

Abstract
Genomes across a wide range of eukaryotic organisms fold into higher-order chromatin domains. Topologically associating domains (TADs) were originally discovered empirically in low-resolution Hi-C heat maps representing ensemble average interaction frequencies from millions of cells. Here, we discuss recent advances in high-resolution Hi-C, single-cell imaging experiments, and functional genetic studies, which provide an increasingly complex view of the genome’s hierarchical structure–function relationship. On the basis of these new findings, we update the definitions of distinct classes of chromatin domains according to emerging knowledge of their structural, mechanistic and functional properties.
TADs are a fundamental feature of genome folding and were co-discovered in 2012 in some of the first genome-wide chromatin-folding maps1–4. TADs were originally defined algorithmically in low-resolution (40 kb) mammalian Hi-C matrices as megabase-scale genomic blocks in which DNA sequences exhibit significantly higher interaction frequency with other DNA sequences within the domain than with those outside of the block (Fig. 1a). Perhaps the most salient feature of TADs is that they are demarcated by boundaries (Fig. 1a,b). A compelling hypothesis proposed to explain these seminal empirical observations was that most of the mammalian genome is folded into adjacent, globular chromatin-interaction domains connected by linear boundaries1–4 (Fig. 1b).
Fig. 1 |. The structural features of topologically associating domains.
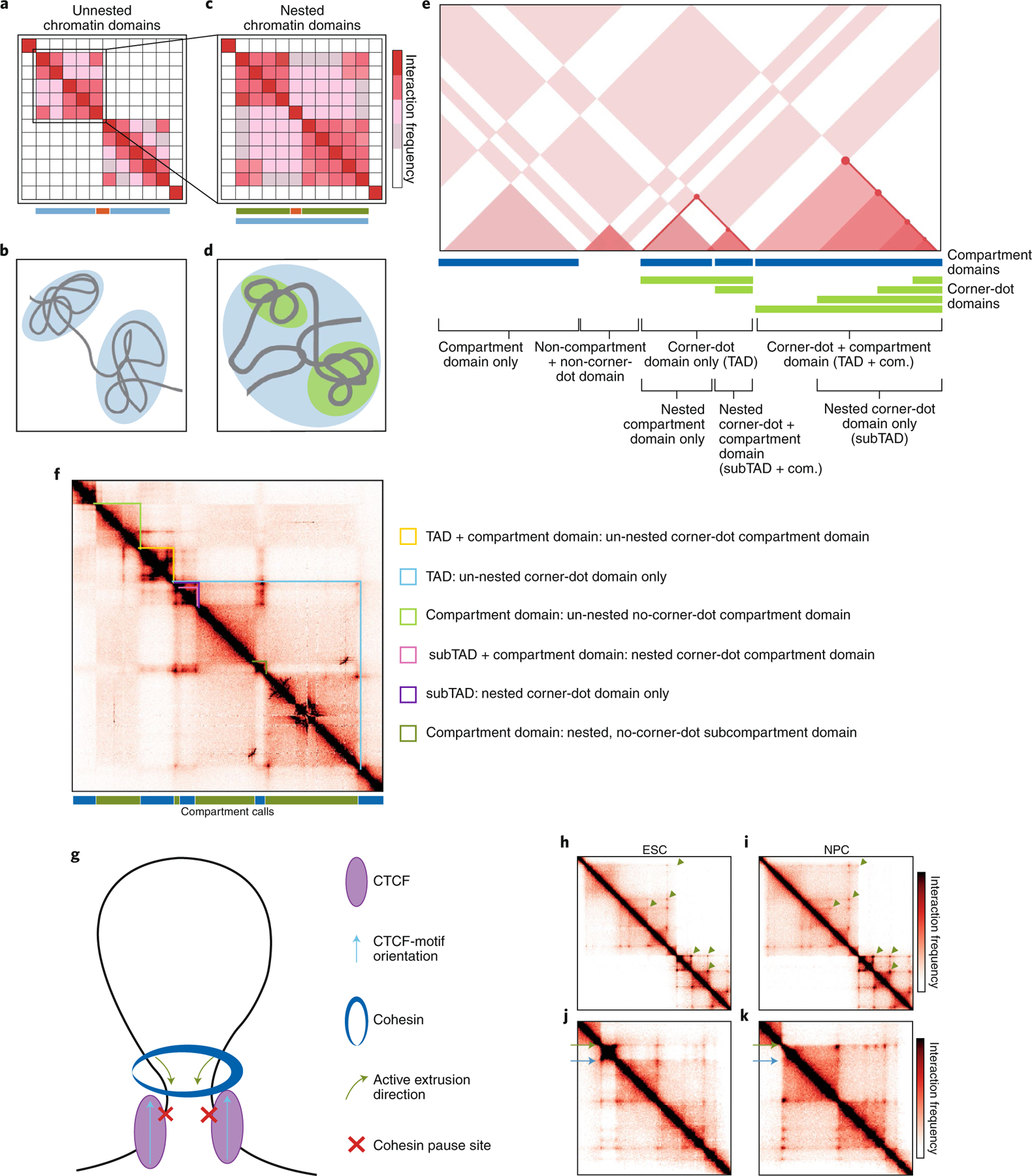
a–d, Heat-map representations (top) and schematized globular interactions (bottom) of TADs (a,b) and nested subTADs (c,d). e, Cartoon representation of different classes of contact domains parsed by their structural features and degree of nesting. f, Identification of contact-domain classes from e in cortical neuron Hi-C data from ref. 37, binned at 10-kb resolution. g, Cohesin translocation extrudes DNA in an ATP-dependent manner into long-range looping interactions that form the topological basis for TAD and subTAD loop domains. h–k, Contact frequency heat maps of high-resolution Hi-C data from ref. 37, performed on embryonic stem cells (ESC, h,j) and neural progenitor cells (nPC; i,k). h,i, Green arrows denote the corners of a subset of the nested chromatin domains evident in this genomic region. j,k, Green arrows annotate a high-insulation-strength, cell-type-invariant TAD boundary. Blue arrows point to a lower-insulation-strength, cell-type-dynamic subTAD boundary.
Another advance came with the observation that smaller, sub-megabase-scale chromatin domains, so-called subTADs, are nested hierarchically within TADs in mammalian Hi-C maps5,6 (Fig. 1c,d). Only a small proportion of nested subTADs were observed in the original low-resolution Hi-C data, but they could be readily detected genome wide after technical advances facilitated the creation of ultra-high-resolution (1–4 kb) architecture maps. Nested subTADs resemble the domain-like structure of TADs and are also demarcated by boundaries. However, subTAD boundaries exhibit weaker insulation strength, as evidenced by their relatively lower capacity to attenuate long-range contacts between domains, and they are also significantly more likely than TADs to exhibit cell-type-dynamic folding properties1,5,7. We and others have hypothesized that weaker cell-type-dynamic subTAD boundaries have distinct structural, molecular or functional properties from those of TAD boundaries, but this possibility remains an open question.
The term ‘contact domain’ is also used in the Hi-C literature and generally serves as an umbrella term to convey the full set of self-associating chromatin domains (TADs, nested subTADs and compartment domains (discussed below)). Moreover, ‘mini-domains’ or ‘microTADs’ have recently been used to describe the smallest-scale chromatin blocks encompassing a single gene unit in mammals8,9 and flies10. Thus, algorithmic identification of chromatin domains has revealed increasingly smaller and finer-scale structures as technical advances have enabled higher-resolution Hi-C matrices. Moreover, a series of functional genetic perturbation experiments have begun to dissect the possible functional roles of domains at key developmentally regulated loci. At least two distinct mechanisms of domain formation have been identified, which has catalyzed substantial discord in the field regarding the existence, roles and true definitions of TADs and their nested derivatives.
In this Perspective, we discuss leading questions that have emerged after the initial discovery of chromatin domains in Hi-C data: (1) How are TADs, subTADs and compartment domains uniquely defined by their structural and mechanistic properties? (2) What are the hypothesized functional differences between compartment domains and TADs/subTADs formed by loop extrusion? (3) Do boundaries demarcating TADs exhibit structurally and functionally distinct properties from those demarcating subTADs? (4) Do TADs and subTADs structurally exist in single cells, or are they solely an aggregate representation of average interaction frequency across millions of cells from the standard Hi-C technique? (5) What is known about the genome’s structure–function relationship? Does transcription drive the formation of compartment domains or TAD/subTAD loop domains? Does any class of chromatin domain deterministically direct transcription?
Loop extrusion is a leading mechanism governing domain formation
Substantial progress has been made toward understanding the mechanisms that govern chromatin-domain formation. Mammalian genomes contain many domains structurally characterized in Hi-C maps by the presence of ‘corner dots’—a punctate group of adjacent pixels with significantly enhanced interaction frequency compared with that of the surrounding local domain structure (Fig. 1e,f). Corner-dot structures are thought to represent long-range looping interactions (schematically drawn in Fig. 1g) that exhibit a persistently high interaction frequency in a large proportion of cells (that is, persistent loops). Chromatin domains that co-localize with corner dots at their apexes have been hypothesized to represent so-called loop domains. Our own qualitative observations of Hi-C maps in mammalian systems have revealed the presence of megabase-scale, un-nested loop domains and nested loop domains (Fig. 1e,f).
Recent reports and forthcoming studies by large consortia have identified 10,000–60,000 corner-dot structures representing persistent loops in various human cell types6,11. Most corner dots are anchored by motifs bound by the architectural protein CTCF12. Specifically, 60–90% of all corner dots with an interpretable CTCF motif in both anchoring fragments display a ‘convergent’ motif orientation6,13 (Fig. 1g). Inversion of CTCF motifs with genome editing disrupts the corner dot and the TAD demarcated by the dot, thus demonstrating that a convergent CTCF-motif orientation is necessary for the formation of loop domains14–16. Moreover, short-term degradation of the CTCF protein results in severe ablation of loop domains17. Thus, a substantial subset of persistent loops represented by corner dots require binding of CTCF to convergently oriented motifs.
A windfall of new data has also recently advanced understanding of how the two convergently oriented CTCF-binding sites establish and maintain spatial proximity. In principle, motif orientation should not matter if loop establishment occurs through simple diffusion in the 3D nuclear space. The seminal model of ‘loop extrusion’ asserts that molecular motors loaded on the genome could track along the DNA sequence, thereby ‘extruding’ the intervening DNA in the process18,19. Computational studies simulating loop extrusion recapitulate loop domains16,20–22, and the authors of these studies predicted the existence of DNA-extruding factors.
Structural maintenance of chromosomes (SMC) complexes, such as cohesin or condensin, have long been thought to potentially serve as loop-anchoring factors, either by stabilizing preformed loops or through an active extrusion mechanism. Peaks of enriched cohesin occupancy on DNA identified via chromatin immunoprecipitation–sequencing co-localize with CTCF-binding sites23–26 but are slightly shifted to the 3′ ends of convergently oriented motifs13,21. This finding was a clue suggesting a tracking mechanism of cohesin–CTCF recruitment. Knockout of the cohesin-release factor WAPL prolongs cohesin residence time on the genome, leads to longer looping interactions that cross conventional TAD boundaries and increases the number of TAD/subTAD loop domains27. Moreover, knockout of the cohesin-loading factors Scc4 and Nipbl, or the Rad21 cohesin subunit, ablates a large fraction of loop domains across multiple mammalian cell types27–29. Direct evidence supporting loop extrusion via SMC complexes has come from single-molecule-imaging studies showing that condensin30,31 and cohesin32–34 can translocate along naked DNA in vitro in an ATP-dependent manner. Thus, loop extrusion, in which SMC complexes pass over divergently oriented CTCF motifs and stall at those in convergent orientation (Fig. 1g), has been proposed as a leading hypothesis for the mechanism of loop-domain formation.
We also define a key subgroup of chromatin domains that neither co-localize with corner dots nor register with compartments (Fig. 1e,f, ‘non-compartment + non-corner-dot domains’). It is important to highlight that, for domains formed by extrusion mechanisms, preferential contacts within the domain (that is, not at corner dots) are hypothesized to be a composite signal of active extrusion events (that is, transient loops in the making)21. Therefore, the possibility that non-compartment + non-corner-dot domains are mechanistically formed by extrusion cannot be ruled out. Furthermore, an important area of active exploration is the discovery and dissection of additional extrusion-blocking factors. Precise annotation of the suite of diverse proteins that influence extrusion rates across the genome would give credence to the hypothesis that boundaries with unique molecular characteristics can give rise to differential extrusion-blocking strength, thus causing corner dots with varying interaction frequency. Alternative mechanisms that could contribute to non-compartment + non-corner-dot domains include (1) loop extrusion against strong transient boundaries (that is, highly dynamic boundaries in individual cells), (2) loop extrusion against weak boundaries present in a high proportion of cells or (3) novel still-unknown mechanisms.
Compartmentalization is a second mechanism contributing to domain formation
A second mechanism that contributes to the establishment or maintenance of chromatin domains in eukaryotes is compartmentalization. Compartments were initially identified in 1-Mb binned Hi-C heat maps by their chromosome-wide ‘plaid’ pattern of ultra-long-range intra-chromosomal and inter-chromosomal contacts35 (Fig. 1e). The empirically defined plaid pattern has been hypothesized to represent the partitioning of the human genome into either A compartments of actively transcribed genes and active histone marks or B compartments with inactive genes and repressive marks35. The initial low-resolution Hi-C maps suggested that multiple megabase-scale TADs are nested within a single contiguous segment of an A or B compartment. However, in high-resolution heat maps, the mammalian genome has recently been discovered to instead partition into at least six substantially smaller subcompartments with various combinations of repressive and active chromatin modifications6. Notably, ultra-high-resolution Hi-C maps in flies have uncovered ‘compartment domains’—fine-grained compartments that perfectly register with domains devoid of corner dots10. Indeed, the number of corner-dot domains in flies is minimal10,36, thus suggesting that compartmentalization may be the primary driver of domain formation, at least in some non-mammalian organisms.
Together, these high-resolution analyses provide evidence that an intriguing subset of chromatin domains across eukaryotes could be classified as ‘compartment domains’, owing to a perfect alignment between the domain-like structure and compartment coordinates and the absence of a corner dot (Fig. 1e,f; ‘compartment domain only’ and ‘nested compartment domain only’ in Box 1). A critical unanswered question is whether and how loop extrusion occurs in organisms in which compartmentalization is the driving chromatin-domain mechanism.
Box 1 |. Chromatin-domain definitions.
| Definitions | Structural observation | Hypothesized mechanism |
|---|---|---|
| Chromatin domain | Small triangles of enhanced contact frequency that tile the diagonal of each contact matrix | |
| Compartmentalization | Plaid pattern in Hi-C maps, allowing alternating A/B designations for genomic intervals that display similar plaid patterns | Co-segregation of chromatin with similar histone marks or bound proteins, possibly formed in part via phase-separation forces |
| Compartment domain | A chromatin domain whose boundaries align with inflection points in A/B compartmentalization signals | |
| Loop | A point of enriched contacts in Hi-C heat maps, appearing as a dot (a series of adjacent pixels with enhanced contact frequency with respect to the local chromatin domain structure) if a loop occurs in many cells at the time of fixation (although not all loops present across a population manifest structurally as dots, owing to the transient nature of extrusion) | Active cohesin extrusion of chromatin, which is paused by proteins bound to the genome, most notably CTCF |
| Loop domain (TAD or subTAD) | A contact domain formed via loop-extrusion mechanisms and often but not always having a dot at the corner (corner-dot domain), owing to the transient nature of extrusion (non-compartment or non-corner-dot domain) | Extrusion pausing manifesting in a domain boundary |
Loop and compartment domains are not mutually exclusive. Their overlap and nesting properties enable the identification of six distinct chromatin domain classes:
TAD + compartment domain: un-nested corner-dot + compartment domain
TAD only: un-nested corner-dot domain only
Compartment domain only: un-nested no-corner-dot compartment domain
Nested subTAD + compartment domain: nested corner-dot compartment domain
Nested subTAD only: nested corner-dot domain only
Compartment domain only: nested no-corner-dot subcompartment domain
Notably, although ‘mini-domains’ or ‘microTADs’ have recently been used to describe the smallest-scale chromatin blocks encompassing a single gene unit in mammals8,9 and flies10, we currently do not define them herein. If further studies indicate that gene unit domains have corner dots and are created by loop extrusion, then we suggest either to continue to define them as nested subTADs or to re-define them as ultra-nested micro-TADs. However, if future studies indicate that gene unit domains are not formed by loop extrusion, then they should be defined in the future according to their mechanism of formation, whether by compartmentalization and/or phase separation or by a novel organizing principle.
What’s in a name? Refining the definitions of TADs and subTADs as loop-extrusion domains mechanistically distinct from compartment domains
One question under intense debate is how to update the historical definitions of TADs and subTADs in light of the recent mechanistic discovery of loop extrusion and the striking competition between compartmentalization and looping mechanisms that underlie domain formation10,27–29. Indeed, cohesin knockdown results in strengthening of existing compartments and finer-scale compartmentalization after loss of corner-dot TADs or subTADs in mammalian systems27–29. These results suggest that loop extrusion and compartmentalization are distinct and competing forces, thus reinforcing the concept that chromatin domains formed by the two mechanisms must be uniquely and clearly defined.
Here, we propose to refine the historical definitions of TADs and subTADs by adding additional qualifiers reflecting new mechanistic discoveries. We define TADs as corner-dot domains formed mechanistically by extrusion that is blocked by boundaries created by architectural proteins (Fig. 1 and Box 1). TADs also remain at the top level of the folding hierarchy and cannot be further nested under larger, on-diagonal corner-dot domains (light-blue corner-dot domain, Fig. 1f). TAD loop domains may also be substratified into those that also perfectly correspond to compartments or do not co-localize with compartments (Fig. 1e,f; ‘TAD only’ and ‘TAD + compartment domain’). We refine the definition of subTADs as corner-dot domains that are nested within larger TADs and are formed mechanistically by extrusion and substratified into those that do or do not additionally co-localize with compartments (Fig. 1e,f; ‘nested subTAD only’ and ‘nested subTAD + compartment domain’). We also highlight the most abstract and poorly understood domain types as those that do not correspond to compartments and are not persistent corner-dot TADs or subTADs. Such domains could still be created by extrusion blocking from weak boundaries or still-unknown mechanisms (‘non-compartment + non-corner-dot domains’, Fig. 1e,f). Evaluating the possible functional or mechanistic differences between loop domains that also co-localize with compartments and loop domains that do not register with compartments is important for future functional and mechanistic dissection.
TADs, subTADs and their boundaries can be structurally distinguished by their nested properties
Another currently debated question is whether contact domains are folded hierarchically or whether the largest, megabase-scale, TADs are simply artifacts of the high spatial noise and low resolution of early Hi-C maps. There is less evidence for nesting in Drosophila than in mammalian systems10,36, thus suggesting that complex hierarchical-domain structures might be less prominent in some organisms. Although more analyses are required to quantitatively resolve the existence of nested domains across species and cell types, strong visual evidence exists of large TADs and smaller nested subTADs in the highest-resolution Hi-C maps published to date in mice37 (green arrowheads in Fig. 1h,i). Thus, in addition to the classification of TADs as compartment and non-compartment loop domains, we hypothesize that stratifying chromatin domains and their boundaries by their nested properties during the design and interpretation of functional and mechanistic experiments is also important (Fig. 1e,f).
Several lines of evidence support the possibility that nested versus un-nested boundaries might have different structural and functional properties. First, megabase-scale TADs are largely cell-type invariant, whereas subTADs exhibit a higher tendency to reconfigure in a cell-type-specific manner1,5,7. In mammalian systems, one subTAD boundary will often co-localize with a cell-type-invariant TAD boundary (green arrows in Fig. 1j,k), whereas the alternative subTAD boundary nested within the larger TAD is often cell-type-specific (blue arrow in Fig. 1j,k). Moreover, because long-range interactions occur more frequently over boundaries demarcating nested versus un-nested domains, subTAD boundaries exhibit mechanistically weaker insulation than TAD boundaries. We hypothesize that extrusion may assemble both TAD and subTAD corner-dot domains, but that the nested cell-type-specific boundaries unique to subTADs might be governed by different densities or types of architectural proteins from those at un-nested invariant boundaries. Interestingly, recent reports have indicated a role for transposable elements in the formation of cell-type-specific boundaries38,39. Thus, an important area for future inquiry will be to unravel the structural, functional and mechanistic differences among boundaries across length scales.
Chromatin domains and boundaries are clearly present but stochastically detected in single cells
Chromatin domains have long been emphasized to be empirically defined from Hi-C maps representing an ensemble average interaction frequency across millions of cells. Do domain-like structures indicative of compartment domains, TADs or subTADs exist in individual eukaryotic nuclei? Seminal single-cell Hi-C studies have provided initial insight into this question, suggesting that even sparse, low-complexity matrices created from individual nuclei are consistent with the possibility that domain-like structures could exist in single cells40,41. In agreement with single-cell Hi-C results, Oligopaint experiments coupled with super-resolution microscopy have confirmed that genomic loci are spatially grouped into domain-like structures in individual mammalian cells42 (Fig. 2a). Importantly, the most frequently detected boundaries in single cells occur at the locations predicted by ensemble Hi-C maps42 (Fig. 2a). Many wild-type single cells also show random placement of domain-like blocks, an observation that is consistent with the established transient nature of the extrusion process and would be expected by imaging a snapshot in time across a population of individual cells in which extrusion was not synchronized. Indeed, the randomized placement of domain-like blocks in single cells, with preference for strong boundaries observed in ensemble Hi-C data, would be expected, given that ensemble Hi-C maps have always shown clear demarcation of TAD blocks as well as low interaction frequencies across boundaries. Our own current working hypothesis is that the precise domain demarcations, which are strongest in ensemble maps and most frequent in single-cell maps, might indeed suggest the true functional boundary elements. Low-frequency demarcation points of blocks in single cells might indeed represent only ‘loops in the making’ and may not be functional boundaries. Thus, imaging studies have attenuated concerns that TADs are only a statistical artifact of Hi-C data by demonstrating that chromatin domains and their boundaries are detectable and tiled across the mammalian genome in single cells.
Fig. 2 |. Chromatin domains and their boundaries are present in single cells.

a,b, Cartoon representations of contact domains identified in single cells via high-resolution imaging42. a, Wild-type cells display a biased preference for boundary locations. b, After knockout (KO) of cohesin, globular domains still exist but do not display the same boundary preference. c, Representative heat maps of the effects of cohesin and Nipbl removal on loop and compartment domains, as portrayed in refs. 28,29.
One unanswered question is whether structural differences exist in single cells between un-nested TADs versus nested subTADs versus compartment domains. Chromatin domains in mammalian systems remain distinctly observable in single cells after cohesin depletion but are distributed across the genome randomly and show a loss in preferential positioning at CTCF sites42 (Fig. 2b). However, because this particular study did not explicitly distinguish between TAD/subTAD loop domains and compartment domains, further classification of the precise class of structures imaged should aid in interpretation of these data. Ensemble Hi-C analyses of genome folding have revealed that loop domains are destroyed and that compartment domains are strengthened and become more fine-grained after knockdown of cohesin27–29 (Fig. 2c). The ensemble strengthening of compartment domains in cohesion-knockdown cells forms the basis for our own working hypothesis that compartment domains would become less random and more synchronized in single cells in a cohesion-knockdown imaging experiment. Data from Bintu et al. are in direct opposition to our working model, because that study shows that domain-like structures remaining after cohesin knockdown are truly random42, a finding not consistent, at face value, with the compartment-domain strengthening from ensemble Hi-C27–29. Thus, the mechanistic and functional nature of chromatin domains that remain in single cells after extrusion disruption remains an important open and unanswered question.
Loop domains exhibit a markedly different cause-and-effect relationship with genome function than compartment domains
A final leading question covered by this Perspective is related to the eukaryotic genome’s structure–function relationship: does form follow function, or does function follow form? Perturbative studies have to date have produced apparently conflicting results, and the functional roles of chromatin domains are highly specific to the genomic context, developmental timing and eukaryotic organism in question. We also highlight that genetic dissection of the effects of key architectural features on genome function will be greatly facilitated by first delineating the compartment domains, un-nested corner-dot TADs and nested corner-dot subTADs. Evidence to date indicates that TADs and subTAD loop domains, compared with A/B compartments, exhibit a distinct functional connection to gene regulation.
There are three emerging mechanisms through which loop domains might influence transcription: (1) direct, strong contact of enhancers and promoters via persistent loops (that is, the enhancer and promoter are at the anchors of the corner-dot domains and co-localize with extrusion boundaries) (Fig. 3a), (2) weak contact of enhancers and promoters via transient extrusion of SMC complexes across the loop domain (that is, the enhancer and promoter are within a loop domain but not co-localized by a boundary, so extrusion factors pass over the elements transiently) (Fig. 3b) and (3) developmental miswiring of enhancers to non-target promoters outside of the TAD or subTAD after genetic destruction of loop-domain boundaries (Fig. 3c). Most the seminal studies have used the historically identified definition of TADs as megabase-scale chromatin domains1, so the class of chromatin domain genetically dissected in each study is unclear. Here, we assume that the large megabase-scale domains identified by Dixon et al. in mammalian cells represent loop domains1. Studies to date provide examples in which compartment domains, in contrast to loop domains, are instructive, decoupled from, or created as a consequence of transcription or the formation of nuclear condensates. For brevity, we discuss the structure–function role of compartments in the Supplementary Note. For the interpretation of future studies, it will be highly important to delineate corner-loop TADs, nested corner-loop subTADs and compartment domains before the genetic dissection of the functional roles of topological features.
Fig. 3 |. Evidence for and against TADs as a critical functional intermediary in the regulation of genes by developmentally active enhancers.

a–c, Schematics of three emerging mechanisms through which loop domains can influence transcription: direct, strong contact of enhancers and promoters via persistent loops (red arcs) at the corners of domains (a), transient, weak contact of enhancers and promoters via transient loop extrusion (blue arcs) across the loop domain (b), and developmental miswiring of enhancers to non-target promoters outside of the TAD or subTAD after genetic destruction of loop domain boundaries (c). d, Representation of the activity readout of a reporter assay after random integration in genomic loci, from refs. 43,69. e, Three published examples of boundary disruption or inversion leading to developmental issues. f, Depiction of a model of long-range transcriptional regulation in which an enhancer’s regulatory contribution trends with its activity signature and Hi-C contact frequency with the target gene57. g, Schematized box plot of measured distances from the enhancer to the Sox2 promoter in actively expressing (left) and inactive (right) cells60. NS, not significant. h, Representation of the relatively modest transcriptional changes observed after cohesin and Nipbl depletion observed in refs. 28,29. RPKM, reads per kilobase per million mapped reads. i, Cartoon of unencumbered development observed after perturbation of a TAD boundary opposing the Shh gene66.
First, several elegant genetic perturbation studies over the past 10 years have together allowed a model to gain traction in which TADs create insulated neighborhoods that demarcate the enhancer search space for target genes (Fig. 3c). Importantly, random insertion of an ectopic transgene sensor across the mouse genome has shown enhancer activation patterns during embryonic development that correlate with some large megabase-scale TADs43 (Fig. 3d). Across numerous studies, genetic disruption of specific TAD boundaries (via experimental intervention or disease) causes ectopic inter-domain contacts between enhancers and non-target promoters and consequent aberrant gene expression44–52 (Fig. 3e). Most notably, studies focused on model systems connected to key developmentally regulated biological phenomena (for example, X-chromosome inactivation and mammalian limb development) have shown a convincing link between TAD-boundary disruption, ectopic enhancer–promoter interactions and alterations in gene expression levels45–49,51,52. Moreover, boundary disruptions have also been reported as strongly correlated to pathologically altered gene expression in human cancers45,51, neurological disorders53, rare congenital disorders50 and diseases of limb development46,54. In these early reports, miswiring of enhancer–promoter interactions across the disrupted boundary has been proposed as the mechanism for pathologically altered gene expression. Thus, evidence continues to grow in support of the model in which boundaries created by TADs generally ensure proper spatiotemporal regulation of gene expression by topologically confining enhancers to their target promoters in the appropriate developmental time window55.
In addition to the architectural roles of TAD/subTAD loop domains in preventing developmental miswiring of enhancer–promoter interactions, corner dots also can directly connect enhancers to promoters via CTCF-dependent and CTCF-independent mechanisms5,13,56 (Fig. 3a,b). Spatial proximity can be achieved during the extrusion process (1) when both the enhancer and promoter are within the same loop domain and transiently come into contact, owing to the movement of the extrusion factor (so-called transient loops) (Fig. 3b) or (2) when the enhancer and promoter anchor the boundaries of a corner-loop domain, and extrusion factors stall against boundaries to form so-called persistent loops (Fig. 3a). A recent high-throughput CRISPR interference screen recruited dCas9–KRAB and guide RNAs to thousands of putative non-coding regulatory elements57. The authors found that a multiplicative contribution of interaction frequency and enhancer activity together serve as the best predictor of enhancer–gene targets (‘ABC model’; Fig. 3f). Imaging studies have also provided evidence that enhancers are spatially proximal to their target promoters in single cells with high expression of the gene58,59. However, whether the contacts imaged in these studies are persistent or transient loops is currently unknown. Moreover, in some cases, enhancers might activate their distal targets without proximity60 (Fig. 3g), but the genome-wide extent of this finding is still under investigation. Finally, loop engineering experiments result in upregulation of gene expression after forced long-range interactions, but the effects of enhancer proximity on gene expression can sometimes be modest61,62. Together, these early data highlight that enhancer–promoter spatial co-localization can contribute to gene expression levels; however there is a great need to systematically dissect the functional roles of transient and persistent loops across genomic contexts in governing transcription.
Beyond locus-specific studies, investigators have also assessed gene expression changes globally after ablation of TAD and subTAD loop domains. Specifically, after depletion of CTCF with an auxin-mediated degron, thousands of loop domains across the genome have been found to be disrupted while compartments are unaffected17. Moreover, acute degradation of subunits of the cohesin complex destroys most loop domains and leads to stronger partitioning of the genome into compartment domains28,29. Despite the severe global ablation of corner-loop domains, these studies have shown unexpectedly modest effects on gene expression. CTCF depletion for 24 hours results in only 370 differentially expressed genes in mouse embryonic stem cells. After only 6 hours of complete cohesin degradation, only 146 genes showed a 1.75-fold change in expression, and only two genes showed a fivefold change in expression (Fig. 3h). The lack of pervasive gene expression changes despite widespread loop-domain dissolution was notable because the authors used precision nuclear run-on sequencing (PRO-seq) for nascent-transcript detection28. Cohesin depletion over a longer 5-day timeframe resulted in more than 1,000 dysregulated genes, but this higher number is likely to be due to secondary effects that occur in long-term perturbation studies29.
Determining whether all enhancer–promoter interactions are disrupted with cohesin knockdown (for example, those in non-compartment + non-corner-dot domains or compartment domains), or whether only those connected via strong corner-dot TADs or subTADs are abolished, will be of high interest. Moreover, for each gene, the functional effect of loop-domain disruption may manifest only in the specific developmental lineage in which nearby enhancers are active, and the topological features are relevant; in each of these studies, only a single cell type or tissue and developmental stage was queried. Indeed, after cohesin removal from mature macrophages, gene expression is preferentially altered after inflammatory signaling induction, thus suggesting that the effects of cohesin removal may be especially evident after induction and establishment of a new gene expression program63. Finally, a very recent study has demonstrated that RNA polymerase II elongation can decrease cohesin binding and disrupt CTCF- or cohesin-mediated loops, thus indicating that transcription can also affect TADs and subTADs64. Recent data have also demonstrated that chemical inhibition of transcriptional elongation can compromise TAD-boundary strength65. Overall, in the case of loop domains, the limited data available to date indicate that loops can influence function, albeit to a modest degree in some cases, and genome function in the form of transcription can also influence looping structure.
Beyond the three general models for the functional role of loop domains, the challenging work to assess the link between each individual boundary and developmentally regulated transcription is now underway. For example, several studies have genetically dissected topological features at the sonic hedgehog (Shh) locus in mouse limb development; these studies are particularly important for the topic of this Perspective because a clear corner-loop domain connects the Shh gene to its ZRS enhancer. In one study, specific deletions of a specific CTCF site or a 35-kb region encompassing the boundary next to ZRS have been found to result in minimal disruption of Shh expression and no clear developmental defects66 (Fig. 3i). Importantly, structural maps show that the contact domain, including the corner dot connecting ZRS to Shh, is possibly still intact with these two deletions, and a minor degree of inter-TAD interactions may occur between ZRS and the adjacent domain (Fig. 3i). In an independent study, two CTCF sites at the ZRS boundary were both deleted, including an additional CTCF site not included in the 35-kb deletion from the other study67. Deletion of both CTCF67 sites led to disruption of the corner-loop domain and a 50% decrease in Shh levels. These results reinforce that boundaries consist of multiple protein-binding sites and that ablation of TAD structures often requires multiple deletions to overcome redundancies that preserve important chromatin topological features52.
Our working model is that chromatin interactions between a gene’s enhancer and promoter must be severely abolished (such as by switching the enhancer into a completely different domain) before an effect on gene expression becomes evident at a precise developmental time. This model was built in part on the basis of a recent systematic dissection of genome structure–function at the Sox9–Kcnj2 locus in mice52. The authors show that the boundary demarcating the TADs around Sox9 and Kcnj2 is ablated only after homozygous disruption of all occupied CTCF sites at the boundary and within adjacent domains, thus highlighting the remarkable redundancy of architectural-protein-binding sites governing TAD structural integrity. Importantly, despite complete fusion of both TADs, only minor alterations in Sox9 and Kcnj2 expression were observed, and there were no apparent phenotypic consequences. Sox9 and Kcnj2 still contacted their target enhancers, presumably because cohesin-based loop extrusion still occurred. In contrast to the TAD-fusion results, the inversion of the boundary or the aberrant placement of the boundary led to gained or lost contacts of Sox9 and Kcnj2 with enhancers, thus leading to pronounced effects on gene expression and severe developmental phenotypes. Together, these results indicate that, at least at this locus, ectopic placement of boundaries can break wild-type enhancer–promoter interactions and redirect enhancers to new target genes, thus leading to severe gene expression changes that give rise to pathologic phenotypes. Simply removing a boundary element is not sufficient to modify endogenous enhancer–promoter contacts, because endogenous interactions are insufficiently abolished.
Given that genetic inversions at boundaries have more pronounced effects on gene expression than genetic perturbations of boundary strength, severe chromosome rearrangements might be hypothesized to have the strongest genome-wide effects on transcription. A recent study has created high-resolution maps of genome folding in a Drosophila species with highly rearranged balancer chromosomes68. The authors show that extensive genome-wide deletions, duplications and inversions in Drosophila can markedly shuffle chromatin-domain placement, but that only minor alterations in gene expression result. As evidence continues to accumulate regarding whether and how extrusion occurs in Drosophila and whether domain-like structures in Drosophila are compartment domains, determining whether the modest effects of domain-like structures on transcription in certain fly species are due to their status as strictly compartment domains will be critical. Another critical point is that balancer chromosomes have been selected for their ability to allow animal viability; therefore, determining how severe chromosomal rearrangements in cases of visible phenotypes would affect gene expression would be interesting. Beyond these exciting questions for future work, we emphasize that a lesson from this work is that not all genes might be regulated through long-range spatial contacts.
Many of the hypotheses proposed here remain to be rigorously tested. One emerging principle is that distinguishing compartment domains from loop domains, and careful quantification of their nested and cell-type-specific properties, will be essential to obtain clear insight into the functionality of chromatin domains and their boundaries. Forthcoming studies pairing population- and single-cell-based data should account for the strengths and weaknesses of both approaches and are expected to yield new insights into the genome’s structure–function relationship. Although early studies in the 3D genome-folding field focused on cell lines, emerging studies across model organisms, early developmental stages, time points across the cell cycle, genetic perturbations and human disease models, will continue to build understanding of how transcription and other genome functions shape and are shaped by the 3D genome.
Supplementary Material
Acknowledgements
We thank members of the 3D genome-folding community for helpful discussions. In particular, we gratefully acknowledge B. Ren for feedback on this work. J.E.P.-C. is supported as a New York Stem Cell Foundation—Robertson Investigator and an Alfred P. Sloan Foundation Fellow. This research was supported by The New York Stem Cell Foundation (J.E.P.-C.), the Alfred P. Sloan Foundation (J.E.P.-C.), the NIH Director’s New Innovator Award from the National Institute of Mental Health (1DP2MH11024701; J.E.P.-C.), a National Institute of Mental Health grant (1R011MH120269; J.E.P.-C.), a 4D Nucleome Common Fund grant (1U01HL12999801; J.E.P.-C), a joint NSF–NIGMS grant to support research at the interface of the biological and mathematical sciences (1562665; J.E.P.-C.), a Brain Research Foundation Fay Frank Seed Grant (J.E.P.-C.) and a National Science Foundation Graduate Research Fellowship under grant DGE-1321851 (J.A.B.).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41588-019-0561-1.
References
- 1.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou C, Li L, Qin ZS & Corces VG Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 48, 471–484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sexton T et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Phillips-Cremins JE et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norton HK et al. Detecting hierarchical genome folding with network modularity. Nat. Methods 15, 119–122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh T-HS et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Preprint at bioRxiv 10.1101/638775 (2019). [DOI] [PMC free article] [PubMed]
- 9.Krietenstein N et al. Ultrastructural details of mammalian chromosome architecture. Preprint at bioRxiv 10.1101/639922 (2019). [DOI] [PMC free article] [PubMed]
- 10.Rowley MJ et al. Evolutionarily conserved principles predict 3D chromatin organization. Mol. Cell 67, 837–852.e837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phanstiel DH et al. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol. Cell 67, 1037–1048.e1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips JE & Corces VG CTCF: master weaver of the genome. Cell 137, 1194–1211 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Z et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Wit E et al. CTCF binding polarity determines chromatin looping. Mol. Cell 60, 676–684 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Guo Y et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162, 900–910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanborn AL et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl Acad. Sci. USA 112, E6456–E6465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nora EP et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasmyth K Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu. Rev. Genet 35, 673–745 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Riggs AD DNA methylation and late replication probably aid cell memory, and type I DNA reeling could aid chromosome folding and enhancer function. Phil. Trans. R. Soc. Lond. B 326, 285–297 (1990). [DOI] [PubMed] [Google Scholar]
- 20.Alipour E & Marko JF Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40, 11202–11212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fudenberg G et al. Formation of chromosomal domains by loop extrusion. Cell Reports 15, 2038–2049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goloborodko A, Marko JF & Mirny LA Chromosome compaction by active loop extrusion. Biophys. J 110, 2162–2168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parelho V et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Rubio ED et al. CTCF physically links cohesin to chromatin. Proc. Natl Acad. Sci. USA 105, 8309–8314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stedman W et al. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27, 654–666 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wendt KS et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Haarhuis JHI et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell 169, 693–707.e614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SSP et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarzer W et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terakawa T et al. The condensin complex is a mechanochemical motor that translocates along DNA. Science 358, 672–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganji M et al. Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stigler J, Çamdere GO, Koshland DE & Greene EC Single-molecule imaging reveals a collapsed conformational state for DNA-bound cohesin. Cell Rep 15, 988–998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davidson IF et al. Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J 35, 2671–2685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanke M, Tahara E, Huis In’t Veld PJ & Nishiyama T Cohesin acetylation and Wapl-Pds5 oppositely regulate translocation of cohesin along DNA. EMBO J 35, 2686–2698 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eagen KP, Aiden EL & Kornberg RD Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl Acad. Sci. USA 114, 8764–8769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonev B et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kruse K et al. Transposable elements drive reorganisation of 3D chromatin during early embryogenesis. Preprint at bioRxiv 10.1101/523712 (2019). [DOI]
- 39.Zhang Y et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet 51, 1380–1388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flyamer IM et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Symmons O et al. Functional and topological characteristics of mammalian regulatory domains. Genome Res 24, 390–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowen JM et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hnisz D et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lupiáñez DG et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narendra V et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Bemmel JG et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat. Genet 51, 1024–1034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraft K et al. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital malformations. Nat. Cell Biol 21, 305–310 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Laugsch M et al. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell 24, 736–752.e712 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Despang A et al. Functional dissection of the Sox9–Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat. Genet 51, 1263–1271 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Sun JH et al. Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell 175, 224–238.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franke M et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Norton HK & Phillips-Cremins JE Crossed wires: 3D genome misfolding in human disease. J. Cell Biol 216, 3441–3452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kagey MH et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fulco CP et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mateo LJ et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen H et al. Dynamic interplay between enhancer-promoter topology and gene activity. Nat. Genet 50, 1296–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexander JM et al. Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. eLife 8, e41769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng W et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim JH et al. LADL: light-activated dynamic looping for endogenous gene expression control. Nat. Methods 16, 633–639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuartero S et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol 19, 932–941 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heinz S et al. Transcription elongation can affect genome 3D structure. Cell 174, 1522–1536.e1522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barutcu AR, Blencowe BJ & Rinn JL Differential contribution of steady-state RNA and active transcription in chromatin organization. EMBO Rep 20, e48068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williamson I et al. Developmentally regulated Shh expression is robust to TAD perturbations. Development 146, dev179523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paliou C et al. Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc. Natl Acad. Sci. USA 116, 12390–12399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghavi-Helm Y et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet 51, 1272–1282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Symmons O et al. The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev. Cell 39, 529–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.