

Abstract
Immune checkpoint blockade (ICB) has become a standard of care in a subset of solid tumors. Although cancer survivorship has extended, rates of durable response of ICB remain poor; furthermore, cardiac adverse effects are emerging, which impact several mechanical aspects of the heart. Cardio-oncology programs implement a clinical assessment to curtail cardiovascular disease progression but are limited to the current clinical parameters used in cardiology. Pharmacogenomics provides the potential to unveil heritable and somatic genetic variations for guiding precision immunotherapy treatment to reduce the risk of immune-related cardiotoxicity. A better understanding of pharmacogenomics will optimize the current treatment selection and dosing of immunotherapy. Here, we summarize the recent pharmacogenomics studies in immunotherapy responsiveness and its related cardiotoxicity and highlight how patient genetics and epigenetics can facilitate researchers and clinicians in designing new approaches for precision immunotherapy. We highlight and discuss how single-cell technologies, human-induced pluripotent stem cells and systems pharmacogenomics accelerate future studies of precision cardio-oncology.
Introduction
Checkpoint inhibitors harness the innate and adaptive immune system to eliminate tumor cells (1). Cytotoxic T-lymphocyte-associated antigen 4, programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1) play a central role in peripheral tolerance (1). Binding of CTLA-4 to B7 molecules on antigen-presenting cells and PD-1 to PD-L1 on the tumor surface or other immune cells will subsequently induce a negative signal to dampen T cell activation (2,3). Monoclonal antibodies that interfere with the PD-1/PD-L1 and CTLA-4/B7 axis, will therefore reinvigorate lymphocyte activity from an anergic state. (Fig. 1). These revolutionizing findings were recognized in the 2018 Nobel Prize in Physiology and Medicine to James P. Allison and Tasuku Honjo for the discovery of CTLA-4 and PD-1 as negative immune regulation targets for cancer therapy. Indeed, ipilimumab has doubled 10-year survival rates in metastatic melanoma (4,5); PD-1/PD-L1 inhibitors have also shown significant clinical impact in melanoma, non-small cell lung, bladder, ovarian and colorectal cancers, among others (6–8).
Figure 1.
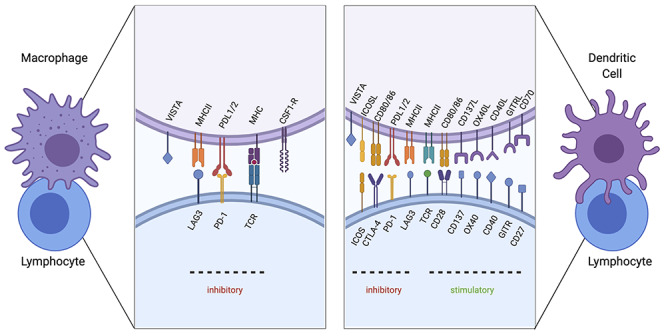
Immune checkpoint signaling under investigation. Validated checkpoint marker interactions between macrophages (left) and dendritic cells (right) with lymphocytes are depicted. TCR interacting with MHC II serves as the first stimulation signal. Macrophages can play an immunosuppressive role by upregulating the surface expression of checkpoint proteins PD-L1 and ligand 2 (PD-L2), which interacts with Programmed cell death proten 1 (PD-1) on lymphocytes. Furthermore, lymphocyte-activation gene 3 (LAG3) will bind to MHC II molecules and reduce monocyte differentiation. Colony-stimulating factor 1 receptor (CSF1R) also functions to differentiate monocytes to macrophages. V-domain immunoglobulin suppressor of T cell activation (VISTA) functions to dampen T-cell activation. A secondary signal between dendritic cells and lymphocytes will determine the stimulatory or inhibitory response. Stimulatory checkpoint pairs include CD28 and CD80/86, CD137/CD137 ligand, OX40/OX40 ligand, CD40/CD40 ligand, CD27/CD70 and glucocorticoid-induced tumor necrosis factor receptor (GITR)/GITR ligand. Inhibitory checkpoint pairs include inducible T cell co-stimulator (ICOS) with ICOS ligand, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4)/CD80 or 86, PD-1/PDL1/2, LAG3/MHC II and VISTA (Created with BioRender.com).
Despite advances in immune checkpoint blockade (ICB), rates of durable response remain poor (9). The estimated response rate of ICB across cancers is 12.46% (10). Predicting the risk of ICB efficacy has remained elusive and focuses primarily on biological indicators of poor immune surveillance (1). A greater obstacle is the alarming rate of adverse effects (up to 50%) that consequently result in the early discontinuation of treatment (11). Although rare, cardiac adverse effects in ICB can be serious and lethal (12,13). They encompass a diverse set of disorders in the heart including myocarditis, pericarditis, arterial vascular disease, venous thromboembolism, pulmonary hypertension, arrhythmias and heart failure (14). Despite not having a ground understanding of the molecular mechanisms governing efficacy and toxicity, an unprecedented number of new drugs are entering clinical trials. As of March 2020, the cancer cell therapy pipeline worldwide includes 1483 active agents, with an increase of 472 from the previous year (15). To date, the U.S. Food and Drug Administration approved seven antibody ICBs highlighted in Table 1 (16). It will be vital to incorporate additional biomarkers, like patient genetics to better understand treatment responsiveness; and to elucidate underlying molecular determinants of cardiotoxicity and detain growing rates of toxicity as more ICB drugs enter the clinic.
Table 1.
Food and Drug Administration-approved ICB therapies
| Drug | Target | Histology | Key trials | Combination |
|---|---|---|---|---|
| Ipilimumab | CTLA-4 | Melanoma | NCT00094653 | Monotherapy |
| Nivolumab | PD-1 | Melanoma | Checkmate-037,069,067,238 | Ipilimumab |
| NSCLC | CheckMate-063, 017,057,227,9LA | Ipilimumab, Ipilimumab + platinum | ||
| RCC | CheckMate-025, 214 | Ipilimumab | ||
| Hodgkin’s lymphoma | CheckMate-205 | Monotherapy | ||
| Head and neck | CheckMate-141 | Monotherapy | ||
| Colorectal | CheckMate-142 | Ipilimumab | ||
| HCC | CheckMate-040 | Ipilimumab | ||
| SCLC | CheckMate-032 | Monotherapy | ||
| Bladder | CheckMate-275 | Monotherapy | ||
| Esophageal SCC | ATTRACTION-3 | Platinum, fluoropyrimidine | ||
| Pembrolizumab | PD-1 | Melanoma | KeyNote-001, 002,006,054/EORTC 1325 | Monotherapy |
| NSCLC | KeyNote-001,010, 024,021,189,407,042 | Pemetrex, cisplatin/carboplatin, paclitaxel | ||
| RCC | KeyNote-426 | Atixinib | ||
| Hodgkin’s lymphoma | KeyNote-087 | Monotherapy | ||
| Head and neck | KeyNote-012,048 | Platinum, fluorouracil | ||
| MCC | CITN-09/KeyNote-017 | Monotherapy | ||
| MSI-H | NCT01876511 | Monotherapy | ||
| Gastric | KeyNote-059 | Monotherapy | ||
| HCC | KeyNote-244 | Monotherapy | ||
| Cervical | KeyNote-158 | Monotherapy | ||
| PMBCL | KeyNote-170 | Monotherapy | ||
| SCLC | KeyNote-158,028 | Monotherapy | ||
| Bladder | KeyNote-045,052,057 | Monotherapy | ||
| Endometrial | KeyNote-146 | Lenvatinib | ||
| Cemiplimab | PD-1 | CSCC | NCT0276048 | Monotherapy |
| Avelumab | PD-L1 | RCC | JAVELIN Renal 101 | Axitinib |
| MCC | JAVELIN Merkel 200 | Monotherapy | ||
| Bladder | JAVELIN Solid Tumor | Monotherapy | ||
| Atezolizumab | PD-L1 | NSCLC | Birch, Poplar, FIR, Oak, IMpower150,130 ,110 | Bevacizumab, carboplatin, and paclitaxel |
| HCC | IMbrave150 | Bevacizumab | ||
| SCLC | IMpower133 | carboplatin, etoposide | ||
| Bladder | IMvigor210 | Monotherapy | ||
| Breast | IMpassion130 | n-ab paclitaxel | ||
| Durvalumab | PD-L1 | NSCLC | PACIFIC | Monotherapy |
| SCLC | CASPIAN | Carboplatin, etoposide | ||
| Bladder | NCT01693562 | Monotherapy |
CSCC, cutaneous squamous cell carcinoma; HCC, hepatocellular carcinoma; MCC, Merkel cell carcinoma; MSI-H, microsatellite instability- high; NSCLC, non-small cell lung cancer; PMBCL, primary mediastinal B-cell lymphoma; RCC, renal cell carcinoma; SCC, squamous cell carcinoma.
Information summarized from https://www.canceresearch.org/.
Pharmacogenomics utilizes genetic variation to explain the distribution of drug responses by differences in pharmacokinetics or pharmacodynamic pathways. Such strategies have been employed to explain anthracycline-induced cardiotoxicity in association to single-nucleotide polymorphisms (SNPs) (17,18). The goal for precision immuno-oncology will be to apply pharmacogenomic methods to offer a non-invasive technique of identifying actionable biomarkers of responsiveness and immune-related toxicity. With increasing surveillance of cardiotoxicity, it should become clearer whether events are either a direct insult from cancer treatment or the exacerbation of underlying disease comorbidities (i.e. diabetes, hypertension and existing cardiovascular conditions). In this review, we will focus on recent evidence describing cardiotoxicity of ICB and how pharmacogenomic studies contribute to clinical management of personalized immunotherapy.
Pharmacogenomics in immunotherapy
Tumor mutation burden (TMB) is the average number of mutations per megabase. A high TMB score is a biomarker of ICB response already utilized in the clinic for nominating non-small cell lung cancer (NSCLC), melanoma and microsatellite instability-high colorectal cancer patients for immunotherapy treatment (5,6). Despite the increasing accessibility of germline and tumor genetic testing, it remains unclear if TMB and canonical oncogene driver mutations serve as reliable indicators of ICB response. Samstein et al. (19) profiled the TMB in 1662 advanced pan-cancer patients treated with ICB. The top 20% mutated tumors associated with a better overall survival in colorectal, head and neck, melanoma, non-small cell and bladder cancers, but not in renal cell, glioma or breast cancer (19). Major differences in the tumor microenvironment (tumor-infiltrating lymphocytes, tumor antigens, clonality, immune cell suppressors, etc.) make it difficult to apply one universal metric across different histologies and capture tumor stochasticity (20,21). Understanding the interaction of patient genetic signatures and the tumor immune microenvironment will be necessary for the development of robust predictors of ICB resistance and represent a critical step towards stratifying patients to ICB responders and non-responders. Genome-wide association studies (GWAS) have already found SNPs in CTLA-4 and PD-L1 that distinguish drug response (22). In light of this, quantitative trait mapping and functional studies have to be performed to support these findings in the near future. In the next sections, we will highlight recent evidence describing genetic or genomic alterations in phosphatase and tensin homolog (PTEN) and human leukocyte antigen (HLA) as example determinants of ICB response and their potential role as pharmacogenomic markers of immune-related cardiotoxicity.
Phosphatase and tensin homolog
PTEN acts as a phosphatase to substrates with serine, threonine and tyrosine residues and canonically negatively regulates phosphatidylinositol 3-kinase (PI3K) signaling (23). PTEN dysfunction results in uncontrolled cell proliferation and induces immune escape and apoptosis of activated T cells through the upregulation of PD-L1. Unsurprisingly, PTEN loss is frequent in tumor resistance to ICB (24–27). This phenomenon can be explained in models of PTEN loss that show significantly less CD8+ T-cell tumor infiltration and functionality compared to tumors with intact PTEN (27,28). Peng et al. (27) linked this loss to vascular endothelial growth factor overexpression and the release of immunosuppressive cytokines. Recent studies have described PTEN as a crucial molecular pathway modulator in regulatory T cells (Tregs) that represses T follicular helper cell and T helper type 1 cell responses. Pten knockout mice show loss of the interleukin-2 receptor leading to the expansion of impaired Tregs (29). Additionally, these mice also showed impaired metabolic programming via reduced reactive oxidative species mitochondrial mass and membrane potential. Together, these studies can clarify the immune signaling mechanism observed in PTEN mutants to explain drug responses. It will be important to assess whether rare germline variants differ from common somatic variants, as germline variants may be more relevant to immune response changes in the heart.
Several clinical trials are testing the efficacy of immunotherapy with canonical variants in the AKT/PIK3CA/PTEN pathway in solid tumors (NCT03772561, NCT03842228), as well as the use of PI3K inhibitors in combination with immunotherapy in metastatic melanoma (NCT03131908), colorectal cancer (NCT03711058), large B cell lymphoma (NCT03484819), lung cancer (NCT03257722) and gynecologic malignancies (NCT03719326).
Human leukocyte antigen
Highly polymorphic or loss of HLA-I loci are also proposed as predictors of ICB response (30,31). HLAs are the cell surface proteins that serve as substrates for antigens and are characterized by a high sequence variation in the peptide-binding domain (32). Tumors with a high mutation burden are likely to create a more diverse set of tumor antigen peptides and in turn elicit a more diverse cytotoxic immune response. For this reason, TMB is a putative predictor of ICB response (33). Tumor antigens lacking somatic variants can also elicit an immune response, though this is limited by central tolerance.
Groups have leveraged new sequencing methods to characterize HLA diversity in ICB-treated patients. Chowell et al. reported homozygosity at the HLA-I locus was associated with a significant reduction of overall survival following ICB treatment [P = 0.003, hazard ration (HR) = 1.38, 95% confidence interval (CI) 1.11–1.70] in melanoma and NSCLC. Additionally, the investigators used T-cell receptor (TCR) sequencing to compare TCR subclones in HLA heterozygotes versus homozygotes. TCR complementary-determining region 3 subclones are responsible for recognizing processed antigens on T-cells and were more abundant in HLA heterozygotes compared to homozygotes. Specifically, patients harboring B44 alleles had better disease outcome (P = 0.01, HR = 0.61, 95% CI = 0.42–0.89) and those with B62 alleles had reduced survival (P = 0.0007; HR = 2.29; 95% CI 1.40–3.74) (34). In a later study, the investigators created an HLA evolutionary divergence (HED) score to calculate the high sequence diversity of HLA-A, B and C alleles and tumor immunogenicity to predict response to anti-PD1 and anti-CTLA-4. Using HED, the investigators reported heterozygous HLA-I patients with a HED in the upper quartile responded better to ICB compared to the lower quartile in two cohorts in late-stage melanoma and one cohort in NSCLC. They found that TMB does not correlate with HED, as HED may only represent variants causing neoantigens recognized by T-cells. However, they did see a significant positive correlation of HED with the number of viral peptides, neopeptides and self-peptides (35). Assessing individuals’ immune-priming score will be an important step implemented in the genetic screening process before administering ICB treatment.
Understanding the frequency and strength of antigen peptides is of equal importance. Cai et al. analyzed 147 lung adenocarcinoma samples from The Cancer Genome Atlas (TCGA) to identify mutant peptides that bind to HLA. The algorithm identified 8804 strong and weak neopeptides. Surprisingly the second-ranked gene with predicted HLA-DRB1 (Major Histocompatibility Complex, Class II, beta chain) binding was the skeletal and cardiac muscle encoding gene, TTN. Canonical lung cancer genes were also highly ranked: KRAS, EGFR, STK11 and TP53 (36). This study used machine learning approaches to predict HLA substrates. A major caveat to this approach of prediction suffers from potential bias in training sets and overfitting. Additionally, larger genes (i.e. TTN) are overrepresented in these studies and are often not corrected by gene cDNA length leading to potential false-positive rates. This is why functional studies, while harboring its own limitations, are essential for establishing causality.
Pharmacogenomics of immunotherapy-related cardiotoxicity
ICB is associated with a spectrum of immune-related adverse events (irAEs): dermatitis, colitis, hepatitis, hypophysitis, pneumonitis and myocarditis (37). The frequency of irAEs is common in patients treated with ipilimumab (64–80%), pembrolizumab (up to 79%) and combination therapies (96%). High grade toxicity occurs in 40–50% of combination treatment cases 40–50% of combination treatment cases (38). Cardiac-related adverse events include myocarditis, pericarditis and arrhythmias (39). Myocarditis in patients treated with ICB is rare irAE (1%); however, it has disproportionally high mortality rates (50%) and warrants attention (12,40–42). The onset of myocarditis occurs within the first month (40,43) and is more frequent in the combination of PD-1 and CTLA-4 inhibitors (40). Managing adverse effects presents several challenges, including patients not receiving their full treatment course. Here, we focus on the recent literature that helps explain how immunotherapy and combination of epigenetic modulators with immunotherapy may trigger the immune system to attack cardiac tissue and how we can utilize ‘-omics’ tools to mitigate adverse reactions in the heart.
Despite the lack of prospective randomized controlled trials to assess ICB mediated cardiotoxicity, case reports offer insight into the underlying mechanism of toxicity. Three hypothetical mechanisms have been proposed to explain ICB mediated myocarditis: (1) TCR recognizes the same antigen in the tumor and the heart, (2) antigens in the heart and tumor have substantial sequence overlap and are recognized by the same TCR and (3) one T-cell has two chimeric TCRs recognizing distinct antigens (44). These hypotheses are reliant on cardiomyocytes’ ability to escape immune surveillance by expression of checkpoint markers. Indeed, PD-1 surface expression is present on human and murine cardiomyocytes (45). An in vivo study of Pd1 knockout mice described the development of cardiomyopathy leading to decreased overall survival (45,46). These models showed thinning of the right ventricle, lowered ventricular systolic function and autoreactive antibodies against cardiomyocytes.
In support of the first hypothesis, Martinez-Calle et al. reported a fatal case of fulminant myocarditis following a single dose of anti-PD1. The patient tested positive for cardiac troponin autoantibodies and had elevated troponin T levels suggesting a pre-existing T memory response that was abrogated by PD-1 blockade (47). TCR recognition in the heart thus results in cytotoxic effect to cardiac tissue.
Evidence of the second hypothesis is reported in two clinical cases of fulminant myocarditis following ipilimumab and nivolumab treatment that show a substantial increase of infiltrating lymphocytes and macrophages in the cardiac muscle Lymphocyte receptor sequence shows a significant overlap of TCR sequences among cardiac, skeletal and tumor infiltrates, suggesting that the antigens in the myocardium and skeletal muscle were recognized by infiltrating lymphocytes. Additionally, one case had a 10-fold increase in PD-L1 expression on the injured cardiac tissue compared to the normal tissue suggesting an exhausted immune phenotype (48). Finally, the third concept of chimeric TCRs is borrowed from reports of TCRs in autoimmune diseases but has yet to be applied in the context of cardiotoxicity (49). Further functional work will be required to understand which hypothesis predominates in cardiac irAEs.
Genetic variation, both somatic and germline, can also provide insight to explain the overlap in TCR signaling between the tumor and cardiomyocytes in ICB-treated patients. Human genetic studies, such as GWAS, have advanced the understanding of the genetic factors in response to irAEs. For instance, CTLA-4 SNPs (rs4553808, rs11571317 and rs231775) were found to associate with irAEs in ipilimumab-treated cases (22). Additionally, susceptibility loci in major histocompatibility complex (MHC) genes in patients of autoimmune disease linked to cardiac-related adverse have begun to show avenues of pharmacogenomic approaches in myocarditis and myopathies (50). Large-scale prospective controlled studies will be necessary to make a robust assessment of SNPs and ICB-mediated cardiotoxicity. After identifying a robust list of loci, it will be necessary to perform quantitative trait locus mapping or molecular experiments to refine the functional consequences of genetic variation associated to cardiotoxicity. These methods are commonly employed following major GWAS such as cardiovascular disease (51) and cancer (52). These studies help further refine genetic susceptibility in complex traits that may involve several genes contributing risk of immunotherapy-related cardiotoxicity.
Epigenetic modulators in pharmacogenomics
As discussed in great detail, the efficacy of immunotherapy is based on the expression of immune checkpoint markers on immune or tumor cell surfaces. In addition to epistatic effects of germline or somatic variation on expression of PD-1 and PD-L1, DNA methylation and chromatic accessibility also play a vital role (53). DNA methyltransferase (DNMT) and histone deacetylase (HDAC) are key enzymes that serve as epigenetic ‘writers’ and ‘readers’, respectively. DNMT are responsible for adding methyl groups to gene promoters and thus silencing gene expression. Whereas, HDACs remove acetyl groups from lysine residues on histones to activate genes. Additionally, bromodomain and extra-terminal motif (BET) modulates interactions of histone acetyl transferases with HDACs with transcription factors involved in gene expression and cell cycle control (53). Inhibitors against these key enzymes are currently explored as modulators of immune-deprived tumors and harbor great interest to combine with ICB (54).
DNMT inhibitors, such as 5-azacitidine and decitabine, are the more common epigenetic modulators used in oncology clinical care. In human cancer cell lines, azacitidine was shown to upregulate interferon signaling and decreased Th1 response (55). A case report of three patients presenting with pericarditis following 1–5 cycles of azacytidine with mild elevation of troponin I levels (56). Patients’ symptoms were resolved following steroid treatment (56). Azacitidine and decitabine are in active Phase I/II trials with ICBs, monitoring for hypersensitivity in these trials is imperative (ClinicalTrials.gov Identifiers: NCT02959437 and NCT02957968).
Class I and II HDACs are the most active regulators of histones in cancer (57). Indeed, vorinostat, Class I and II HDAC inhibitor, was approved in 2006 for cutaneous T-cell lymphoma and showed minimal high-grade adverse effects (4%) (58). HDACs have been shown to play a role in macrophage polarization (59). Specifically, a novel Class IIa HDAC inhibitor was previously reported to play a direct role in polarizing macrophages to highly phagocytic and pro-inflammatory phenotype and synergized with anti-PD1 treatment in a breast cancer model resistant to ICB monotherapy (60). HDACis have also shown promise to deplete myeloid-derived suppressor cells (61). HDACis in combination with ICB are actively in Phase I and Phase II clinical trials in numerous solid tumors. Safety and tolerability outcomes are anticipated.
JQ1 is among the first generation of BET inhibitors synthesized and tested in the clinic. Pre-clinical studies of JQ1 in NSCLC murine models showed decreased Treg tumor infiltration and expression of PD-1 and CTLA-4 (62). However, the first-in-human Phase I study of BETi had an early termination due to dose-limiting toxicity (63). Additionally, BETi are actively being tested for tolerability in multiple myeloma (NCT03068351), progressive lymphoma (NCT01949883) and solid tumors (NCT02259114). Monitoring cardiotoxicity effects in epigenetic modulators with ICB is highly suggested and will be important for understanding how epigenetic modulation affects normal tissue homeostasis.
Clinical implementation of pharmacogenomics
Standard guidelines on ICB-mediated cardiotoxicity do not currently exist. To date, cardio-oncology programs implement individual procedures for the management of this clinical niche (64,65). For patients diagnosed with cardiotoxicity, permanent discontinuation of ICB and administration of corticosteroids followed by a tapering dose of glucocorticoids after improved signs are recommended (13). The European Society of Medical Oncology published their consensus for the management of cardiotoxicity and outline strategies for prevention, screening, monitoring and treatment. Specifically for ICB, an emphasis on acquiring baseline cardiac evaluation (electrocardiogram, troponin, B-type natriuretic peptide or N-terminal-pro-brain natriuretic peptide, C-reactive protein, viral titer, global longitudinal strain and cardiac magnetic resonance imaging) and carefully assessing patients with a history of cardiovascular disease is addressed (66). Detection and management of ICB-mediated cardiotoxicity is challenging and will require a multidisciplinary team as well as new methodologies to assess risk, especially in patients with a history of autoimmune disease, pre-existing cardiovascular disease or patients treated with targeted or immunotherapy combinations. This will require a shift in the infrastructure to the cardio-oncology standard of care to incorporate pharmacogenomic markers informed by cardiotoxicity registries. The Clinical Pharmacogenetics Implementation Consortium, PharmGKB and RadioGenomics Consortium are examples of consortiums that are focusing on GWAS and translational cellular screens to evaluate drug responses and adverse effects (67–69). Additionally, they help set guidelines for translating genotyping tests in cancer patients receiving immunotherapy.
Discussion and future directions
Single-cell sequencing technology
Tumor heterogeneity and plasticity is a large contributor to the marginal outcomes of ICB treatment (70). Single-cell RNA sequencing (scRNA-seq) technology is enabling the ‘reverse translation’ of clinical findings and confirming such observations in a controlled experimental environment. For example, Goswami et al. performed mass cytometry and single-cell sequencing on glioblastoma patients and identified CD73hi macrophages that persisted following treatment of anti-PD1 and anti-CTLA-4. Furthermore, they found that mice deficient in CD73hi macrophages were sensitized to combination ICB treatment (71). This reverse translational approach lead to the development of a new clinical trial aimed at combining immune checkpoint therapy with the targeting of the CD73 pathway (72). Several potential limitations about scRNA-seq have been addressed (73). Currently, short-read sequencing is the dominant sequencing platform used. While this method is efficient, without paired genotyping makes it difficult to know whether variants or transcript isoforms are responsible for differential expression. A partial sampling of the transcript as well as dimension reduction steps used in the analysis results in a bias for abundant genes and poses a challenge for the identification of rare transcripts (74). Also, established protocols have not been implemented leading to a lack of golden standard markers used to represent a specific-cell type or appropriate statistical cutoffs. The majority of protocols require fresh samples, which are often difficult for investigators to acquire (75). Given these limitations, The Human Tumor Atlas Network is leading the initiative to incorporate spatial and single-cell sequencing longitudinal sampling and comprehensive clinical data that will enrich our understanding of tumor immunology and personalized immunotherapy (76).
Single-cell sequencing has also been used to describe the various resident cell populations in the heart (77,78). Understanding cellular homeostasis is a fundamental resource for identifying transitions of cellular states in the heart following ischemic injury and heart failure. Notably, Nomura et al. (79) revealed a subset of cardiomyocytes that highly expressed p53 and oxidative programming by NRF2 and ERK that was critical for the development of heart failure following ischemic stress (79). Vascular tissue is also characterized as highly heterogenous and transient in a disease environment. Wirka et al. harnessed atherosclerotic tissue in mice and patients and used single-cell sequencing to identify a unique cell population, ‘fibrocytes’. Overexpression of transcription factor 21 promotes smooth muscle modulation that was protective for coronary artery disease (80). These studies are revealing the essential role of immune cell and cell cycle control in cardiovascular disease sequelae. Importantly, immune checkpoints often have redundant pathways and could help explain toxicity outcomes. To affirm this, single-cell studies specific to ICB-induced cardiotoxicity are required.
Conceivably, assessment of gene expression and phenotypic heterogeneity at the single-cell level will inform personalized therapeutic decision-making. The profiling of patient plasma samples or indirect profiling of patient-driven engineered heart tissues obtained through biopsies could in principle be used to predict treatment responses and monitor cardiotoxicity resulting from immunotherapy.
Human-induced pluripotent stem cells
Acquiring cardiac samples for studying cardiotoxicity is both invasive and rare. Human-induced pluripotent stem cells (hiPSCs) have served as robust model systems, e.g. in targeted therapies (HER2 inhibitors, anthracycline and tyrosine kinase inhibitor) induced toxicity (81–83). Kitani et al. tested trastuzumab (HER2/neu receptor inhibitor) treatment in vitro on hiPSC-differentiated cardiomyocytes (hiPSC-CMs) that resulted in decreased contractility, deformation distance, calcium handling and altered mitochondrial metabolic signaling. Additionally, treatment of 5′ adenosine monophosphate-activated protein kinase-activating compounds rescued contractility properties in hiPSC-CM from patients who received trastuzumab and experienced ejection fraction decline of >20% (82). These results suggest that altered energy metabolism pathway in cardiomyocytes influences the development of trastuzumab-induced cardiac dysfunction and is a targetable pathway for managing cardiac dysfunction. Additionally, this study reveals the use of a hiPSC-CM in vitro assay for studying antibody treatment-mediated toxicity.
Human iPSC-CMs are attractive models for cardio-oncology because they express that the majority of ion channels and sarcomere proteins found in the adult heart, can spontaneously contract, and implement quick differentiation protocol. However, once differentiated, hiPSC-CMs have physiological limitations that differ from the human heart, such as high resting potential, low sodium channel density and underdeveloped T-tubules (81). Despite these considerations, hiPSC-CM from patients experiencing toxicity will have a large impact on additional pharmacogenomic targets. The oncology research field has leveraged genetic engineering strategies to genetic dependencies across cancer types using genome-scale CRISPR-Cas (84). By pooling gRNA, such high-throughput assays can assess the loss-of-function effects of oncogenes to explain differences in cell viability and drug response (85–87). Similar approaches are feasible and instrumental for identifying novel pharmacogenomic targets in cardio-oncology.
Systems pharmacogenomics
Biological entities are involved in intricate and complex interactions, thereby forming highly complex systems. Understanding cancer and cardiotoxicity from the point-of-view of how subcellular systems and molecular ‘interactome’ network perturbations underlie disease initiation and progression is the essence of the fields of systems biology and network medicine as demonstrated in recent studies (88–90). As drug targets (including immunotherapy) do not operate in isolation from the complex cellular systems of proteins that comprise the molecular machinery of the cell with which they associate, we believe that each drug–gene interaction must be examined in an appropriate integrative context (91). The main hypothesis of systems pharmacogenomics is that subcellular networks gradually rewire throughout disease initiation, progression and maintenance, and drug responses, leading to progressive shifts of local and global network properties and systems states, all of which, in turn, underlie disease-causing factors and responsiveness (Fig. 2). Obviously, genomic alterations, such as amplification, deletion, translocation and mutations, are the primary events of drug responses. But such events can only be selected in cells if they encode the appropriate changes or perturbations in the human interactome and systems properties of the affected cells. Therapeutic interventions need to be designed to deal with perturbations of subcellular systems properties and have little to do, functionally speaking, with genomic alterations only.
Figure 2.
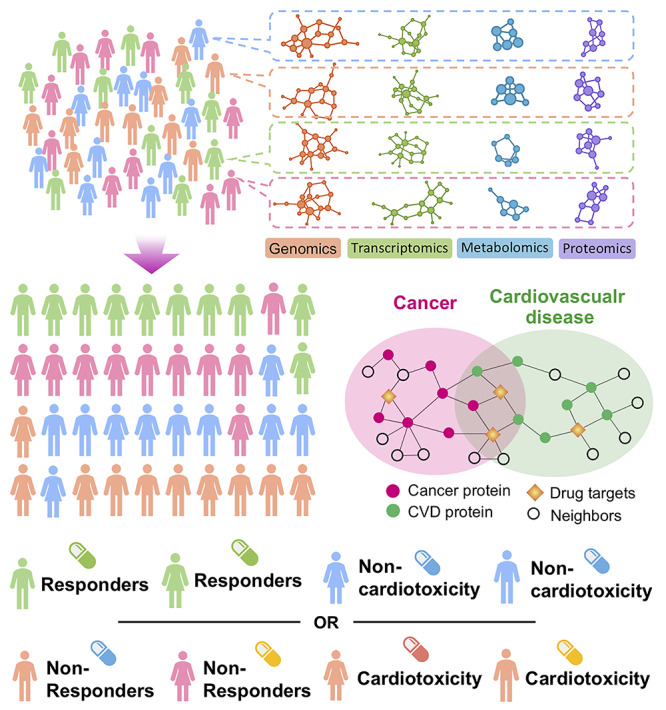
A diagram illustrating the proof-of-concept of systems pharmacogenomics approaches for precision immunotherapy and immune-related cardiotoxicity. A diagram illustrating how multi-omics measurements can be utilized in personalized immunotherapy and evaluate cardiotoxicity. Network-based integrative analysis (89,90) of genomic, transcriptomic, metabolomic and proteomic profiles of individuals will help to identify clinically actionable subgroups to assign personalized treatments. Overlapping biological pathways shared by cancer and cardiovascular disease will reveal complementary drug target vulnerabilities that will allow for the stratification of patients receiving ICB by response and cardiotoxicity.
We posit that an integrated, systems pharmacogenomics approach (Fig. 2), which incorporates large-scale existing patient multi-omics profiles (e.g. genomics, transcriptomics, proteomics, and metabolomics) from TCGA, The Cancer Imaging Archive, Clinical Proteomic Tumor Analysis Consortium and other open data resources into the human protein–protein interactome, will offer a novel and effective way in developing personalized cardio-oncology for cancer patients receiving immunotherapy or other cancer treatments if broadly applied. Bioinformatics and systems biology approaches have shown possibilities in identifying pathways, which, in turn, provide actionable biomarkers to minimize cardiotoxicity during cancer treatment and cardiovascular risk for cancer survivors (92–94). ‘Accelerate the development of guidelines for monitoring and management of patient symptoms to minimize side effects of therapy’ is one of the 10 recommendations from the 2016 Cancer Moonshot initiative (95). Cardio-oncology is a multidisciplinary field, which requires the creative and energetic involvement of biologists, physicians, technology developers, data scientists, patient groups and others. Cardiovascular specialists and oncologists can build on their experience, leveraging pharmacogenomics clinical specialists, and use precision medicine to facilitate discovery science and improve the efficiency of clinical research, with the goal of providing more precise information to improve the health of individuals with immunotherapies or other cancer treatments.
Conclusions
In summary, cancer immunotherapy with ICBs enables immune activation, thereby inducing anti-tumor responses in a variety of solid tumors. However, immunotherapy-induced cardiovascular toxicity is life-threatening and a proposed Achilles’ heel of this class of therapeutic agents although it is rare. Pharmacogenomics studies of anthracycline-induced cardiotoxicity have been well described, while the genetic variants that predispose to immunotherapy-related cardiotoxicity are in its infancy.
The unique integration of genomic signatures, protein biomarkers and imaging markers using systems biology or mathematical models would offer new risk prediction tools and novel immune targets for emerging development of precision immunotherapy and further reducing immune-related cardiotoxicity (96,97). Comprehensive identification of patients’ specific predispositions, to individualize immunotherapy strategies and therefore yield the greatest clinical benefits at the lowest impact of heart and cardiovascular systems, is the ultimate goal of precision cardio-oncology and immunotherapy.
Conflict of Interest statement: The authors declare no conflict of interest.
Funding
National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) (Award Number K99HL138272 and R00HL138272 to F.C.); VeloSano Pilot Program (Cleveland Clinic Taussig Cancer Institute) (to F.C.).
Author contributions
F.C. conceived the study. J.A.C performed data analysis. F.C., J.A.C and C.E. wrote and critically revised the manuscript.
References
- 1.. Ribas A. and Wolchok J.D. (2018) Cancer immunotherapy using checkpoint blockade. Science, 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.. Leach D.R., Krummel M.F. and Allison J.P. (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science, 271, 1734–1736. [DOI] [PubMed] [Google Scholar]
- 3.. Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L.J., Malenkovich N., Okazaki T., Byrne M.C. et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med., 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.. Lipson E.J. and Drake C.G. (2011) Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res., 17, 6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.. Schadendorf D., Hodi F.S., Robert C., Weber J.S., Margolin K., Hamid O., Patt D., Chen T.-T., Berman D.M. and Wolchok J.D. (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol., 33, 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.. Brahmer J.R., Tykodi S.S., Chow L.Q.M., Hwu W.-J., Topalian S.L., Hwu P., Drake C.G., Camacho L.H., Kauh J., Odunsi K. et al. (2012) Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med., 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.. Gettinger S., Horn L., Jackman D., Spigel D., Antonia S., Hellmann M., Powderly J., Heist R., Sequist L.V., Smith D.C. et al. (2018) Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. J. Clin. Oncol., 36, 1675–1684. [DOI] [PubMed] [Google Scholar]
- 8.. Overman M.J., McDermott R., Leach J.L., Lonardi S., Lenz H.J., Morse M.A., Desai J., Hill A., Axelson M., Moss R.A. et al. (2017) Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol., 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.. Hellmann M.D., Paz-Ares L., Bernabe Caro R., Zurawski B., Kim S.-W., Carcereny Costa E., Park K., Alexandru A., Lupinacci L., Mora Jimenez E. et al. (2019) Nivolumab plus Ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med., 381, 2020–2031. [DOI] [PubMed] [Google Scholar]
- 10.. Haslam A. and Prasad V. (2019) Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open, 2, e192535–e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.. Friedman C.F., Proverbs-Singh T.A. and Postow M.A. (2016) Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol., 2, 1346–1353. [DOI] [PubMed] [Google Scholar]
- 12.. Moslehi J.J., Salem J.E., Sosman J.A., Lebrun-Vignes B. and Johnson D.B. (2018) Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet, 391, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.. Zhou Y.-W., Zhu Y.-J., Wang M.-N., Xie Y., Chen C.-Y., Zhang T., Xia F., Ding Z.-Y. and Liu J.-Y. (2019) Immune checkpoint inhibitor-associated cardiotoxicity: current understanding on its mechanism, diagnosis and, management. Front. Pharmacol., 10, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.. Herrmann J. (2020) Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat. Rev. Cardiol., 17, 474–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.. Yu J.X., Upadhaya S., Tatake R., Barkalow F. and Hubbard-Lucey V.M. (2020) Cancer cell therapies: the clinical trial landscape. Nat. Rev. Drug Discov. doi: 10.1038/d41573-020-00099-9. [DOI] [PubMed] [Google Scholar]
- 16.. Vaddepally R.K., Kharel P., Pandey R., Garje R. and Chandra A.B. (2020) Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel), 12, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.. Leong S.L., Chaiyakunapruk N. and Lee S.W.H. (2017) Candidate gene association studies of anthracycline-induced cardiotoxicity: a systematic review and meta-analysis. Sci. Rep., 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.. Brown S.A., Sandhu N. and Herrmann J. (2015) Systems biology approaches to adverse drug effects: the example of cardio-oncology. Nat. Rev. Clin. Oncol., 12, 718–731. [DOI] [PubMed] [Google Scholar]
- 19.. Samstein R.M., Lee C.-H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A. et al. (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet., 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.. Dagogo-Jack I. and Shaw A.T. (2018) Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol., 15, 81–94. [DOI] [PubMed] [Google Scholar]
- 21.. Hegde P.S. and Chen D.S. (2020) Top 10 challenges in cancer immunotherapy. Immunity, 52, 17–35. [DOI] [PubMed] [Google Scholar]
- 22.. Shek D., Read S.A., Ahlenstiel G. and Piatkov I. (2019) Pharmacogenetics of anticancer monoclonal antibodies. Cancer Drug Resist., 2, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.. Yehia L., Ngeow J. and Eng C. (2019) PTEN-opathies: from biological insights to evidence-based precision medicine. J. Clin. Invest., 129, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.. Zhao J., Chen A.X., Gartrell R.D., Silverman A.M., Aparicio L., Chu T., Bordbar D., Shan D., Samanamud J., Mahajan A. et al. (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med., 25, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.. Sharma M.D., Shinde R., McGaha T.L., Huang L., Holmgaard R.B., Wolchok J.D., Mautino M.R., Celis E., Sharpe A.H., Francisco L.M. et al. (2015) The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv., 1, e1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.. Roh W., Chen P.L., Reuben A., Spencer C.N., Prieto P.A., Miller J.P., Gopalakrishnan V., Wang F., Cooper Z.A., Reddy S.M. et al. (2017) Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med., 9, eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.. Peng W., Chen J.Q., Liu C., Malu S., Creasy C., Tetzlaff M.T., Xu C., McKenzie J.A., Zhang C., Liang X. et al. (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov., 6, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.. Bucheit A.D., Chen G., Siroy A., Tetzlaff M., Broaddus R., Milton D., Fox P., Bassett R., Hwu P., Gershenwald J.E. et al. (2014) Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res., 20, 5527–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.. Shrestha S., Yang K., Guy C., Vogel P., Neale G. and Chi H. (2015) Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol., 16, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.. McGranahan N., Rosenthal R., Hiley C.T., Rowan A.J., Watkins T.B.K., Wilson G.A., Birkbak N.J., Veeriah S., Van Loo P., Herrero J. et al. (2017) Allele-specific HLA loss and immune escape in lung cancer evolution. Cell, 171, 1259–1271.e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.. Rosenthal R., Cadieux E.L., Salgado R., Bakir M.A., Moore D.A., Hiley C.T., Lund T., Tanić M., Reading J.L., Joshi K. et al. (2019) Neoantigen-directed immune escape in lung cancer evolution. Nature, 567, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.. Reche P.A. and Reinherz E.L. (2003) Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J. Mol. Biol., 331, 623–641. [DOI] [PubMed] [Google Scholar]
- 33.. Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T., Ivanova Y., Hundal J., Arthur C.D., Krebber W.J. et al. (2014) Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature, 515, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.. Chowell D., Morris L.G.T., Grigg C.M., Weber J.K., Samstein R.M., Makarov V., Kuo F., Kendall S.M., Requena D., Riaz N. et al. (2018) Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science, 359, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.. Chowell D., Krishna C., Pierini F., Makarov V., Rizvi N.A., Kuo F., Morris L.G.T., Riaz N., Lenz T.L. and Chan T.A. (2019) Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat. Med., 25, 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.. Cai W., Zhou D., Wu W., Tan W.L., Wang J., Zhou C. and Lou Y. (2018) MHC class II restricted neoantigen peptides predicted by clonal mutation analysis in lung adenocarcinoma patients: implications on prognostic immunological biomarker and vaccine design. BMC Genomics, 19, 582–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.. Postow M.A., Sidlow R. and Hellmann M.D. (2018) Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med., 378, 158–168. [DOI] [PubMed] [Google Scholar]
- 38.. Motzer R.J., Tannir N.M., McDermott D.F., Arén Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthélémy P., Porta C., George S. et al. (2018) Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med., 378, 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.. Upadhrasta S., Elias H., Patel K. and Zheng L. (2019) Managing cardiotoxicity associated with immune checkpoint inhibitors. Chronic Dis. Transl. Med., 5, 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.. Mahmood S.S., Fradley M.G., Cohen J.V., Nohria A., Reynolds K.L., Heinzerling L.M., Sullivan R.J., Damrongwatanasuk R., Chen C.L., Gupta D. et al. (2018) Myocarditis in patients treated with immune checkpoint inhibitors. J. Am. Coll. Cardiol., 71, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.. Al-Kindi S.G. and Oliveira G.H. (2018) Reporting of immune checkpoint inhibitor-associated myocarditis. Lancet, 392, 382–383. [DOI] [PubMed] [Google Scholar]
- 42.. Palaskas N., Lopez-Mattei J., Durand J.B., Iliescu C. and Deswal A. (2020) Immune checkpoint inhibitor myocarditis: pathophysiological characteristics, diagnosis, and treatment. J. Am. Heart Assoc., 9, e013757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.. Postow M.A., Chesney J., Pavlick A.C., Robert C., Grossmann K., McDermott D., Linette G.P., Meyer N., Giguere J.K., Agarwala S.S. et al. (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med., 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.. Brown S.-A., Ray J.C. and Herrmann J. (2020) Precision cardio-oncology: a systems-based perspective on cardiotoxicity of tyrosine kinase inhibitors and immune checkpoint inhibitors. J. Cardiovasc. Transl. Res., 13, 402–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.. Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N. et al. (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science, 291, 319–322. [DOI] [PubMed] [Google Scholar]
- 46.. Wang J., Okazaki I.M., Yoshida T., Chikuma S., Kato Y., Nakaki F., Hiai H., Honjo T. and Okazaki T. (2010) PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol., 22, 443–452. [DOI] [PubMed] [Google Scholar]
- 47.. Martinez-Calle N., Rodriguez-Otero P., Villar S., Mejías L., Melero I., Prosper F., Marinello P., Paiva B., Idoate M. and San-Miguel J. (2018) Anti-PD1 associated fulminant myocarditis after a single pembrolizumab dose: the role of occult pre-existing autoimmunity. Haematologica, 103, e318–e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.. Johnson D.B., Balko J.M., Compton M.L., Chalkias S., Gorham J., Xu Y., Hicks M., Puzanov I., Alexander M.R., Bloomer T.L. et al. (2016) Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med., 375, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.. Cusick M.F., Libbey J.E. and Fujinami R.S. (2012) Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol., 42, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.. Hoefsmit E.P., Rozeman E.A., Haanen J.B.A.G. and Blank C.U. (2019) Susceptible loci associated with autoimmune disease as potential biomarkers for checkpoint inhibitor-induced immune-related adverse events. ESMO Open, 4, e000472–e000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.. Yao C., Chen G., Song C., Keefe J., Mendelson M., Huan T., Sun B.B., Laser A., Maranville J.C., Wu H. et al. (2018) Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun., 9, 3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.. Gong J., Mei S., Liu C., Xiang Y., Ye Y., Zhang Z., Feng J., Liu R., Diao L., Guo A.-Y. et al. (2017) PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res., 46, D971–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.. Delmore J.E., Issa G.C., Lemieux M.E., Rahl P.B., Shi J., Jacobs H.M., Kastritis E., Gilpatrick T., Paranal R.M., Qi J. et al. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.. Topper M.J., Vaz M., Marrone K.A., Brahmer J.R. and Baylin S.B. (2020) The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol., 17, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.. Bezu L., Chuang A.W., Liu P., Kroemer G. and Kepp O. (2019) Immunological effects of epigenetic modifiers. Cancers (Basel), 11, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.. Newman M., Malla M. and Gojo I. (2016) Azacitidine-induced pericarditis: a case series. Pharmacotherapy, 36, 443–448. [DOI] [PubMed] [Google Scholar]
- 57.. Li Y. and Seto E. (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med., 6, a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.. Mann B.S., Johnson J.R., Cohen M.H., Justice R. and Pazdur R. (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist, 12, 1247–1252. [DOI] [PubMed] [Google Scholar]
- 59.. Groot A.E. and Pienta K.J. (2018) Epigenetic control of macrophage polarization: implications for targeting tumor-associated macrophages. Oncotarget, 9, 20908–20927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.. Guerriero J.L., Sotayo A., Ponichtera H.E., Castrillon J.A., Pourzia A.L., Schad S., Johnson S.F., Carrasco R.D., Lazo S., Bronson R.T. et al. (2017) Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature, 543, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.. Wang H.F., Ning F., Liu Z.C., Wu L., Li Z.Q., Qi Y.F., Zhang G., Wang H.S., Cai S.H. and Du J. (2017) Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother., 66, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.. Adeegbe D.O., Liu Y., Lizotte P.H., Kamihara Y., Aref A.R., Almonte C., Dries R., Li Y., Liu S., Wang X. et al. (2017) Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov., 7, 852–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.. Postel-Vinay S., Herbschleb K., Massard C., Woodcock V., Soria J.C., Walter A.O., Ewerton F., Poelman M., Benson N., Ocker M. et al. (2019) First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer, 109, 103–110. [DOI] [PubMed] [Google Scholar]
- 64.. Bhatia N., Santos M., Jones L.W., Beckman J.A., Penson D.F., Morgans A.K. and Moslehi J. (2016) Cardiovascular effects of androgen deprivation therapy for the treatment of prostate cancer: ABCDE steps to reduce cardiovascular disease in patients with prostate cancer. Circulation, 133, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.. Montazeri K., Unitt C., Foody J.M., Harris J.R., Partridge A.H. and Moslehi J. (2014) ABCDE steps to prevent heart disease in breast cancer survivors. Circulation, 130, e157–e159. [DOI] [PubMed] [Google Scholar]
- 66.. Curigliano G., Lenihan D., Fradley M., Ganatra S., Barac A., Blaes A., Herrmann J., Porter C., Lyon A.R., Lancellotti P. et al. (2020) Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol., 31, 171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.. Barbarino J.M., Whirl-Carrillo M., Altman R.B. and Klein T.E. (2018) PharmGKB: a worldwide resource for pharmacogenomic information. Wiley Interdiscip. Rev. Syst. Biol. Med., 10, e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.. Kerns S.L., Fachal L., Dorling L., Barnett G.C., Baran A., Peterson D.R., Hollenberg M., Hao K., Narzo A.D., Ahsen M.E. et al. (2020) Radiogenomics consortium genome-wide association study meta-analysis of late toxicity after prostate cancer radiotherapy. J. Natl. Cancer Inst., 112, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.. Relling M.V. and Klein T.E. (2011) CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin. Pharmacol. Ther., 89, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.. Galluzzi L., Chan T.A., Kroemer G., Wolchok J.D. and López-Soto A. (2018) The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med., 10, eaat7807. [DOI] [PubMed] [Google Scholar]
- 71.. Goswami S., Walle T., Cornish A.E., Basu S., Anandhan S., Fernandez I., Vence L., Blando J., Zhao H., Yadav S.S. et al. (2020) Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med., 26, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.. Sharma P. and Allison J.P. (2020) Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol., 20, 75–76. [DOI] [PubMed] [Google Scholar]
- 73.. Paik D.T., Cho S., Tian L., Chang H.Y. and Wu J.C. (2020) Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 17, 457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.. Hwang B., Lee J.H. and Bang D. (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med., 50, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.. Suvà M.L. and Tirosh I. (2019) Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol. Cell, 75, 7–12. [DOI] [PubMed] [Google Scholar]
- 76.. Rozenblatt-Rosen O., Regev A., Oberdoerffer P., Nawy T., Hupalowska A., Rood J.E., Ashenberg O., Cerami E., Coffey R.J., Demir E. et al. (2020) The Human Tumor Atlas Network: charting tumor transitions across space and time at single-cell resolution. Cell, 181, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.. DeLaughter D.M., Bick A.G., Wakimoto H., McKean D., Gorham J.M., Kathiriya I.S., Hinson J.T., Homsy J., Gray J., Pu W. et al. (2016) Single-cell resolution of temporal gene expression during heart development. Dev. Cell, 39, 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.. Tucker N.R., Chaffin M., Fleming S.J., Hall A.W., Parsons V.A., Bedi K.C., Jr., Akkad A.D., Herndon C.N., Arduini A., Papangeli I. et al. (2020) Transcriptional and cellular diversity of the human heart. Circulation. doi: 10.1161/CIRCULATIONAHA.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.. Nomura S., Satoh M., Fujita T., Higo T., Sumida T., Ko T., Yamaguchi T., Tobita T., Naito A.T., Ito M. et al. (2018) Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun., 9, 4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.. Wirka R.C., Wagh D., Paik D.T., Pjanic M., Nguyen T., Miller C.L., Kundu R., Nagao M., Coller J., Koyano T.K. et al. (2019) Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med., 25, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.. Sharma A., McKeithan W.L., Serrano R., Kitani T., Burridge P.W., Álamo J.C., Mercola M. and Wu J.C. (2018) Use of human induced pluripotent stem cell–derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc., 13, 3018–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.. Kitani T., Ong S.-G., Lam C.K., Rhee J.-W., Zhang J.Z., Oikonomopoulos A., Ma N., Tian L., Lee J., Telli M.L. et al. (2019) Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation, 139, 2451–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.. Wang H., Sheehan R.P., Palmer A.C., Everley R.A., Boswell S.A., Ron-Harel N., Ringel A.E., Holton K.M., Jacobson C.A., Erickson A.R. et al. (2019) Adaptation of human iPSC-derived cardiomyocytes to tyrosine kinase inhibitors reduces acute cardiotoxicity via metabolic reprogramming. Cell Syst., 8, 412–426.e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.. Behan F.M., Iorio F., Picco G., Gonçalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M. et al. (2019) Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature, 568, 511–516. [DOI] [PubMed] [Google Scholar]
- 85.. Chan E.M., Shibue T., McFarland J.M., Gaeta B., Ghandi M., Dumont N., Gonzalez A., McPartlan J.S., Li T., Zhang Y. et al. (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature, 568, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.. Kim E., Ilic N., Shrestha Y., Zou L., Kamburov A., Zhu C., Yang X., Lubonja R., Tran N., Nguyen C. et al. (2016) Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov., 6, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.. Berger A.H., Brooks A.N., Wu X., Shrestha Y., Chouinard C., Piccioni F., Bagul M., Kamburov A., Imielinski M., Hogstrom L. et al. (2016) High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell, 30, 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.. Cheng F., Desai R.J., Handy D.E., Wang R., Schneeweiss S., Barabasi A.L. and Loscalzo J. (2018) Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun., 9, 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.. Cheng F., Hong H., Yang S. and Wei Y. (2017) Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Brief. Bioinform., 18, 682–697. [DOI] [PubMed] [Google Scholar]
- 90.. Cheng F., Lu W., Liu C., Fang J., Hou Y., Handy D.E., Wang R., Zhao Y., Yang Y., Huang J. et al. (2019) A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun., 10, 3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.. Cheng F., Kovacs I.A. and Barabasi A.L. (2019) Network-based prediction of drug combinations. Nat. Commun., 10, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.. Menche J., Sharma A., Kitsak M., Ghiassian S.D., Vidal M., Loscalzo J. and Barabasi A.L. (2015) Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science, 347, 1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.. Guney E., Menche J., Vidal M. and Barabasi A.L. (2016) Network-based in silico drug efficacy screening. Nat. Commun., 7, 10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.. Ghiassian S.D., Menche J., Chasman D.I., Giulianini F., Wang R., Ricchiuto P., Aikawa M., Iwata H., Muller C., Zeller T. et al. (2016) Endophenotype network models: common Core of complex diseases. Sci. Rep., 6, 27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.. Ledford H. (2016) Cancer experts unveil wishlist for US government 'moonshot'. Nature, 537, 288–289. [DOI] [PubMed] [Google Scholar]
- 96.. Mandawat A., Williams A.E. and Francis S.A. (2017) Cardio-oncology: the role of big data. Heart Fail. Clin., 13, 403–408. [DOI] [PubMed] [Google Scholar]
- 97.. Liu C., Ma Y., Zhao J., Nussinov R., Zhang Y., Cheng F. and Zhang Z. (2020) Computational network biology: data, models, and applications. Phys. Rep., 846, 1–66. [Google Scholar]