

Abstract
Background
Statins are the cornerstone of pharmacotherapy for atherosclerotic cardiovascular disease. While these drugs are generally safe, treatment adherence is not optimal in a considerable proportion of patients because of the adverse effects on skeletal muscles in the forms of myopathy, myalgia, muscular pain, nocturnal muscle cramping, weakness, and rare rhabdomyolysis.
Methods
For the purpose of this narrative review, we searched for the literature suggesting the involvement of the ubiquitin–proteasome system in the development of statin–induced myopathy.
Results
Statins have been shown to up–regulate the expression of the muscle–specific ubiquitin–proteasome system as the major non–lysosomal intracellular protein degradation system. It has been postulated that statins may provoke instability in the myocyte cell membrane when subjected to eccentric exercise stress, triggering activation of intracellular proteolytic cascades and changes in protein degradation machinery. This is accompanied by the up–regulation of a series of genes implicated in protein catabolism, in addition to those of the ubiquitin–proteasome system.
Conclusions
Based on the available literature, it seems that the involvement of ubiquitin–proteasome system is potentially implicated in the pathophysiology of statin–induced myopathy.
Keywords: Statins, Myopathy, Ubiquitin‐proteasome system
The ubiquitin‐proteasome system
The levels of intracellular proteins are the result of the balance between synthesis and degradation processes, both fundamental for the correct functioning of the cell. In particular, protein degradation is finely regulated taking place through two main routes. The first is represented by lysosomes, vesicular organelles containing acidic hydrolases with different specificities, responsible for the disposal of endogenous or exogenous proteins internalized by endocytosis and pinocytosis. 1 , 2 The other proteolytic pathway involves ubiquitin, a polypeptide of 76 amino acids, which acts as a marker for degradation, and a multienzymatic complex, the proteasome. The ubiquitin‐proteasome system (UPS) does not require compartmentalization, and the catalytic complex acts ubiquitously (nucleus, cytoplasm, and in association with the endoplasmic reticulum), with ATP dependent and ATP independent mechanisms that involve numerous adjuvant molecules and that have been merged into the so‐called ‘signalosome’. 3
The UPS acts on different classes of both short and long half‐life proteins, and intervenes in the regulation of numerous cellular processes, being responsible for about 90% of extra‐lysosomal degradation, such as cell cycle control by proteolysis of specific regulatory proteins, cell growth, and proliferation through the degradation of oncoproteins and proteins of the signal transduction pathway, DNA repair, regulation of transcription, regulation of immune and inflammatory responses, processing of the antigens presented in association with the major class I histocompatibility complex (MHC I), and degradation of mutated or damaged proteins (DRiPs).
Because the proteasome intervenes in processes of vital importance for the cell, its inhibition leads to cell death and its malfunctioning can be the basis of numerous pathological events. 4 , 5 The proteins that must be degraded by this system are covalently labelled with ubiquitin. The marking is intended to label the substrate to be degraded to the multicatalytic enzyme complex. The degradation involves also many different protein substrates and is based on a very ingenious and efficient recognition and marking mechanism. 4
The ubiquitin attack on the substrate takes place through the formation of the isopeptide bond that is generated between the ubiquitin caboxide and the amino group of a Lys residue on the substrate to be degraded.
The first ubiquitin molecule can be covalently attached to the others, leading to the formation of ubiquitin chains (Figure 1). It has been observed that chains consisting of four or more ubiquitin units constitute the signal for proteasoma 26S. The ubiquitin attack is an ATP dependent process catalyzed by three enzymes: E1, E2, and E3, which also confer specificity to the process. The first reaction, catalyzed by E1, is responsible for the activation of ubiquitin: the C‐terminal of Gly 76 of ubiquitin, activated in an ATP‐dependent reaction, is linked to the Cys of the active site of the enzyme E1. The activated ubiquitin is transferred to E2, in whose active site there is a Cys residue. Through a transferase activity, E2 transfers ubiquitin to the E3 ligase that binds ubiquitin to Lys residues of the target protein, forming an isopeptide bond with the C‐terminal of Gly 76. In some cases, E2 transfers ubiquitin directly to the target protein without the intervention of E3. 1 , 2 , 3 , 4 , 5
FIGURE 1.
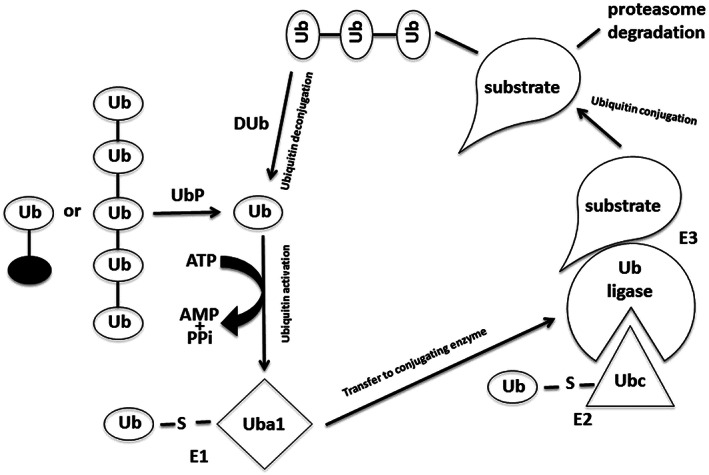
Protein degradation pathway by ubiquitin‐proteasome system. Ub ubiquitin; UbP, ubiquitin protease; DUb, deubiquitinating enzyme.
The enzyme system E1, E2, and E3 acts cyclically, allowing the formation of a ‘cluster’ of ubiquitin molecules because of the progressive union of an ubiquitin monomer with the Lys 48 of the previous ubiquitin. The specificity for the substrate in the ubiquitination system is because of the combined action of the different E2 transferases and of the different E3 ligases, which in combination recognize the protein destined for degradation.
Degradation begins with the recognition of specific signals by the E2/E3 enzymes on the protein. Following signal recognition, the substrate is labelled with ubiquitin, thus directing it to the proteasome. The signals present on the protein that induce the ubiquitin attack are the N‐terminal sequence, the phosphorylation of PEST sequences, the ‘destruction box’, and the loss of conformation. 1 , 2 , 3 , 4 , 5
The presence of particular amino acids in the N‐terminal of the protein is a universal signal. It has been shown that proteins with N‐terminal residues loaded (Lys or Arg) or voluminous (Phe, Leu, Tyr, and Trp) are rapidly ubiquitinated and degraded; on the contrary, proteins with small N‐terminal residues are quite stable.
The degradation signal requires the presence of a lysine close to the N‐terminal residue to assemble the ubiquitin ‘cluster’. The phosphorylation of specific sequences (Pro‐Glu‐Ser‐Thr, PEST) or of ‘destruction‐box’ (nine amino acids) represents the signal that determines the labelling with ubiquitin of target proteins. This signal is used by some proteins that regulate the cell cycle (e.g. the cicline), by p53, and by the transcription factor inhibitor nuclear factor kB (NF‐kB, IkB). 1 , 2 , 3 , 4 , 5
Proteins damaged by oxidation, mutations, or cellular stress, such as molecules that lose their three‐dimensional structure or are located in a wrong cellular compartment become excellent substrates for the ubiquitination process. The signals that activate this process are not yet perfectly clear but certainly involve the exposure of hydrophobic regions that are normally not exposed by proteins in their native structure. For example, proteins only partially or incorrectly synthesized in the endoplasmic reticulum are called ‘Detective Ribosomal Products’ (DRiPs); about 30% of the newly synthesized proteins—because they do not show up in their correct native conformation—are transferred by means of a specific transport from the endoplasmic reticulum to the cytoplasm and degraded by the UPS. In some cases, improperly shaped proteins are associated with assistance proteins (called chaperones) that mediate recognition with the ubiquitination enzymes. 1 , 2 , 3 , 4 , 5
The proteasome is a multicatalytic enzymatic complex located in the cytoplasm and in the nucleus of eukaryotic cells. It constitutes the main system of protein degradation and possesses different types of proteolytic exopeptidasic and endopeptidasic activities. Because of its functionality, the enzyme requires ATP and the ubiquitin labelling of the substrate to be degraded. The complex is able to realize the compartmentalization of proteolysis because the region responsible for the proteolytic activity (proteolytic chamber) is confined within the complex structure called ‘core’ 20S that can be associated with different types of regulators (19S or 11S/PA28).
The compartmentalization is essential for the regulation of proteolysis and avoids the erroneous degradation of proteins that must remain intact and functional. Time and space control of degradation is regulated not only by ubiquitin labelling but also by changes in cellular localization of the proteasome. In fact, using specific localization signals, the proteasome can be addressed in different compartments of the cytoplasm or of the nucleus. The main activities carried out by the proteasome and its regulators are the recognition and binding of proteins linked to ubiquitin, the activity of assembling and disassembling proteins, and the ability to move proteins inside the proteolytic chamber, different peptidal activities used to degrade damaged and regulatory proteins. This proteolytic complex therefore plays a key role in the maintenance of protein homeostasis, in the cell cycle, in transcription, and in the immune response as it generates the peptides that bind the molecules of the MHC‐I. 1 , 2 , 3 , 4 , 5
Statin treatment and cardiovascular risk
It has been several decades since the awareness of the existence of a close association between high plasma levels of low‐density lipoprotein (LDL) cholesterol and cardiovascular risk and the fact that its reduction is associated to a significant reduction in the risk to develop cardiovascular disease. In particular, the results of clinical studies conducted to test the efficacy of the inhibitors of 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase (statins), indicate a direct relationship between the reduction of LDL‐cholesterol levels and the reduction of cardiovascular events. 6 The results of the meta‐analysis of Baigent et al. 6 clarified very well the proportionality of the relationship between LDL cholesterolemia and cardiovascular events, highlighting that, with each reduction of 1 mmol/L or 39 mg/dL of LDL levels, there is a 12% reduction in mortality, essentially linked to a reduction in mortality from coronary causes, and a 21% reduction in cardiovascular events. This reduction seems to be similar in all patients regardless of age, sex, and the presence of cardiovascular diseases and baseline cholesterol levels.
The more recent literature has shown that the cardiovascular risk reduction associated to lipid‐lowering therapy is proportional to baseline cardiovascular risk of the patient and inversely to the achieved LDL values. In particular, a meta‐analysis of individual participant data from randomized trials involving at least 1000 participants and at least 2 years treatment duration of more vs. less intensive statin regimens (five trials; 39 612 individuals; median follow‐up 5.1 years) and of statin vs. control (21 trials; 129 526 individuals; median follow‐up 4.8 years) was carried out. Across all 26 trials, all‐cause mortality was reduced by 10% per 1·0 mmol/L LDL reduction (RR 0.90, 95% CI 0.87–0.93; P < 0.0001), largely reflecting significant reductions in deaths because of coronary heart disease (RR 0.80, 99% CI 0.74–0.87; P < 0.0001) and other cardiac causes (RR 0.89, 99% CI 0.81–0.98; P = 0.002), with no significant effect on deaths because of stroke (RR 0.96, 95% CI 0.84–1.09; P = 0.5) or other vascular causes (RR 0.98, 99% CI 0.81–1.18; P = 0.8). 7
Based on these results, European and North American guidelines for cardiovascular disease prevention, recommend reaching an LDL‐C level less than 115 mg/dL for subjects with low‐to‐moderate added cardiovascular risk, less than 100 mg/dL for subjects with high cardiovascular risk, and less than 70 mg/dL for subjects with very high cardiovascular disease risk. 8 , 9 , 10
In spite of novel lipid‐lowering therapies, 11 , 12 statins remain the most evidence‐based and cost‐effective approach to reduce LDL cholesterolemia and to reduce cardiovascular event risk. Besides, these drugs have pleiotropic cholesterol‐independent activities that contribute to their cardioprotective action. 13 , 14 , 15 , 16 , 17 , 18
In spite of these benefits, the efficacy of statins is often limited by the relatively low tolerability of more intense treatments, leading to dramatically reduced persistence. 19 Muscle‐related adverse events are among the most known and frequent causes of statin treatment interruption.
Statins' muscle‐related adverse events
Statins are generally effective and safe drugs; however, the main side effects could limit their prescription, in particular as regard liver injury and skeletal muscle toxicity (i.e. myopathy, myalgia, muscular pain, nocturnal muscle cramping, weakness, and rare rhabdomyolysis). 20 , 21 In 2001, cerivastatin was withdrawn from the market worldwide because of 31 rhabdomyolisis‐related deaths. 22
Although the incidence of statin‐induced myopathy is low in clinical trials, muscle symptoms are common in clinical practice. This is probably because of the fact that patients at high risk of statin‐related adverse events linked to predisposing factors like drugs affecting statin metabolism or co‐morbidities, have usually been excluded in clinical trials. 23 The presence of a negative drucebo effects has also been suggested to explain a considerable part of statin‐associated muscle symptoms in clinical practice. 24
Epidemiological data assess that myalgia represents from 6% to 14% of all adverse events associated with statin use occurring in 5% to 7% of patients. 25 , 26 , 27 Myopathy occurs with an incidence of 195 cases per 100 000 patient‐years (frequency <0.1% in patients on monotherapy with statins). 23 Lastly, in the post‐marketing surveillance, data from the Food and Drug Administration's (FDA) adverse events reporting system (AERS) records that the incidence of rhabdomyolysis is approximately 0.70 per 100 000 patient‐years with 0.15 deaths per 1 million prescriptions. 23 , 27
Creatine kinase (CK) is commonly used as serum marker to define skeletal muscle damage and its severity. However, myopathy can occur without CK elevations. 28 Conversely, intense physical exercise increases CK levels but without chronic muscle pain. 29 For these reasons, CK is a non‐sensitive biomarker of the induction of statin‐induced myotoxicity. However, because of the lack of specific biomarkers, CK is currently used to characterize myotoxicity related to statin‐therapy.
On the other hand, the terminology for describing muscle toxicity related to statin use is not yet harmonized. The American College of Cardiology/American Heart Association/National Lung Institute Clinical Advisory (ACC/AHA/NHLBI) defines myopathy as muscle pain, soreness, and frailty with or without abnormal serum CK and myalgia as muscle aches or weakness without CK elevation. Myositis has been described as muscle aches with CK enzyme level elevation (CK elevated but ≤10 times the ULN). 30 , 31 At the same time, according to National Lipid Association (NLA) myopathy includes the presence of previous symptoms plus a CK (>10 times the e limit of normal, ULN). 32
With respect to rhabdomyolysis, the most severe myotoxic effects statin‐related, the ACC characterizes this event with CK level 10 times the ULN, combined frequently with brown urine and urinary myoglobin. 30 , 31 NLA refers to rhabdomyolysis when CK level is over 10 000 IU/L plus an elevation in serum creatinine. 32
Despite the previous definitions given by different medical societies, the FDA does not give a specific differentiation between myopathy, myalgia, and myositis which are defined as muscle pain, weakness, and an elevation of the plasma CK value (>10 times the ULN). 33 It refers to rhabdomyolysis when CK is 50 times the ULN (or greater than 10 000 IU/L) with acute renal failure because of myoglobin precipitation in the kidney tubules. 34
The consequences of muscle symptoms as well as reduced drug compliance with possible discontinuation of therapy can lead to increased cardiovascular risk, impairment of the quality of life and limitation of physical activity. 27
Variable factors may increase the risk of statin‐related myopathy, both endogenous and exogenous. 25 The main endogenous factors are advanced age (75–80 years), gender (women are more predisposed), low body mass index, co‐morbidities (hypothyroidism, diabetes mellitus, liver disease, chronic renal failure, metabolic muscle disease, CYP450 polymorphism), vitamin D or carnitine palmityl transfer deficiency, and Asian ethnicity (Asians commonly require lower statin doses). 35 , 36 , 37
Exogenous factors that have been associated with statin‐related myopathy are: surgery, heavy alcohol consumption, vigorous physical exercise, drugs or foods that can interfere with the pharmacokinetics of statins. 35
Related to that, statins with the exception of pravastatin (metabolized by sulfation), are subject to hepatic phase 1 metabolism mediated by CYP450 enzymes. 38 In particular, the CYP3A4 isoform is responsible of the biotransformation of atorvastatin, lovastatin and simvastatin; fluvastatin is metabolized via the CYP2C9 system while rosuvastatin is a CYP2C9 and CYP2C19 substrate. 38
CYP3A4 inhibitors such as azole antifungals, anticoagulants (warfarin), immunosuppressants (cyclosporine), macrolide antibiotics, non‐dihydropyridine calcium channel blockers (verapamil and diltiazem), HIV‐protease inhibitors, antidepressants (nefazodone), amiodarone, excessive grapefruit juice or cranberry juice raise serum concentrations of statins increasing the risk of myopathy. 39 , 40
At the same time, significant increases in the concentrations of fluvastatin and rosuvastatin have been observed following co‐administration with fluconazole, amiodarone, cimetidine, fluvoxamine, trimethoprim‐sulfamethoxazole and ticlopidine. 39 , 40
Other drugs such as fibrates also increase the risk of myopathy by inhibiting the glucuronidation of statins. 41 While fenofibrate is a mild inhibitor, concomitant use of a statin and gemfibrozil increases the incidence all statin‐related myopathies and rhabomyolysis by 40%. 42 Gemfibrozil competes with statins for the hepatic microsomal enzymes uridine diphosphate glucuronosyl transferase (UGT)1A1 and inhibits human hepatic uptake transporter organic anion transporter 2 (OATP)2 increasing statins' plasma concentrations. 43 , 44
Another factor which affects pharmacokinetics of statins is the inhibition of the multidrug‐resistance protein (MRP)‐2; it alters the egress of hydrophilic molecules that cause myopathy in the same way as lipophilic ones. 44
There are multiple recommendations given by medical associations to prevent statins‐related adverse events.
According to ACC/AHA, statin treatment should be particularly monitored in patients with the aforementioned risk factors. The cholesterol treatment guidelines advice to start moderate or high dose of any statin in elderly patients (>75 age), evaluating both clinical atherosclerotic cardiovascular disease (ASCVD) benefits and co‐morbidities or drug interactions. For patients younger than 75 years tolerant to statins, it is not necessary to follow specific recommendations, except for pharmacokinetic factors. 30 , 45 Relating to CK levels, ACC/AHA envisage the measurement of its levels before starting statins treatment. If skeletal muscle symptoms appear during therapy, CK levels should be remeasured and reconciled with the parameters before treatment initiation. If CK levels exceed 10 times the baseline levels or statin myositis is suspected, statin treatment should be stopped immediately. If the CK level is less than five times ULN levels, patients should be monitored without discontinuation of therapy; if necessary statin switching or lower dosage could be evaluated until the normalization of the CK levels. 45 The European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) recommend starting statin treatment in older people only if at least one other cardiovascular risk factor is present in addition to age. Based on CK levels, they recommend not to start treatment in those patients who have five‐fold levels more at ULN. 46
Instead, the NLA Muscle Safety Expert Panel proposed a questionnaire for patients treated with statins for monitoring symptoms. 32 If a patient has CK levels three times the ULN or above and intense myalgia, statin should be discontinued for 2–4 weeks and eventually start again at a lower dose in co‐administration with another cholesterol lowering drug (colesevelam, ezetimibe). 32 Therefore, patients with a high CV risk should resume statin‐therapy.
Conversely, the International Atherosclerosis Society (IAS) sustains that statin prescription to patients over 80 years of age should be reserved for those who have a cardiovascular risk of at least 15% upon the 10‐year based on the Framingham risk algorithm. 47 Several algorithms for the diagnosis and management of myalgia have been created. They exclude confusing criteria of myalgia (e.g. hypothyroidism, extreme physical exercises, and vitamin D deficiency) and generally suggest to start with a low‐dose or hydrophilic statin with or without ezetimibe. 47 , 48 Indeed, some recent studies showed that myalgia and myopathy are likely dose‐related.
The Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) and the Pravastatin or Atorvastatin Evaluation and Infection Therapy‐Thrombolysis in Myocardial Infarction 22 (PROVE IT‐TIMI 22) trials compared three groups of patients, one treated with high a dose of atorvastatin, the second treated with placebo, and the third treated with low doses of drug. From the comparison of the patients results, the incidence of myopathy in the high‐dose statin group is 0.6% higher than the other two groups. 49 , 50
If statin treatment is not tolerated, an alternative option can be taken such as low‐dosage drug, intermittent doses using long half‐life statins like atorvastatin and rosuvastatin (14 and 19 h, respectively), switching to a different statin with another metabolism (e.g. from CYP450‐dependent to CYP‐450 independent) or solubility in monotherapy or with another hypolipidemic agent. 51 , 52 One last advice is to set up a therapy with nutraceuticals (e.g. rice fermented red, niacin) and CoQ10 supplementation even if data about that is still inconclusive or scarce. 53 , 54 , 55 , 56 , 57 However, if muscle symptoms persist even after discontinuation of statins, it is recommended to measure the CK levels and to pursue a muscle biopsy evaluating possible immunomediate necrosis that can be eventually treated with immunosuppressants. 58
Role of ubiquitin proteasome in statin‐induced myopathy
Statin‐related myopathy risk factors have been identified and mechanisms proposed, but there is not yet a unified pathophysiological understanding. The mechanisms seem to be interdependent: an increased statin systemic exposure which increases skeletal muscle exposure leads to an intracellular skeletal myocyte susceptibility and perturbation of muscle function. 59 Studies suggest multiple hypotheses (Table 1). First of all, structural abnormalities appeared in the biopsy of skeletal muscle of statins‐treated patients, even in asymptomatic subjects. 60 It has been postulated that statins alter energy metabolism by reducing beta‐oxidation of fatty acids and by increasing the amount of intracellular lipids with vacuoles filled of lipids and fibre atrophy. 28 However, in vitro studies show that morphological alterations could be reduced with mevalonic acid pretreatment, assuming that this side effect is attributable to the inhibition of HMG‐CoA. 61 Consequently, the depletion of intracellular cholesterol could lead to the instability of the myocyte cell membrane.
TABLE 1.
Proposed hypotheses on the pathogenesis of statin‐induced myopathy.
| Paper | Type of study | Mechanisms proposed |
|---|---|---|
| Guijarro et al., 1998 62 | Cell line | Interfering with the isoprenylation of intracellular selenocysteine‐GTP‐binding proteins (i.e. Ras, Rac and Rho), which promote cell growth and attenuate the apoptosis of myofibers |
| Phillips et al., 2002 28 | Human study (4 patients) | Altered energy metabolism by reducing beta‐oxidation of fatty acids and by increasing the amount of intracellular lipids with vacuoles filled of lipids and fibre atrophy |
| Willoughby et al., 2003 76 | Human study (9 patients) | Up‐regulation of the UPS during eccentric exercise has been related to increased muscle injury, decreased muscle strength, and a decrease in myofibrillar protein |
| Sandri et al., 2004 80 | Cell line | Blocking IGF‐1 signalling promoting FoxO dephosphorylation, nuclear localization, and transcription of the atrogin‐1 gene |
| Ludwig et al., 2005 81 | Cell line | The depletion of intracellular cholesterol resulting from inhibition of HMG‐CoA could lead to the instability of the myocyte cell membrane |
| Needham et al., 2007 66 | Human study (8 patients) | Modulating the immune system by up‐regulating the expression of MHC‐I and the mediators of inflammation leads to muscle symptoms |
| Oh et al., 2007 67 | Human study (133 statin‐intolerant patients and 158 controls) | Genetic polymorphisms, in particular those of the coenzyme Q2 gene, involved in the biochemical activity of ubiquinone, and CYP450 related to drugs‐metabolism |
| Hanai et al.,2007 65 | Cell line | Increasing in atrophy‐related genes (atrogenes) such as atrogin‐1, results in enhanced degradation of skeletal muscle protein via the ubiquitin proteasome pathway |
| Marcoff et al., 2007 64 | Systematic Review | Interfering with the isoprenylation of intracellular selenocysteine‐GTP‐binding proteins (i.e. Ras, Rac and Rho), which promote cell growth and attenuate the apoptosis of myofibers |
| Catapano et al., 2012 44 | Systematic Review | Inhibition of the multidrug‐resistance protein (MRP)‐2; it alters the egress of hydrophilic molecules that cause myopathy |
| Bouitbir et al., 2019 68 | Systematic Review | Statin‐induced myopathy could be only the result of HMG‐CoA reductase (HMGCR) gene inhibition. |
In addition to blocking cholesterol metabolism, statins also reduce products of the mevalonic pathway, thereby increasing the vulnerability of skeletal muscle cells as well as alteration of sterol metabolism. 28 Mevalonate is the precursor of prenylated isoprenoids including ubiquinone/coenzyme Q10 involved in electron transport in oxidative phosphorylation. 62 Even if low dosages of statins reduce serum levels of ubiquinone, intramuscular levels remain unaltered. 63 In particular, a study conducted in cultured rat vascular smooth muscle cells has shown that statins interfere in a dose‐dependent manner with the isoprenylation of intracellular selenocysteine‐GTP‐binding proteins (i.e. Ras, Rac, and Rho), which promote cell growth and attenuate the apoptosis of myofibers. 62 , 64
The atrophy of skeletal muscle cells could indicate the ability of statins to induce the expression of atrogin‐1, which is responsible for the destruction of some muscle proteins. In other studies, statin‐induced myopathy appeared to be immune‐mediated. 65 In fact, in some patients, statins are able to modulate the immune system by upregulating the expression of MHC‐I and some inflammatory mediators. 66 This is also supported by the fact that the muscle symptoms have been reduced in patients treated with immunosuppressants.
It has also been suggested that statin‐related myopathic effects are related to genetic polymorphisms, in particular those of the coenzyme Q2 gene, involved in the biochemical activity of ubiquinone, and CYP450 related to drugs‐metabolism. 67
It is not definitively clear if the effect of statin treatment is a consequence of HMG‐CoA reductase inhibition or another nonspecific drug effect. Fish knockdown of the HMG‐CoA reductase (HMGCR) gene showed a muscle response similar to that seen in other studies, strongly questioning that statin‐induced myopathy could be only the result of HMG‐CoA reductase inhibition. 68
Nevertheless, some studies have documented a controversial role of UPS inhibition in particular clinical conditions, such as Alzheimer disease. 69
The impairment of the UPS has been indicated as one possible mechanism for muscle wasting and atrophy 70 , 71 , 72 , 73 , 74 , 75 as well as statin‐induced muscle myopathy during exercise. Up‐regulation of the UPS during eccentric exercise has been related to increased muscle injury, decreased muscle strength, and a decrease in myofibrillar protein 76 (Figure 2). Urso et al. investigated the role of ubiquitin proteasome pathway gene expression in skeletal muscle after exercise and statin use. In this study, individuals did eccentric exercises with one leg before and after placebo or high‐dose atorvastatin (80 mg/d) treatment. The unexercised leg was used as a control. Among subjects taking statin treatment alone, only five genes were differentially expressed in comparison to the unexercised leg or the exercised leg of the placebo group, showing that statins do not impact significantly on skeletal muscle gene expression. On the other hand, eccentric exercise alone induced 80 genes that were differentially expressed compared with the non‐exercised leg of the placebo group. Authors concluded that eccentric exercise along with statin treatment had the most significant effect on transcription factors and genes involved in the ubiquitin proteasome pathway when compared with eccentric exercise or statin use alone. This study showed that the statin‐induced myopathy could be explained by alterations of the expression of different genes because of the combination of statins and exercise. 77
FIGURE 2.
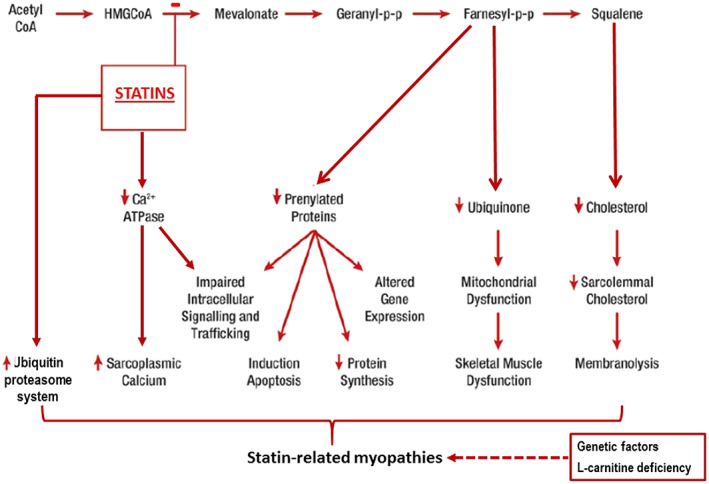
Potential mechanisms implicated in the pathophysiology of statin‐associated myopathies.
The relationship between statins and UPS has also been investigated in studies exploring the role of atrogin‐1 that is a ubiquitin protein ligase significantly induced by any stimulus that promote muscle atrophy and that may be a key effector in muscle degradation during catabolic states. 78
Authors hypothesized that statin myotoxicity could be similar to the atrophic response seen in other muscle‐wasting diseases. 79 Increases in atrophy‐related genes (atrogenes) such as atrogin‐1, results in enhanced degradation of skeletal muscle protein via the ubiquitin proteasome pathway. 64
Statin treatment blocks IGF‐1 signalling promoting FoxO dephosphorylation, nuclear localization, and transcription of the atrogin‐1 gene. 80 Authors demonstrated that the lack of geranylgeranyl induced by statin inhibition of HMGCoA reductase is responsible for at least part of this toxicity. They showed that geranylgeranyl precursors introduction in both muscle cell cultures and in zebrafish embryos treated with lovastatin blocks expression of atrogin‐1 and avoids myotoxicity. On the contrary, blocking the enzymes that couple geranylgeranyl precursors onto intracellular proteins produces the same effects observed for lovastatin in terms of cell morphology and atrogin‐1 expression. These results suggest that statins may exert their toxic effects in muscle by inhibiting the function of a geranylgeranyl‐conjugated protein that directly or indirectly results in atrogin‐1 expression. 81
Recent data show that L‐carnitine ((3R)‐3‐hydroxy‐4‐(trimethylazaniumyl)butanoate), an endogenous transporter of fatty acids into the mitochondrial matrix, biosynthesized within the human body from L‐lysine and S‐adenosyl‐methionine, could partially counteract the UPS negative effects. 82 Carnitine plays a key role in importing acetyl‐CoA into mitochondria. Excess mitochondrial acetyl‐CoA is produced when an oversupply of fuel (predominantly glucose or fatty acids) enters the cell and mitochondrial oxidative capacity is saturated. The elevated concentration of acetyl‐CoA may facilitate acetylation of lysine residues on mitochondrial enzymes, modulating their activity. Enzyme acetylation may inhibit fuel utilization when energy is in excess, favouring lipogenesis. 83 However, preclinical models suggest that L‐carnitine supplementation could down‐regulate UPS genes in skeletal muscle. 84 In particular, supplementation of carnitine markedly decreases the expression of MuRF1 and concentrations of ubiquitinated proteins in skeletal muscle of rats, indicating a diminished degradation of myofibrillar proteins by the UPS. 85 This effect could be mediated by activation of the IGF‐1/PI3K/Akt signalling pathway which in turn might contribute to the observed down‐regulation of MuRF1 and muscle protein ubiquitination. 85 L‐carnitine supplementation seems to have a protective effect against soleus muscle atrophy caused by hind limb suspension and decreased E3 ligase messenger RNA expression, suggesting the possibility that L‐carnitine protects against muscle atrophy, at least in part, through UPS inhibition. 86 These results could be of particular interest, because L‐carnitine could exert some anti‐inflammatory activity in humans, reducing the serum levels of some cytokines (Interleukin 6, tumour necrosis factor alpha, C‐reactive protein) often increased in myopathies. 87 On the other side, some papers raise doubts about the usefulness of carnitine in some clinical conditions. 88 , 89
The role of carnitine in cancer is also not well established. Recent findings have suggested that carnitine system could be involved in the metabolic flexibility of cancer cells. 90 On the contrary, the UPS down‐regulation and autophagy activation trigger apoptosis in cancer cells. 91
In addition, Vitamin D supplementation could exert some positive effects on prevention of muscle mass loss preventing muscle mass losses. 92 , 93 At the same time, essential aminoacids have also documented positive effects on preclinical models of statin‐induced myopathy. 94 , 95 Other studies have shown that statin‐induced creatine impairment could play a role in statin myopathy, suggesting creatine supplementation as a potential approach to prevent muscle‐related adverse effects of statins and improve statin tolerance. 96
Conclusions
In summary, it has been hypothesized that the introduction of statins into the myocyte cell membrane may provoke instability when subjected to eccentric exercise stress, provoking activation of intracellular proteolytic cascades and changes in protein degradative machinery. This is consistent with the up‐regulation of a series of genes implicated in protein catabolism, in addition to those of the UPS. However, since there is a lack of in vivo studies, further research should be performed to elucidate the impact of statins on protein catabolism inducing myotoxicity.
Conflict of interest
Dr. Cicero has given talks, furnished scientific consultancies and/or participated in trials sponsored by Amgen, Angelini, Mylan, Sharper, and Sanofi. Dr. von Haehling has been a paid consultant for and/or received honoraria payments from Bayer, Boehringer Ingelheim, BRAHMS, Chugai, Grünenthal, Helsinn, Hexal, Novartis, Respicardia, Roche, Sorin, and Vifor; owns shares in Actimed. He reports research support from IMI and the German Center for Cardiovascular Research (DZHK). Dr. Banach has served on the speakers bureau of Abbott/Mylan, Abbott Vascular, Actavis, Akcea, Amgen, Biofarm, KRKA, MSD, Sanofi‐Aventis, Servier and Valeant, and has served as a consultant to Abbott Vascular, Akcea, Amgen, Daichii Sankyo, Esperion, Lilly, MSD, Resverlogix, Sanofi‐Aventis; Grants from Sanofi and Valeant. Other authors have no competing interests to declare.
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. 97
Sahebkar A., Cicero A. F. G., Di Giosia P., Pomilio I., Stamerra C. A., Giorgini P., Ferri C., von Haehling S., Banach M., and Jamialahmadi T. (2020) Pathophysiological mechanisms of statin‐associated myopathies: possible role of the ubiquitin‐proteasome system, Journal of Cachexia, Sarcopenia and Muscle, 11, 1177–1186, doi: 10.1002/jcsm.12579
References
- 1. Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005;6:79. [DOI] [PubMed] [Google Scholar]
- 2. Grune T. Oxidative stress, aging and the proteasomal system. Biogerontology 2000;1:31–40. [DOI] [PubMed] [Google Scholar]
- 3. Schwartz PA, Ciechanover A. The ubiquitin‐proteasome pathway and pathogenesis of human diseases. Annu Rev Med 1999;50:57–74. [DOI] [PubMed] [Google Scholar]
- 4. Ciechanover A. The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J 1998;17:7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hershko A, Ciechanover A. The ubiquitin system. Annual Reviews 4139 El Camino Way, PO Box 10139, Palo Alto, CA 94303–0139, USA; 1998.
- 6. Baigent C. Cholesterol Treatment Trialists'(CTT) Collaborators: Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 7. Baigent C, Blackwell L, Emberson J, Holland L, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. 9753, 376, Lancet; 20101670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Revista espanola de cardiologia (English ed) 2017;70:115. [DOI] [PubMed] [Google Scholar]
- 9. Jellinger PS, Handelsman Y, Rosenblit PD, Bloomgarden ZT, Fonseca VA, Garber AJ, et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract 2017;23:1–87. [DOI] [PubMed] [Google Scholar]
- 10. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2019;73:e285–e350. [DOI] [PubMed] [Google Scholar]
- 11. Sahebkar A, Watts GF. New therapies targeting apoB metabolism for high‐risk patients with inherited dyslipidaemias: what can the clinician expect? Cardiovasc Drugs Ther 2013;27:559–567. [DOI] [PubMed] [Google Scholar]
- 12. Banach M, Aronow WS, Serban C, Sahabkar A, Rysz J, Voroneanu L, et al. Lipids, blood pressure and kidney update 2014. Pharmacol Res 2015;95–96:111–125. [DOI] [PubMed] [Google Scholar]
- 13. Parizadeh SMR, Azarpazhooh MR, Moohebati M, Nematy M, Ghayour‐Mobarhan M, Tavallaie S, et al. Simvastatin therapy reduces prooxidant‐antioxidant balance: results of a placebo‐controlled cross‐over trial. Lipids 2011;46:333–340. [DOI] [PubMed] [Google Scholar]
- 14. Sahebkar A, Kotani K, Serban C, Ursoniu S, Mikhailidis DP, Jones SR, et al. Statin therapy reduces plasma endothelin‐1 concentrations: a meta‐analysis of 15 randomized controlled trials. Atherosclerosis 2015;241:433–442. [DOI] [PubMed] [Google Scholar]
- 15. Sahebkar A, Serban C, Mikhailidis DP, Undas A, Lip GYH, Muntner P, et al. Association between statin use and plasma d‐dimer levels: a systematic review and meta‐analysis of randomised controlled trials. Thromb Haemost 2015;114:546–557. [DOI] [PubMed] [Google Scholar]
- 16. Chruściel P, Sahebkar A, Rembek‐Wieliczko M, Serban MC, Ursoniu S, Mikhailidis DP, et al. Impact of statin therapy on plasma adiponectin concentrations: a systematic review and meta‐analysis of 43 randomized controlled trial arms. Atherosclerosis 2016;253:194–208. [DOI] [PubMed] [Google Scholar]
- 17. Sahebkar A, Serban C, Ursoniu S, Mikhailidis DP, Undas A, Lip GYH, et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen: systematic review and meta‐analysis of Randomised placebo‐controlled trials. Thromb Haemost 2016;115:520–532. [DOI] [PubMed] [Google Scholar]
- 18. Serban C., Sahebkar A., Ursoniu S., Mikhailidis D.P., Rizzo M., Lip G.Y.H., et al. A systematic review and meta–analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Scientific Reports 2015;5,art. no. 09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deshpande S, Quek RG, Forbes CA, de Kock S, Kleijnen J, Gandra SR, et al. A systematic review to assess adherence and persistence with statins. Curr Med Res Opin 2017;33:769–778. [DOI] [PubMed] [Google Scholar]
- 20. Thompson PD, Clarkson P, Karas RH. Statin‐associated myopathy. JAMA 2003;289:1681–1690. [DOI] [PubMed] [Google Scholar]
- 21. Joy TR, Hegele RA. Narrative review: statin‐related myopathy. Ann Intern Med 2009;150:858–868. [DOI] [PubMed] [Google Scholar]
- 22. Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. New Engl J Med 2002;346:539–540. [DOI] [PubMed] [Google Scholar]
- 23. Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006;97:S52–S60. [DOI] [PubMed] [Google Scholar]
- 24. Penson PE, Mancini GBJ, Toth PP, Martin SS, Watts GF, Sahebkar A, et al. Introducing the 'Drucebo' effect in statin therapy: a systematic review of studies comparing reported rates of statin‐associated muscle symptoms, under blinded and open‐label conditions. J Cachexia Sarcopenia Muscle 2018;9:1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamilton‐Craig I. Statin‐associated myopathy. Med J Aust 2001;175:486–489. [DOI] [PubMed] [Google Scholar]
- 26. Black DM. A general assessment of the safety of HMG CoA reductase inhibitors (statins). Curr Atheroscler Rep 2002;4:34–41. [DOI] [PubMed] [Google Scholar]
- 27. Ucar M, Mjörndal T, Dahlqvist R. HMG‐CoA reductase inhibitors and myotoxicity. Drug Saf 2000;22:441–457. [DOI] [PubMed] [Google Scholar]
- 28. Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, Kimura BJ, et al. Statin‐associated myopathy with normal creatine kinase levels. Ann Intern Med 2002;137:581–585. [DOI] [PubMed] [Google Scholar]
- 29. Thompson PD, Gadaleta PA, Yurgalevitch S, Cullinane E, Herbert PN. Effects of exercise and lovastatin on serum creatine kinase activity. Metabolism 1991;40:1333–1336. [DOI] [PubMed] [Google Scholar]
- 30. Pasternak RC, Smith SC, Bairey‐Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol 2002;40:567–572. [DOI] [PubMed] [Google Scholar]
- 31. Wilkinson MJ, Laffin LJ, Davidson MH. Overcoming toxicity and side‐effects of lipid‐lowering therapies. Best Pract Res Clin Endocrinol Metab 2014;28:439–452. [DOI] [PubMed] [Google Scholar]
- 32. McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the national lipid association statin safety assessment task force. Am J Cardiol 2006;97:S89–S94. [DOI] [PubMed] [Google Scholar]
- 33. Norata GD, Tibolla G, Catapano AL. Statins and skeletal muscles toxicity: from clinical trials to everyday practice. Pharmacol Res 2014;88:107–113. [DOI] [PubMed] [Google Scholar]
- 34. Maji D, Shaikh S, Solanki D, Gaurav K. Safety of statins. Indian J Endocrinol Metab 2013;17:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toth PP, Harper CR, Jacobson TA. Clinical characterization and molecular mechanisms of statin myopathy. Expert Rev Cardiovasc Ther 2008;6:955–969. [DOI] [PubMed] [Google Scholar]
- 36. Sweis D, Wong IC. A survey on factors that could affect adverse drug reaction reporting according to hospital pharmacists in Great Britain. Drug Saf 2000;23:165–172. [DOI] [PubMed] [Google Scholar]
- 37. McPherson R, Frohlich J, Fodor G, Genest J. Canadian Cardiovascular Society position statement–recommendations for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease. Can J Cardiol 2006;22:913–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Therapeut 2006;80:565–581. [DOI] [PubMed] [Google Scholar]
- 39. Worz CR, Bottorff M. The role of cytochrome P450‐mediated drug‐drug interactions in determining the safety of statins. Expert Opin Pharmacother 2001;2:1119–1127. [DOI] [PubMed] [Google Scholar]
- 40. Kellick KA, Bottorff M, Toth PP. A clinician's guide to statin drug‐drug interactions. J Clin Lipidol 2014;8:S30–S46. [DOI] [PubMed] [Google Scholar]
- 41. Corsini A, Bellosta S, Davidson MH. Pharmacokinetic interactions between statins and fibrates. Am J Cardiol 2005;96:44–49. [DOI] [PubMed] [Google Scholar]
- 42. Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007;49:1753–1762. [DOI] [PubMed] [Google Scholar]
- 43. Prueksaritanont T, Richards KM, Qiu Y, Strong‐Basalyga K, Miller A, Li C, et al. Comparative effects of fibrates on drug metabolizing enzymes in human hepatocytes. Pharm Res 2005;22:71–78. [DOI] [PubMed] [Google Scholar]
- 44. Catapano AL. Statin‐induced myotoxicity: pharmacokinetic differences among statins and the risk of rhabdomyolysis, with particular reference to pitavastatin. Curr Vasc Pharmacol 2012;10:257–267. [DOI] [PubMed] [Google Scholar]
- 45. Stone NJ, Robinson JG, Lichtenstein AH, Merz CNB, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2889–2934. [DOI] [PubMed] [Google Scholar]
- 46. Catapano AL, Reiner Ž, De Backer G, Graham I, Taskinen M‐R, Wiklund O, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011;217:1–44. [DOI] [PubMed] [Google Scholar]
- 47. Strandberg TE, Kolehmainen L, Vuorio A. Evaluation and treatment of older patients with hypercholesterolemia: a clinical review. JAMA 2014;312:1136–1144. [DOI] [PubMed] [Google Scholar]
- 48. Jacobson TA, ed. Toward “pain‐free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clinic Proceedings. n/a: Elsevier; 2008. [DOI] [PubMed] [Google Scholar]
- 49. de Lemos JA, Blazing MA, Wiviott SD, Lewis EF, Fox KA, White HD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004;292:1307–1316. [DOI] [PubMed] [Google Scholar]
- 50. Ose L, Davidson MH, Insull W, Wu M, Tate AC, Melino MR, et al. Lipid‐altering efficacy and safety of simvastatin 80 mg/day: long‐term experience in a large group of patients with hypercholesterolemia. Clin Cardiol 2000;23:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keating AJ, Campbell KB, Guyton JR. Intermittent nondaily dosing strategies in patients with previous statin‐induced myopathy. Ann Pharmacother 2013;47:398–404. [DOI] [PubMed] [Google Scholar]
- 52. Wierzbicki AS, Viljoen A, Hardman TC, Mikhailidis DP. New therapies to reduce low‐density lipoprotein cholesterol. Curr Opin Cardiol 2013;28:452–457. [DOI] [PubMed] [Google Scholar]
- 53. Ruscica M, Gomaraschi M, Mombelli G, Macchi C, Bosisio R, Pazzucconi F, et al. Nutraceutical approach to moderate cardiometabolic risk: results of a randomized, double‐blind and crossover study with Armolipid Plus. J Clin Lipidol 2014;8:61–68. [DOI] [PubMed] [Google Scholar]
- 54. Sahebkar A, Saboni N, Pirro M, Banach M. Curcumin: an effective adjunct in patients with statin‐associated muscle symptoms? J Cachexia Sarcopenia Muscle 2017;8:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodriguez J, Pierre N, Naslain D, Bontemps F, Ferreira D, Priem F, et al. Urolithin B, a newly identified regulator of skeletal muscle mass. J Cachexia Sarcopenia Muscle 2017;8:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Banach M., Patti A.M., Giglio R.V., Cicero A.F.G., Atanasov A.G., Bajraktari G., et al. The Role of Nutraceuticals in Statin Intolerant Patients. Journal of the American College of Cardiology 2018;72(1):96–118 [DOI] [PubMed] [Google Scholar]
- 57. Banach M., Serban C., Ursoniu S., Rysz J., Muntner P., Toth P.P., et al. Statin therapy and plasma coenzyme Q10 concentrations–A systematic review and meta-analysis of placebo–controlled trials. Pharmacological Research 2015;99:329–336, art. no. 2870 [DOI] [PubMed] [Google Scholar]
- 58. Mohassel P, Mammen AL. Statin‐associated autoimmune myopathy and anti‐HMGCR autoantibodies. Muscle Nerve 2013;48:477–483. [DOI] [PubMed] [Google Scholar]
- 59. Turner RM, Pirmohamed M. Statin‐related myotoxicity: a comprehensive review of pharmacokinetic, Pharmacogenomic and Muscle Components. J Clin Med 2020;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, Allemann C, et al. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol 2006;210:94–102. [DOI] [PubMed] [Google Scholar]
- 61. Ludwig A, Friedel B, Metzkow S, Meiners S, Stangl V, Baumann G, et al. Effect of statins on the proteasomal activity in mammalian endothelial and vascular smooth muscle cells. Biochem Pharmacol 2005;70:520–526. [DOI] [PubMed] [Google Scholar]
- 62. Guijarro C, Blanco‐Colio LM, Ortego M, Alonso C, Ortiz A, Plaza JJ, et al. 3‐Hydroxy‐3‐methylglutaryl coenzyme a reductase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ Res 1998;83:490–500. [DOI] [PubMed] [Google Scholar]
- 63. Laaksonen R, Ojala J‐P, Tikkanen MJ, Himberg J‐J. Serum ubiquinone concentrations after short‐and long‐term treatment with HMG‐CoA reductase inhibitors. Eur J Clin Pharmacol 1994;46:313–317. [DOI] [PubMed] [Google Scholar]
- 64. Marcoff L, Thompson PD. The role of coenzyme Q10 in statin‐associated myopathy: a systematic review. J Am Coll Cardiol 2007;49:2231–2237. [DOI] [PubMed] [Google Scholar]
- 65. J‐i H, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, et al. The muscle‐specific ubiquitin ligase atrogin‐1/MAFbx mediates statin‐induced muscle toxicity. J Clin Invest 2007;117:3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up‐regulation of MHC‐I associated with statin therapy. Neuromuscul Disord 2007;17:194–200. [DOI] [PubMed] [Google Scholar]
- 67. Oh J, Ban MR, Miskie BA, Pollex RL, Hegele RA. Genetic determinants of statin intolerance. Lipids Health Dis 2007;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bouitbir J, Sanvee GM, Panajatovic MV, Singh F, Krähenbühl S. Mechanisms of statin‐associated skeletal muscle‐associated symptoms. Pharmacol Res 2019;154:104201. [DOI] [PubMed] [Google Scholar]
- 69. Hegde AN, Smith SG, Duke LM, Pourquoi A, Vaz S. Perturbations of ubiquitin‐proteasome‐mediated proteolysis in aging and Alzheimer's disease. Front Aging Neurosci 2019;11:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Raz V, Raz Y, Paniagua‐Soriano G, Roorda JC, Olie C, Riaz M, et al. Proteasomal activity‐based probes mark protein homeostasis in muscles. J Cachexia Sarcopenia Muscle 2017;8:798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun L, Si M, Liu X, Choi JM, Wang Y, Thomas SS, et al. Long‐noncoding RNA Atrolnc‐1 promotes muscle wasting in mice with chronic kidney disease. J Cachexia Sarcopenia Muscle 2018;9:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Polge C, Cabantous S, Deval C, Claustre A, Hauvette A, Bouchenot C, et al. A muscle‐specific MuRF1‐E2 network requires stabilization of MuRF1‐E2 complexes by telethonin, a newly identified substrate. J Cachexia Sarcopenia Muscle 2018;9:129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Komatsu R, Okazaki T, Ebihara S, Kobayashi M, Tsukita Y, Nihei M, et al. Aspiration pneumonia induces muscle atrophy in the respiratory, skeletal, and swallowing systems. J Cachexia Sarcopenia Muscle 2018;9:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, et al. Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle 2017;8:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ebner N, Anker SD, von Haehling S. Recent developments in the field of cachexia, sarcopenia, and muscle wasting: highlights from the 11th Cachexia Conference. J Cachexia Sarcopenia Muscle 2019;10:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Willoughby DS, Taylor M, Taylor L. Glucocorticoid receptor and ubiquitin expression after repeated eccentric exercise. Med Sci Sports Exerc 2003;35:2023–2031. [DOI] [PubMed] [Google Scholar]
- 77. Urso ML, Clarkson PM, Hittel D, Hoffman EP, Thompson PD. Changes in ubiquitin proteasome pathway gene expression in skeletal muscle with exercise and statins. Arterioscler Thromb Vasc Biol 2005;25:2560–2566. [DOI] [PubMed] [Google Scholar]
- 78. Hoffman EP, Nader GA. Balancing muscle hypertrophy and atrophy. Nat Med 2004;10:584. [DOI] [PubMed] [Google Scholar]
- 79. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 80. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cao P, Hanai J‐I, Tanksale P, Imamura S, Sukhatme VP, Lecker SH. Statin‐induced muscle damage and atrogin‐1 induction is the result of a geranylgeranylation defect. FASEB J 2009;23:2844–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pekala J, Patkowska‐Sokola B, Bodkowski R, Jamroz D, Nowakowski P, Lochynski S, et al. L‐carnitine‐metabolic functions and meaning in humans life. Curr Drug Metab 2011;12:667–678. [DOI] [PubMed] [Google Scholar]
- 83. Ghanta S, Grossmann RE, Brenner C. Mitochondrial proteinacetylation as a cell‐intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl‐lysine modifications. Crit Rev Biochem MolBiol 2013;48:561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Keller J, Ringseis R, Koc A, Lukas I, Kluge H, Eder K. Supplementation with l‐carnitine downregulates genes of the ubiquitin proteasome system in the skeletal muscle and liver of piglets. Animal 2012;6:70–78. [DOI] [PubMed] [Google Scholar]
- 85. Keller J, Couturier A, Haferkamp M, Most E, Eder K. Supplementation of carnitine leads to an activation of the IGF‐1/PI3K/Akt signalling pathway and down regulates the E3 ligase MuRF1 in skeletal muscle of rats. Nutr Metab 2013;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jang J, Park J, Chang H, Lim K. l‐Carnitine supplement reduces skeletal muscle atrophy induced by prolonged hindlimb suspension in rats. Appl Physiol Nutr Metab 2016;41:1240–1247. [DOI] [PubMed] [Google Scholar]
- 87. Haghighatdoost F, Jabbari M, Hariri M. The effect of L‐carnitine on inflammatory mediators: a systematic review and meta‐analysis of randomized clinical trials. Eur J Clin Pharmacol 2019;1–10. [DOI] [PubMed] [Google Scholar]
- 88. Adeva‐Andany MM, Calvo‐Castro I, Fernandez‐Fernandez C, Donapetry‐Garcıa C, Pedre‐Pineiro AM. Significance of L‐carnitine for human health. IUBMB Life 2017;69:578–594. [DOI] [PubMed] [Google Scholar]
- 89. Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell‐intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl‐lysine modifications. Crit Rev Biochem Mol Biol 48:561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Melone MAB, Valentino A, Margarucci S, Galderisi U, Giordano A, Peluso G. The carnitine system and cancer metabolic plasticity. Cell Death Dis 2018;9:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yiting T, Yangyang G, Judong L, Wenhao S, Wei Z, Cuicui M, et al. Downregulation of ubiquitin inhibits the proliferation and radioresistance of non‐small cell lung cancer cells in vitro and in vivo. Sci Rep 2015;5:9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ostadmohammadi V, Milajerdi A, Ghayour‐Mobarhan M, Ferns G, Taghizadeh M, Badehnoosh B, et al. The effects of vitamin D supplementation on glycemic control, lipid profiles and C‐reactive protein among patients with cardiovascular disease: a systematic review and meta‐analysis of randomized controlled trials. Curr Pharm Des 2019;25:201–210. [DOI] [PubMed] [Google Scholar]
- 93. Abrams GD, Feldman D, Safran MR. Effects of vitamin D on skeletal muscle and athletic performance. J Am Acad Orthop Surg 2018. Apr 15;26:278–285. [DOI] [PubMed] [Google Scholar]
- 94. D'Antona G, Tedesco L, Ruocco C, Corsetti G, Ragni M, Fossati A, et al. Peculiar formula of essential amino acids prevents rosuvastatin myopathy in mice. Antioxid Redox Signal 2016;25:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Corsetti G, Pasini E, Romano C, Calvani R, Picca A, Marzetti E, et al. Body weight loss and tissue wasting in late middle‐aged mice on slightly imbalanced essential/non‐essential amino acids diet. Front Med 2018;5:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Balestrino M, Adriano E. Creatine as a candidate to prevent statin myopathy. Biomolecules 2019;9:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]