P., Dennery P. A., Oxidative stress and inflammation modulate Rev-erb α signaling in the neonatal lung and affect circadian rhythmicity. Antioxid. Redox Signal. 21, 17–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motomura Y., Kitamura H., Hijikata A., Matsunaga Y., Matsumoto K., Inoue H., Atarashi K., Hori S., Watarai H., Zhu J., Taniguchi M., Kubo M., The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4+ T cells. Nat. Immunol. 12, 450–459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin Y., Wang S., Zhou Z., Guo L., Yu F., Wu B., Bmal1 regulates circadian expression of cytochrome P450 3a11 and drug metabolism in mice. Commun. Biol. 2, 378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wirtz S., Popp V., Kindermann M., Gerlach K., Weigmann B., Fichtner-Feigl S., Neurath M. F., Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12, 1295–1309 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabb5202/DC1