

Abstract
The CTC1-STN1-TEN1 (CST) complex is essential for telomere maintenance and resolution of stalled replication forks genome-wide. Here, we report the 3.0-angstrom cryo–electron microscopy structure of human CST bound to telomeric single-stranded DNA (ssDNA), which assembles as a decameric supercomplex. The atomic model of the 134-kilodalton CTC1 subunit, built almost entirely de novo, reveals the overall architecture of CST and the DNA-binding anchor site. The carboxyl-terminal domain of STN1 interacts with CTC1 at two separate docking sites, allowing allosteric mediation of CST decamer assembly. Furthermore, ssDNA appears to staple two monomers to nucleate decamer assembly. CTC1 has stronger structural similarity to Replication Protein A than the expected similarity to yeast Cdc13. The decameric structure suggests that CST can organize ssDNA analogously to the nucleosome’s organization of double-stranded DNA.
CTC1-STN1-TEN1 (CST) is a protein complex essential for telomere replication (1–4) and as a DNA polymerase alpha-primase (Pol-α) cofactor (5), and it functions genome-wide to recover stalled replication forks (2, 6–9) and facilitate DNA damage repair (10–13). Consequently, mutations in CST are the basis of human genetic diseases such as Coats plus syndrome and dyskeratosis congenita (14–19).
Although it preferentially binds short telomeric single-stranded DNA (ssDNA) (20–23), CST can also bind less specifically to longer ssDNA (2, 4). An intact heterotrimeric CST complex is necessary for its DNA-binding function (4, 15, 24), but limited understanding of mammalian CST architecture has hampered the determination of its DNA-binding region(s). Structures of human components are limited to STN1 and TEN1 (24), and solving the structure of the largest subunit CTC1 has been technically challenging, with only a single domain being determined (25). The yeast Cdc13 protein associates with Stn1 and Ten1 and has therefore been proposed as a CTC1 homolog, despite Cdc13 and mammalian CTC1 being unrelated in sequence. Hence, it has been unclear if Cdc13 and CTC1 share structural homology.
Cryo–electron microscopy (cryo-EM) structure of human CST decameric supercomplex
We solved the structure of purified recombinant human CST protein (hereafter termed “DNA-free CST”) to 6.3-Å resolution using single-particle cryo-EM (fig. S1). To improve the resolution of the structure, we added a minimal telomeric ssDNA [3xTEL, (TTAGGG)3] (4) to the purified CST protein and unexpectedly discovered a symmetric complex that was considerably larger than the monomeric CST (Fig. 1A and fig. S2, A and B). Subsequent cryo-EM processing revealed that the symmetric complex was a decameric supercomplex (10 CST monomers) with D5 symmetry, which was reconstructed at 3.0-Å global resolution (Fig. 1A and fig. S2C). The CST monomers were computationally extracted from the supercomplex and further sorted to obtain a final set of data, which led to the cryo-EM map of a CST monomer still at 3.0-Å global resolution (Fig. 1B and figs. S2D and S3 to S5) but with the map quality substantially improved (fig. S5A). This enabled us to dock all the available crystal structures of the domains of human TEN1, STN1 (24), and a central OB (oligonucleotideoligosaccharide–binding fold) domain of CTC1 (25) with high confidence (Fig. 1B). Furthermore, we were able to build de novo the remaining unsolved body of CTC1 [894 residues, excluding the one CTC1 OB domain previously solved (25)] (fig. S5B).
Fig. 1. Cryo-EM structure of human CST decameric supercomplex and its architecture.
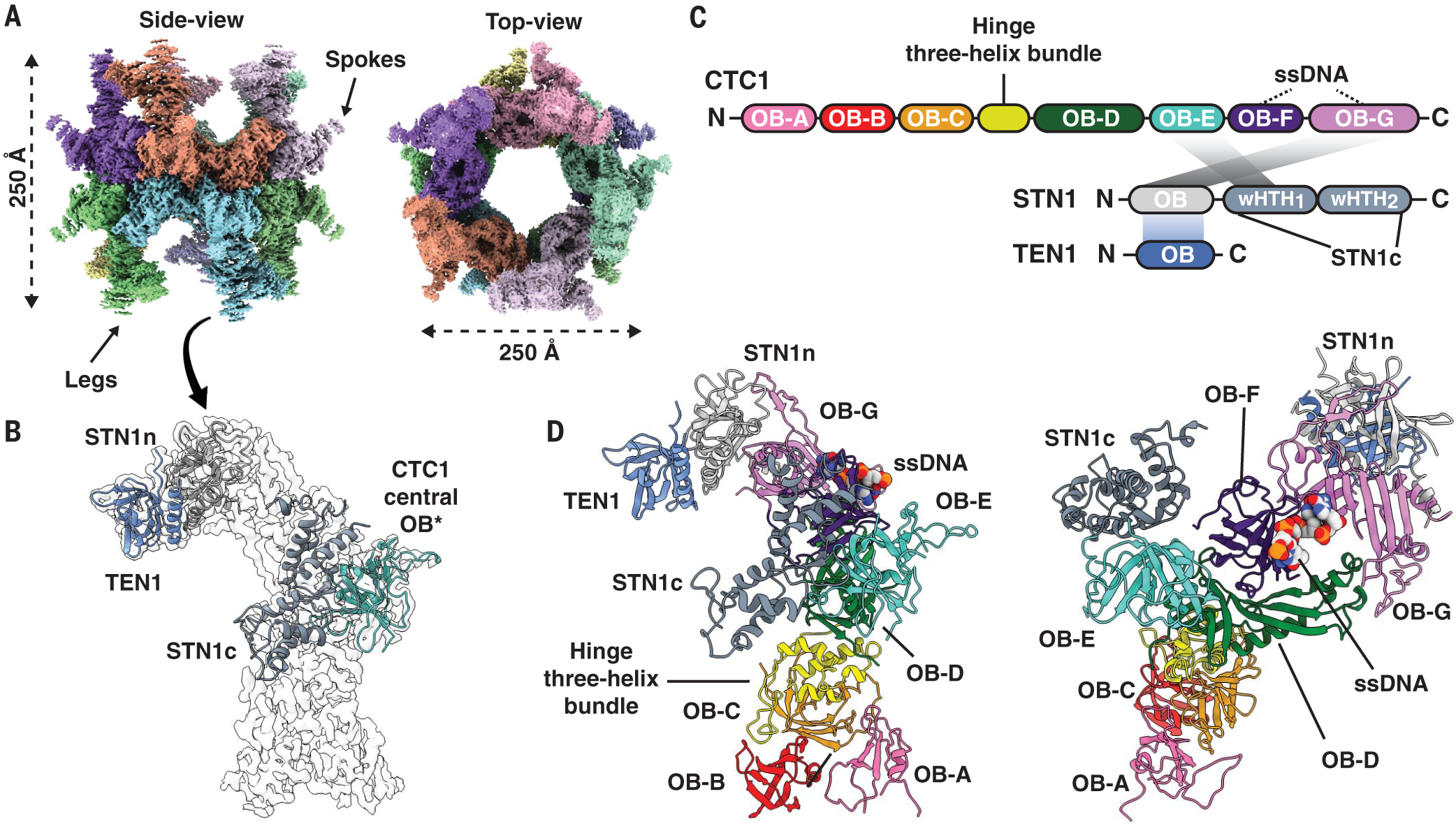
(A) Cryo-EM density of decameric CST complex colored by segmented CST monomers. (B) Docking of available atomic models of a CTC1 OB-domain [*reported as central domain OB-fold (25), PDB 5W2L], STN1n (N-terminal half, PDB 4JOI:A), STN1c (C-terminal half, PDB 4JQF), and TEN1 (PDB 4JOI:C). (C) Structure-based schematic of CST domain architecture and intermolecular interactions between subunits. The individual OB domains of CTC1 are rainbow colored. (D) CTC1 architectural organization of seven OB domains (A to G) and the identified bound-ssDNA (space-filled model). Spokes in (A) are STN1c, whereas the legs are CTC1 OB-A, -B, and -C.
Human chromosome ssDNA telomeric overhangs are 50 to 200 nucleotides (nt) long, much larger than 3xTEL, so we tested CST binding to 15xTEL (90 nt). CST bound 15xTEL with sixfold higher affinity than 3xTEL (fig. S6, A to C, and table S1), and decameric CST supercomplexes were readily apparent by negative-stain EM (fig. S6D). Thus, the decamer can form with both long and short telomeric ssDNA molecules.
Overall architecture of human CST
Model building revealed the overall architecture of the human CST heterotrimer (Fig. 1, C and D; fig. S5B; and table S2). CTC1 is composed of seven tandem OB domains (OB-A through G; Fig. 1, C and D). The human CST complex has a subunit stoichiometry of 1:1:1, unlike the nonuniform stoichiometry reported for the Candida glabrata CST complex (26).
The C terminus of CTC1 (OB-D through G) serves as a hub for STN1 and TEN1 assembly (Fig. 1D). A single STN1 protein has two separate interaction sites with CTC1, with the STN1 N-terminal half (STN1n) interacting with CTC1 OB-G and the C-terminal half (STN1c) with CTC1 OB-E (Fig. 2A). These two halves of STN1 are connected by an unstructured peptide linker of seven residues (Fig. 2A). In contrast to the related ssDNA-binding protein, Replication Protein A (RPA) (27, 28), there is no triple-helix bundle stabilizing the heterotrimeric CST complex (Fig. 2B). Instead, TEN1 binding to CTC1 is bridged by STN1n [similar to the model of the Tetrahymena CST (29, 30)] (Fig. 2B), with STN1n binding to a highly conserved interaction patch on CTC1 OB-G (fig. S7A).
Fig. 2. CST intersubunit interactions and CTC1 molecular motifs.
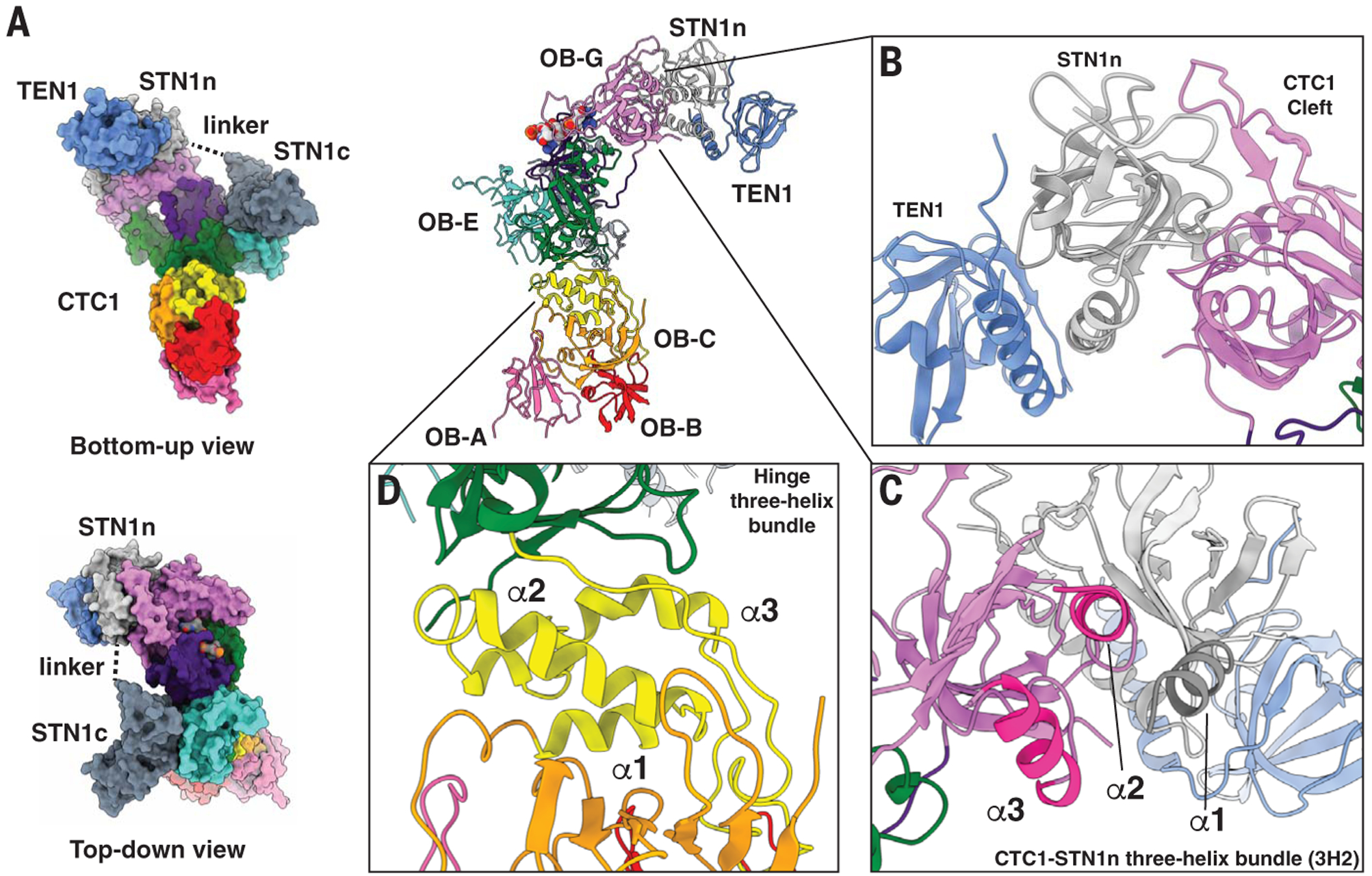
(A) STN1 N-terminal and C-terminal halves—STN1n and STN1c—interact separately with CTC1 by means of a flexible peptide linker. (B) STN1 and TEN1 do not interact with CTC1 using a trimeric helix-bundle like human RPA; instead, STN1 directly interacts with a highly conserved patch on CTC1 (Cleft) and bridges TEN1 to CTC1. (C) CTC1-STN1n three-helix bundle that is involved in CTC1 and STN1 assembly. α1 (highlighted dark gray) is from STN1n and α2 and α3 (highlighted bright pink) are from CTC1 OB-G. (D) The hinge three-helix bundle (annotated α1, α2, and α3) connects OB-A, -B, and -C to the rest of the C-terminal OB-domains of CTC1.
The first winged helix-turn-helix (wHTH) domain of STN1c interacts with CTC1 OB-E (Fig. 1D). However, no strong conservation of residues occurs on the interaction patch of CTC1 OB-E (fig. S7A), suggesting that STN1c-CTC1 interaction could be weaker than STN1n-CTC1 interaction, as reported for the Tetrahymena CST complex (30). Supporting this hypothesis, we found that STN1n alone was able to interact with CTC1, but STN1c could not (fig. S7, B and C). In addition, TEN1 interaction with CTC1 was maintained with STN1n but lost when only STN1c was present. STN1n and CTC1 interact through two regions—CTC1 “cleft region” (the conserved patch on CTC1; Fig. 2B and fig. S7A) and a new CTC1-STN1n three-helix bundle (Fig. 2C). The importance of the cleft and the three-helix bundle for CTC1-STN1 association was confirmed by mutagenesis (fig. S7, D and E).
CTC1 OB folds E, F, and G are arranged spatially on OB-D, which acts like a scaffold, resulting in these four OBs forming a ringlike structure (Fig. 1D). Structural homology analysis of individual CTC1 OB domains found CTC1 to be most similar to RPA and Teb1 (an RPA-like paralog in Tetrahymena) (fig. S8), with CTC1 OB-F most similar to Teb1’s OB-B (31) and OB-G similar to OB-C of RPA70 or Teb1 (27). CTC1 OB-G also has a conserved zinc ribbon motif like that of the OB-C domains of RPA and Teb1 (27, 31) (fig. S9). The scaffolding OB-D has no convincing structural homologies, but given its distinctive extended OB-fold structure (Fig. 1D), it could be an evolved form of the more compact and conventional OB-fold (32). Notably, despite the long-standing suggestion that yeast Cdc13 and mammalian CTC1 are homologs (33), we found weaker structural homologies to Cdc13 than with the best RPA70 homology matches (based on DALI structural homology Z-score, fig. S10).
We also found an intramolecular three-helix bundle bridging OB-C and OB-D (Fig. 2D). This three-helix bundle (termed hinge three-helix bundle) effectively segregates OB-A, -B, and -C from the C-terminal OB domains (Fig. 1, C and D). CST OB-C serves as a scaffold for both OB-A and OB-B (Fig. 1D). Because of extensive flexibility of OB-A, we could only de novo build a poly(alanine) model (~45 residues) for it, with the overall backbone of OB-A clearly showing the OB-fold topology. Structural homology searches of OB-B and OB-C reveal them to be most similar to Ustilago maydis RPA70 OB-A and OB-B (28) (fig. S8). The multiple structural homologies of human CTC1 to various domains of RPA70 suggest that CTC1 may have evolved from RPA (figs. S8 and S10).
Disease mutations in CTC1 that have been shown to interfere with Pol- α binding (15) are located on CTC1 OB-B (A227 and V259) and on scaffold OB-D (V665) (fig. S11). Given that Pol-α has a bilobal architecture (34), the catalytic and primase lobes of Pol- α could engage CST at separate sites.
CST ssDNA-binding anchor site
Four nucleotides, TAGG, were clearly visible in the cryo-EM map of the complex and not in the DNA-free CST cryo-EM map (Fig. 3, A to C; see table S3 for cross-correlation analysis, fig. S12A), suggesting that these interact with the protein most strongly or with highest occupancy. The rest of the ssDNA is likely flexible or bound to CST in multiple binding modes, which is consistent with CST being able to bind multiple configurations of ssDNA dynamically (4, 7, 23). Hereafter, we identify the site of ssDNA binding on CTC1 as the ssDNA anchor patch.
Fig. 3. Telomeric ssDNA-binding anchor site of CST.
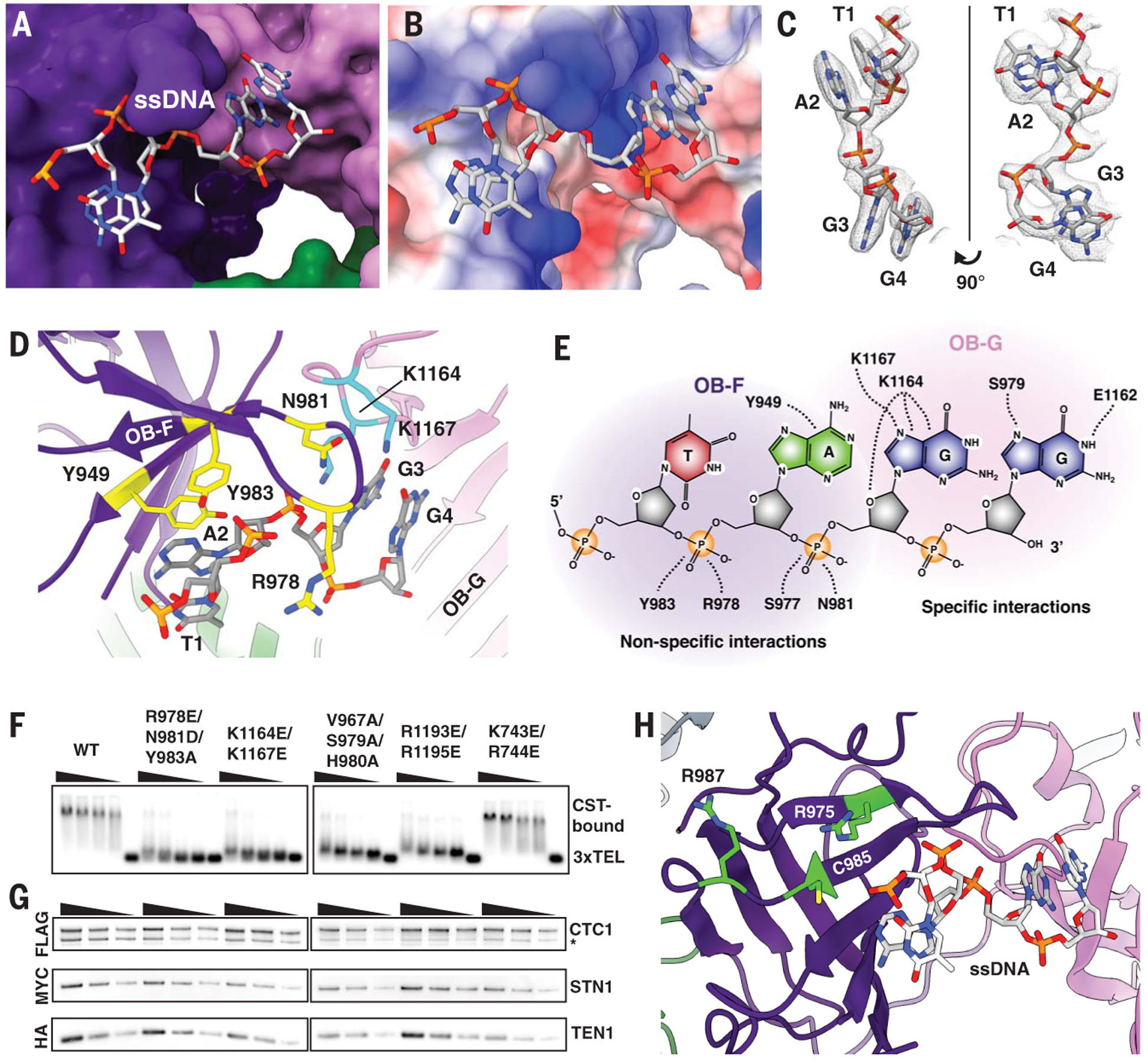
(A) A 4-nt segment of the single-stranded telomeric DNA is located on CTC1. (B) Coulombic surface analysis (36) reveals that the ssDNA anchor site is highly positively charged (blue; red is negatively charged surface). (C) Cryo-EM density of the ssDNA molecule built with the sequence assigned as TAGG (5′-T1-A2-G3-G4–3′). The numbering is based on the visible ssDNA, not the full-length ssDNA. (D) CTC1 residues involved in ssDNA binding are shown in yellow and cyan from OB-F (yellow on purple) and OB-G (cyan on pink), respectively. (E) Schematic of CST ssDNA-binding anchor site across CTC1 OB-F and OB-G. (F) Gel-shift assay showing that CST DNA-binding mutants predicted from the atomic model no longer bind telomeric ssDNA (TTAGGG)3. Wedges indicate twofold dilutions of CST starting at 50 nM, with the fifth lane of each group having no protein added. K743E/R744E mutant does not directly bind DNA and was used as a control to test if charge swaps in the vicinity might be sufficient to destabilize DNA binding. (G) CST DNA-binding mutants can still form heterotrimeric CST complex as shown by tandem immunoprecipitation pull-down assays [FLAG/hemagglutinin (HA)] from exogenously expressed FLAG-CTC1, MYC-STN1, and HA-TEN1. Asterisk (*) indicates protein degradation product. Wedges indicate a twofold dilution that is used to ensure that Western-blot band intensities are in the linear detection range. (H) Human CTC1 disease mutations (15) that abolish ssDNA binding (lime green residues) are located near the ssDNA anchor site. Abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; G, Gly; H, His; K, Lys; L, Leu; N, Asn; R, Arg; S, Ser; V, Val; W, Trp; and Y, Tyr.
Several positively charged residues of CTC1 are involved in the interaction with ssDNA at the anchor patch (CTC1 R978, K1164, and K1167), as well as additional aromatic and neutral-polar residues (CTC1 Y949, N981, and Y983) (Fig. 3D and E). This anchor patch uses several kinds of interaction between CTC1 and ssDNA: for example, R978 and N981 hydrogen bonding to the negatively charged ssDNA phosphate backbone (fig. S12B); K1164 and K1167 hydrogen bonding to ssDNA bases (fig. S12C); and Y949 p-p stacking with the A2 base, which in turn is stacked on the T1 base (fig. S12D). The tyrosine-base-base stack is reminiscent of the stacking arrangements seen in several OB fold–nucleic acid interactions, e.g., the human POT1-ssDNA structure (20). The nonspecific interactions mostly involve CTC1 OB-F, whereas specific interactions are in OB-G (Fig. 3E); the modeled 4-nt ssDNA spans these two OB domains, suggesting that ssDNA binding stabilizes CTC1 architecture (Fig. 3D). In addition, these ssDNA-interacting residues are highly conserved across mammalian CTC1 homologs (fig. S13).
To validate the observed protein-DNA interactions, we performed mutagenesis on sets of CTC1 residues in the ssDNA anchor patch—R987E/N981D/Y983A (anchor site on OB-F), K1164E/K1167E (anchor site on OB-G), V967A/S979A/H980A (structural integrity residues on OB-F), and R1193E/R1195E (structural integrity residues on OB-G) (see fig. S14 for structural mapping of additional tested mutants). Each of these sets of mutations abolished CST DNA-binding activity, whereas the K743E/R744E negative control mutation did not (Fig. 3F). In all of these mutants, CST still forms a heterotrimer complex (Fig. 3G). Previously identified CTC1 disease mutations (R975G, C985Δ and R987W) that have been shown (15) to affect CST-ssDNA binding are also in the vicinity of the ssDNA anchor patch (Fig. 3H).
Assembly mechanism and pathways of decameric CST supercomplex
We identified interactions that appear to mediate decameric supercomplex assembly. The sites can be categorized into two oligomerization classes (Fig. 4, A to C): (i) dimerization (dihedral dimerization) and (ii) tetramerization (two subclasses: adjacent and diagonal). For CST dimerization, three conserved residues at the interface are N745, L843, and R1175. R1175 is particularly interesting, given that it is also within range (<5 Å) for interaction with the phosphodiester groups of T1 or A2 of the opposite dihedral dimer’s telomeric ssDNA (Fig. 4A), which suggests ssDNA binding can also stabilize CST dihedral dimerization. Consistent with this prediction, CST with the CTC1 R1175E mutation showed a 26-fold reduction in DNA-binding ability with 3xTEL ssDNA but no effect when tested with a nonspecific T18 (poly-T) ssDNA (fig. S15).
Fig. 4. Molecular interactions underlying CST decameric supercomplex formation and testing the dimer stapling model.
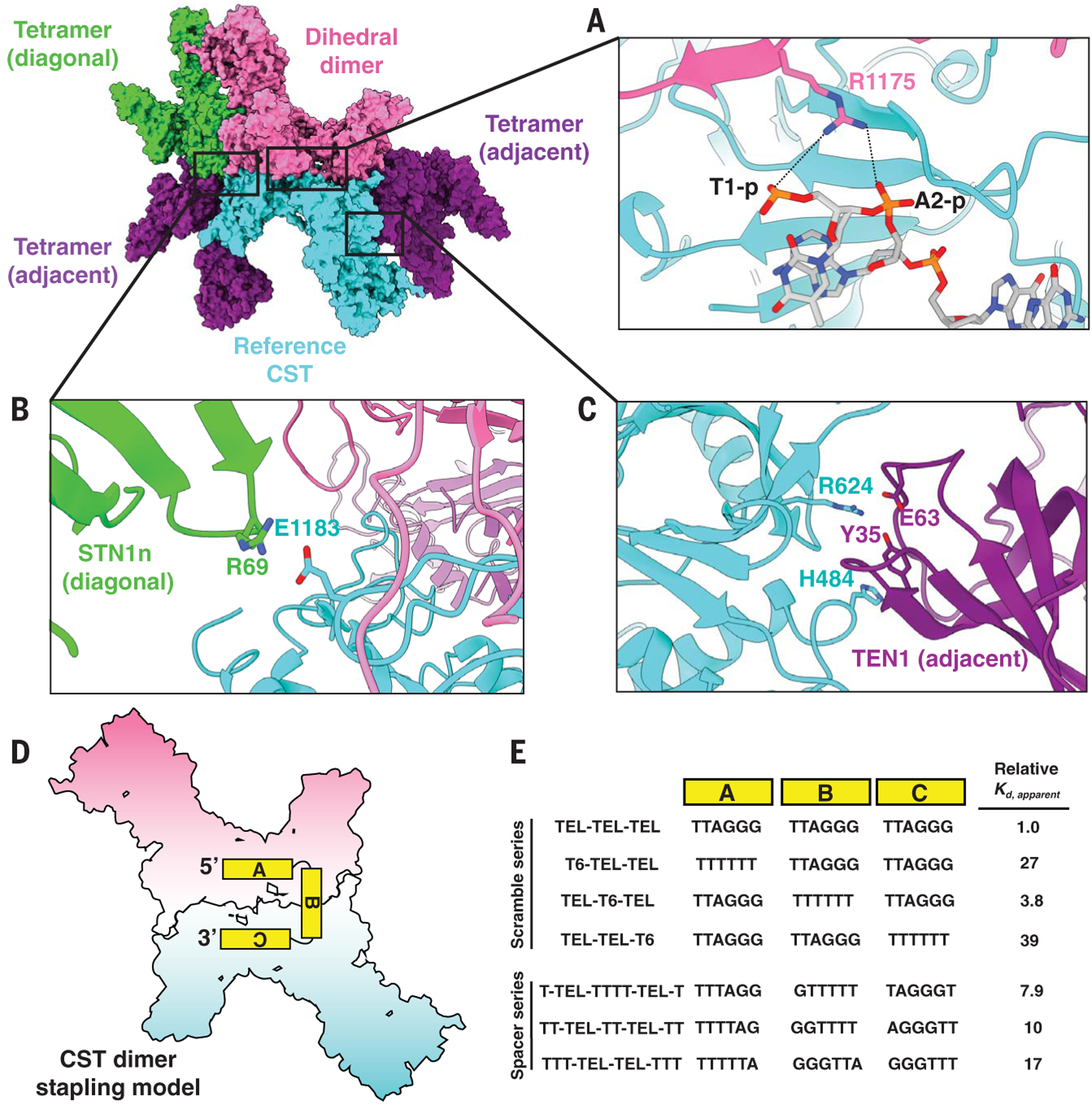
(A to C) The reference CST (cyan) is flanked by four CST complexes—a dihedral dimer (opposite, pink) and three tetrameric partners (one diagonal neighbor, green, and two adjacent neighbors, purple). (A) CTC1 R1175 from the dihedral dimer neighbor (pink) is pointing toward ssDNA bound to the reference CTC1 (cyan), with the black dashed lines representing feasible ionic interactions between R1175 and phosphodiester groups of the ssDNA. (B and C) Identified intermolecular interactions between CTC1, STN1, and TEN1 at interfaces of the decameric supercomplex. (D) CST dimer stapling model with an 18-nt ssDNA molecule. The two monomers are separately colored as pink and cyan for visual clarity. (E) Changes in CST DNA-binding affinity (Kd,apparent) relative to that of 3xTEL with oligo-T substitution of block A, B, or C of 3xTEL (Scramble series). The molecular distance between the TTAGGG sequences of blocks A and C was also varied, and the impact on CST relative DNA-binding affinity was measured (Spacer series). TEL-TEL-TEL oligo is also known as 3xTEL. The relative DNA-binding affinity values are reported to two significant figures; measured values and error analysis are in table S1.
For tetramerization, CTC1 interacts with its diagonally opposite neighbor’s STN1n (CTC1 E1183) (Fig. 4B) and adjacent neighbor’s TEN1 (CTC1 H484 and R624) (Fig. 4C). The proximity of the two ssDNA anchor patches across dihedral dimers (fig. S16A) suggested that a single ssDNA molecule of three TTAGGG repeats could “staple” together two monomers into a dihedral dimer, with the first and last repeat engaged by the CST monomers while the middle repeat served as a linker (Fig. 4D). Consistent with this model, we found that replacing individual TTAGGG modules with T6 reduced CST DNA-binding affinity for either the first or last repeat but was tolerated for the middle repeat (Fig. 4E and fig. S16, B and C). As an additional test, shortening the middle repeat sequence (using oligo-T sequence instead of TTAGGG) to <6 nt negatively affected CST-DNA binding (Fig. 4E and fig. S16, D and E), consistent with the measured molecular distance (~20 Å, fig. S16A) between the 5′ and 3′ ends of the neighboring ssDNA molecules in the dihedral CST dimer.
A comparison of the DNA-free CST model and the monomeric CST model extracted from the decameric supercomplex (for example, compare fig. S1D to fig. S2D) suggested that STN1c has two alternate docking sites on CTC1. To investigate this, we turned to a cryo-EM dataset that had a high population of monomeric CST with telomeric ssDNA added (fig. S17) and found two distinct conformations—one with a “head” density and the other with an “arm” density—albeit at a lower model resolution of ~9 Å (Fig. 5, A and B). Because STN1c is in the “arm” conformation in the decameric CST, and the STN1c “head” conformation would sterically hinder formation of the decamer (by obstructing dihedral dimerization), we propose that switching from “head” to “arm” docking position for STN1c is an important first step for CST to form a decameric supercomplex. STN1c switching is consistent with our finding that STN1c is less stably bound to CTC1 than STN1n (fig. S7, B and C).
Fig. 5. Assembly mechanism and pathway model of CST decameric supercomplex.
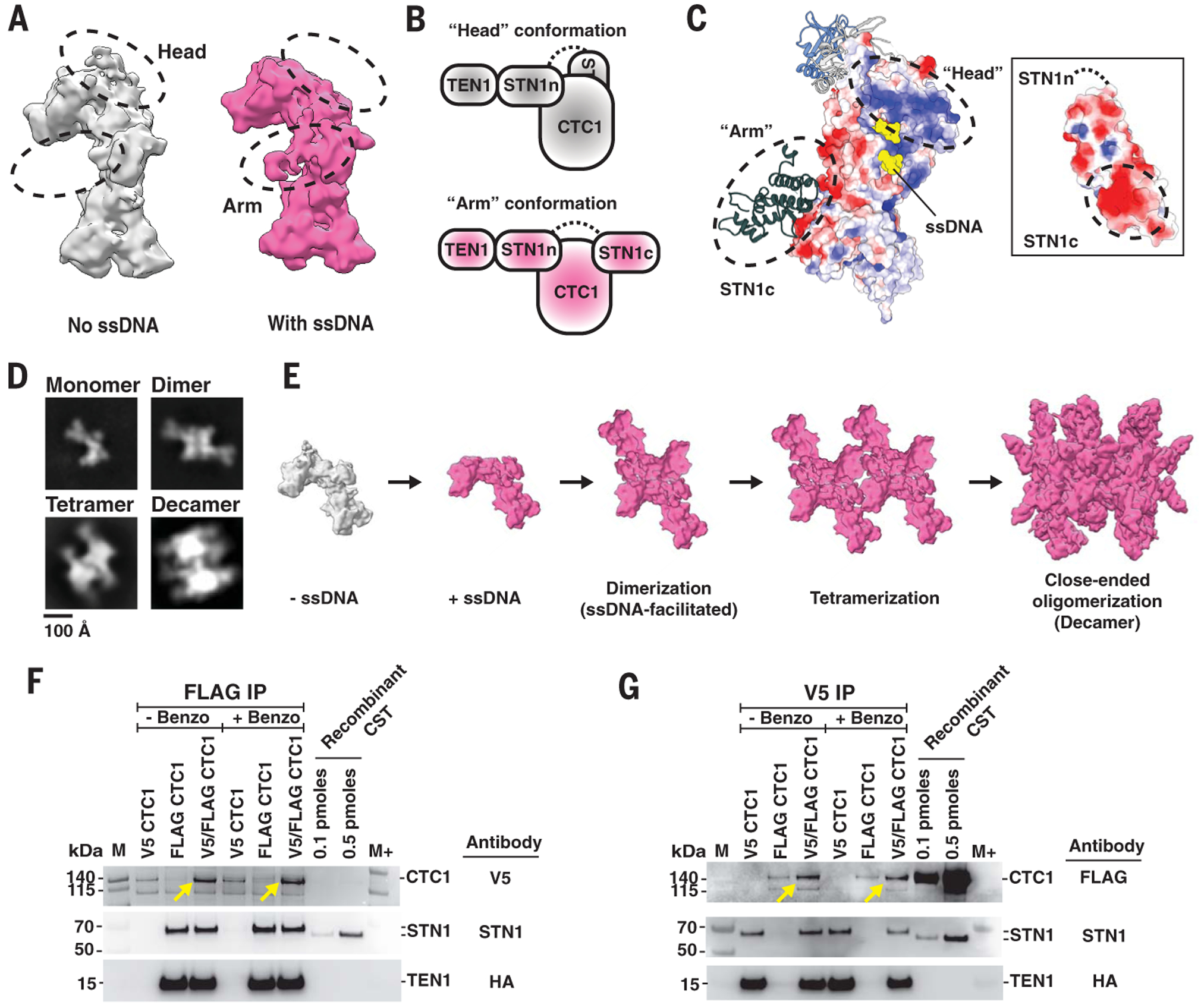
(A) Cryo-EM densities of two conformations of monomeric CST with the differences indicated by dashed black circles. The two conformations—“head” (colored gray) and “arm” (colored pink)—are assigned as CST without and with ssDNA bound, respectively. (B) Cartoon models of CST “head” and “arm” conformations depicted by conformational changes of STN1c docking site on CTC1. The black dashed line represents the unstructured polypeptide region between STN1n and STN1c. (C) Coulombic surface analysis reveals a highly positively charged patch on CTC1 OB-G, where STN1c lies when in “head” conformation. Reciprocally, a highly negatively charged surface is shown on STN1c (see inset). (D) Two-dimensional class averages of negative-stained CST incubated with 3xTEL ssDNA showed multiple oligomeric species of CST, which are assigned as monomer, dimer, tetramer, and decamer. (E) Proposed model of assembly pathway of CST decameric supercomplex upon ssDNA introduction. CST binding of ssDNA prevents STN1c from binding to its original site (“head” conformation, gray), allowing CST to form dimers before progressing to tetramers, and eventually leading to a close-ended decameric supercomplex. (F and G) Immunoprecipitation (IP) of orthogonally tagged CTC1 molecules coexpressed in cells. (F) FLAG IP of HEK293T cell extracts that were cotransfected with V5-CTC1, FLAG-CTC1, or both, and with TEN1 and STN1. Western blot with antibody against V5 showed that FLAG-IP of FLAG-CTC1 also coimmunoprecipitated V5-CTC1 (yellow arrows). (G) Coimmunoprecipitation of FLAG-CTC1 was also observed with V5-IP of V5-CTC1 (yellow arrows). STN1 and TEN Western blots were done to determine the presence of CST heterotrimeric complex assembly. M and M+ indicate protein ladder PageRuler and PageRuler Plus, respectively.
Surface-charge analysis revealed a highly positively charged surface on CTC1 OB-G, where the STN1c is expected to dock in the “head” conformation (Fig. 5C), and similar analysis revealed a reciprocal patch of high negative charge on STN1c (Fig. 5C, inset). This suggested that charge-charge interactions could mediate the transition from monomeric to decameric CST, explaining how a longer ssDNA, with extended binding to the OB-G’s negative patch, can trigger this transition. The charge-charge interactions also suggested that increased salt concentration could mediate the transition in the absence of ssDNA. Indeed, we found a large increase in decameric CST population without addition of ssDNA in a nonphysiological salt concentration of 800 mM NaCl (fig. S18).
Finally, we used negative-stain EM single-particle analysis to identify subcomplexes of the decamer that would give hints to its assembly pathway(s). We observed two subcomplexes, dimers and tetramers (Fig. 5D), which are plausible intermediates in an assembly pathway based on ssDNA-stapled dimers such as the following: CST assembles first as a dihedral dimer before forming a lateral tetramer involving two dihedral dimers, and sequential addition of dimers eventually closes the symmetric circle (decamer) by continuing the lateral oligomerization (Fig. 5E).
Evidence for higher-order CST assemblies in vivo
CST monomers interact specifically to form the decamer, burying a great deal of exposed protein surface area (~2100 Å2 per monomer), and ssDNA has a specific role in triggering decamer assembly. These features indicate that formation of the CST decamer is thermodynamically favorable and that the monomer is built to form the decamer. To confirm that CST forms oligomers in cells, we turned to an orthogonal epitope tag pull-down approach. V5-tagged and FLAG-tagged CTC1 were coexpressed in human embryonic kidney 293T (HEK293T) cells, along with STN1 and TEN1. Pull-down using anti-FLAG beads immunoprecipitated V5-CTC1, as well as FLAG-CTC1, and the reciprocal experiment with anti-V5 beads similarly recovered the CTC1 with both epitope tags (Fig. 5, F and G, and fig. S19, A and B, for IP controls). Notably, the pull-down result was not sensitive to DNA and RNA degradation with benzonase (Fig. 5, F and G, and fig. S19C), so the higher-order complexes were not loosely tethered by nucleic acid. We conclude that a substantial fraction of CST resides in a higher-order protein complex, consistent with decamers existing in vivo.
Discussion
Our structure of the decameric human CST supercomplex bound with telomeric ssDNA provides the platform for understanding mechanisms of various CST functions in DNA replication and DNA damage repair, not only at telomeres but also genome-wide (7, 8, 11, 13, 35). The atomic-resolution details revealed in this structure enabled us to identify amino acids responsible for the numerous interactions that are key for CST functions, i.e., the intricate heterotrimer assembly, ssDNA-binding anchor site, and decamer assembly. Moreover, the structure provides a molecular model to understand the underlying mechanisms of CST mutants in human diseases such as Coats plus and dyskeratosis congenita. Finally, we speculate that the decamer could be a nucleosome-equivalent for G-rich ssDNA, compacting it and competing with G-quadruplex structures both at stalled replication forks and at telomeres.
Supplementary Material
ACKNOWLEDGMENTS
We thank Z. Yu, D. Matthies and R. Huang (Janelia Research Campus), P. Blerkom and J. Kieft (University of Colorado Anschutz), and G. Morgan and C. Page (University of Colorado Boulder) for microscope setup and data collection. We thank F. Asturias (University of Colorado Anschutz) and T. Terwilliger (Los Alamos National Laboratory) for discussion. We thank T. Nahreini (University of Colorado Boulder) and BioFrontiers Institute Computing Core for their support and assistance. We thank D. Youmans for input regarding immunoprecipitation experiments. In addition, we thank the members of the Cech and Wuttke laboratories for their suggestions.
Funding: This work was funded in part by grants from NIH to T.R.C. (R01GM099705), D.S.W. (R01GM059414), and C.J.L. (K99GM131023) and from NSF to D.S.W. (MCB 1716425). A.T.B. is supported by a fellowship provided by NIH-University of Colorado Boulder (T32GM008759). T.R.C. is an HHMI Investigator.
Footnotes
Competing interests: T.R.C. is on the board of directors of Merck and a consultant for STORM Therapeutics.
Data and materials availability: Cryo-EM maps and the de novo–built model are deposited in Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) with the following accession numbers: CST-3xTEL monomer (EMD-21567 and PDB ID 6W6W), CST-3xTEL decamer (EMD-21561), CST-3xTEL oligomer-mixture (Arm–EMD-21565 and head–EMD-21566), and DNA-free CST (EMD-21563). Plasmids encoding human STN1, TEN1, and CTC1 (wild-type and K1175E mutant) are available from T.R.C. under a material transfer agreement with the University of Colorado Boulder.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Surovtseva YV et al. , Mol. Cell 36, 207–218 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyake Y et al. , Mol. Cell 36, 193–206 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Wang F et al. , Cell Rep. 2, 1096–1103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen LY, Redon S, Lingner J, Nature 488, 540–544 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Casteel DE et al. , J. Biol. Chem 284, 5807–5818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M et al. , Nucleic Acids Res. 47, 5243–5259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhattacharjee A, Wang Y, Diao J, Price CM, Nucleic Acids Res. 45, 12311–12324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chastain M et al. , Cell Rep. 16, 1300–1314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart JA et al. , EMBO J. 31, 3537–3549 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kratz K, de Lange T, J. Biol. Chem 293, 14384–14392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirman Z et al. , Nature 560, 112–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greetham M, Skordalakes E, Lydall D, Connolly BA, J. Mol. Biol 427, 3023–3030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Stewart J, Price CM, Cell Cycle 13, 3488–3498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart JA, Wang Y, Ackerson SM, Schuck PL, Front. Biosci 23, 1564–1586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen LY, Majerská J, Lingner J, Genes Dev. 27, 2099–2108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armanios M, Pediatr. Blood Cancer 59, 209–210 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Anderson BH et al. , Nat. Genet 44, 338–342 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Amir M et al. , Front. Mol. Biosci 6, 41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu P, Chang S, Aging Cell 12, 1100–1109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei M, Podell ER, Cech TR, Nat. Struct. Mol. Biol 11, 1223–1229 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Loayza D, Parsons H, Donigian J, Hoke K, de Lange T, J. Biol. Chem 279, 13241–13248 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Baumann P, Cech TR, Science 292, 1171–1175 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Hom RA, Wuttke DS, Biochemistry 56, 4210–4218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryan C, Rice C, Harkisheimer M, Schultz DC, Skordalakes E, PLOS ONE 8, e66756 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shastrula PK, Rice CT, Wang Z, Lieberman PM, Skordalakes E, Nucleic Acids Res. 46, 972–984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lue NF et al. , PLOS Genet. 9, e1003145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A, EMBO J. 21, 1855–1863 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan J, Pavletich NP, Genes Dev. 26, 2337–2347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan B et al. , Nat. Struct. Mol. Biol 22, 1023–1026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang J et al. , Science 350, aab4070 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng Z et al. , Proc. Natl. Acad. Sci. U.S.A 108, 20357–20361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theobald DL, Mitton-Fry RM, Wuttke DS, Annu. Rev. Biophys. Biomol. Struct 32, 115–133 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rice C, Skordalakes E, Comput. Struct. Biotechnol. J 14, 161–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Núñez-Ramírez R et al. , Nucleic Acids Res. 39, 8187–8199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen LY, Lingner J, Nucleus 4, 277–282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen EF et al. , J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.