

Abstract
Familial Mediterranean fever (FMF) is the most common auto‐inflammatory disease. It is transmitted as autosomal recessive trait with mutations in MEditerranean FeVer (MEFV) gene. Despite a typical clinical expression, many patients have either a single or no mutation in MEFV. The current work is aimed to revisit the genetic landscape of FMF disease using high‐coverage whole genome sequencing. In atypical patients (carrying a single or no mutation in MEFV), we revealed many rare variants in genes associated with auto‐inflammatory disorders, and more interestingly, we discovered a novel variant ( a 2.1‐Kb deletion) in exon 11 of IL1RL1 gene, present only in patients. To validate and screen this patient‐specific variant, a tandem of allele‐specific PCR and quantitative real‐time PCR was performed in 184 FMF patients and 218 healthy controls and we demonstrated that the novel deletion was absent in controls and was present in more than 19% of patients. This study sheds more light on the mutational landscape of FMF. Our discovery of a disease‐specific variant in IL1RL1 gene may constitute a novel genetic marker for FMF. This finding suggesting a potential role of the IL33/ST2 signalling in the disease pathogenicity highlights a new paradigm in FMF pathophysiology.
Keywords: Familial Mediterranean Fever, IL1RL1, MEFV, Whole Genome Sequencing
1. INTRODUCTION
Auto‐inflammatory diseases (AIDs) are a distinct group of disorders characterized by an unprovoked systemic inflammation without the presence of high titre of autoantibodies nor antigen‐specific T cells. 1 , 2 Most of the AIDs are monogenic and are caused by highly penetrant mutations in single genes encoding proteins involved in the innate immunity, but complex and polygenic AIDs with significant environmental influence have also been identified. 3
Familial Mediterranean fever (FMF) is the most common Mendelian auto‐inflammatory disease, characterized by uncontrolled activation of the innate immune system, resulting in recurrent brief episodes of fever and serositis with chest, abdominal, joints and muscles pain. 4 Predominantly, FMF affects people from Mediterranean and Middle Eastern ethnic origins (1/200‐1/1000). 5
The causing gene of FMF is the MEditerranean FeVer (MEFV) gene. 5 , 6 The MEFV gene encodes 781 amino acids pyrin (or marenostrin) protein, which is mostly expressed in neutrophils, eosinophils, monocytes, dendritic cells and fibroblasts. 7 , 8 The exact physiological role of pyrin protein is not clear; however, it is suggested to play a role in apoptosis, inflammation, cytokine production and innate immune response. The MEFV gene, located on chromosome 16p13.3, is approximately 14.6 kb long and contains 10 exons. The gene can harbour multiple mutations in different exons; however, exon 2 and exon 10 are two mutational hot spots, with exon 10 having the largest number of mutations. The five founder mutations are p.Met694Val, p.Met694Ile, p.Val726Ala and p.Met680Ile present in exon 10 and p.Glu148Gln in exon 2, together they represent more than 80% of the disease‐causing mutations. 9
The analysis of the typical FMF patients revealed an autosomal recessive model of inheritance. 5 The disease can segregate either in homozygous or in a compound heterozygous modality. However, it is observed that a substantial number of FMF patients are either heterozygous or carry no MEFV mutation. The possibility of pseudo‐dominance is considered in rare cases but it is yet to be proven and it could not explain the large number of clinical FMF cases. 10 , 11 The hypothesis of digenic or oligogenic inheritance is gaining attention and could explain the divergence of clinical FMF with single or no mutation in MEFV gene from the typical paradigm of recessive inheritance. 12 , 13 The presence of mutations in modifier genes associated with inflammation or interactions between MEFV mutation and modifying allele in genes involved in known auto‐inflammatory diseases, as reported in a limited number of studies, could also be responsible for the large spectrum of FMF phenotypes. 14 , 15
The lack of comprehensive genetic analyses of FMF patients with single or no mutated allele in MEFV gene is prompting us to investigate the genetic landscape of FMF disease in a large cohort of FMF patients with different MEFV mutational profiles using both Sanger and whole genome sequencing (WGS).
2. METHODS
2.1. Patients and controls
The study population consisted of 402 unrelated Lebanese subjects including 184 FMF patients (102 males and 82 females with median age 17 ± 5 years) recruited from several medical centres in Beirut, Lebanon, and 218 gender and ethnicity matched healthy controls recruited among subjects visiting the hospitals for routine health check‐up and who were free from any chronic inflammatory and autoimmune disease. Blood sample collection and storage was managed by the Medical Center CEMEDIPP and the American University of Science and Technology in Beirut, Lebanon. The diagnosis of FMF in our patients was made according to the established criteria of both Sohar (Tel Hashomer criteria) 5 and Livneh. 16 More stringent clinical diagnosis criteria were used to establish the diagnosis of FMF in patients with a single disease‐causing MEFV variant or with no identified MEFV variants. The 184 FMF patients were randomly selected from a large cohort of patients for whom Sanger sequencing of 10 exons of MEFV gene was performed, and who, based on copies of MEFV mutated allele, were stratified into three groups: (a) zero mutation: patients without any mutation in MEFV gene; (b) single mutation: patients with only one mutation in MEFV gene; and (c) double mutation: patients with two MEFV mutations. In order to increase the chance to identify novel and/or modifier genes for FMF, we purposely enriched our study cohort with more patients with a single or no variant in MEFV gene. We performed WGS on 50 patient samples (11 patients with double MEFV mutation, 19 patients with a single mutation and 20 patients with no MEFV mutation) randomly selected from the 3—Sanger sequencing—defined subcategories and that of 26 healthy control subjects.
The study protocol was approved by ethics committee of Sidra Medicine, Doha, Qatar (Protocol number # 1511002018). All study subjects signed a written informed consent prior to be enrolled in the study.
2.2. Sample preparation and whole genome sequencing (WGS)
Peripheral blood samples were collected from patients and controls in EDTA tubes and genomic DNA was extracted by standard salt‐precipitation methods. 17 WGS was carried on DNA of 50 FMF cases along with 26 controls with a HiSeq 2500 sequencer (30× average coverage) at Sidra Medicine, Qatar. Paired‐end libraries were generated from 1 μg of genomic DNA using an Illumina TruSeq DNA PCR‐Free Sample Preparation Kit. Genomic DNA was sheared using a Covaris system. Isolated DNA fragment ends were blunted, A‐tailed and ligated with sequencing adaptors with index sequences. Excess adapters and enzymes were removed using AMPure beads (Beckman Coulter Genomics). Indexed libraries were size‐selected to the 350 bp range using bead‐based capture, and the concentration of amplifiable fragments was determined by qPCR, relative to sequencing libraries with a known concentration. Normalized libraries were clustered on a c‐BOT machine, and 125 bp paired‐end sequencing was performed on the HiSeq 2500 system.
2.3. WGS data analysis
Paired‐end raw fastq files were mapped to the reference human genome, build GrCh37, using BWA‐MEM aligner: 0.7.12‐r1039, 18 GATK Haplotype caller was used for variant calling on individual samples. GATK Genotype GVCFs option was used for joint calling across individual samples. Variant calling was performed using recommended best practices of GATK version 3.7. Joint variant file was further gone through with GATK variant quality score recalibration (VQSR) step. 19 The annotation of variants was performed by using SNPEFF (version: 4.3r, GRCh37.75 Reference Build) and dbNFSP 3.0. 20 Ingenuity® Variant Analysis ([https://www.qiagenbioinformatics.com/products/ingenuity‐variant‐analysis)] from QIAGEN, Inc”) was used to filter variants based on various parameter: (a) Variants with low‐call quality (<20), low coverage (<10), which failed in VQSR filter and which were present in low complexity region were excluded, (b) variants with allele frequencies more than 1% in public database including 1000G phase3, 21 gnomAD version 2.1.1 22 and ExAc project release 1 23 were excluded unless established as a pathogenic variant, (c) homozygous, heterozygous or compound heterozygous variants which were present in cases and absent in controls were selected and (d) only non‐synonymous, frameshift, non‐sense and splice site variants, which could be potential deleterious based on CADD version 1.3 score (>20) and functional predictions by SIFT version 5.1.1 and Polyphen‐2 version 2.2r398, were selected. 24 , 25 , 26 Furthermore, variants, which were either related to auto‐inflammatory diseases including FMF or which were reported to interact with known genes associated with auto‐inflammatory diseases, were chosen.
For copy number variant (CNV) analysis, we used three structural variant callers: Delly version 0.7.8, Speedseq version 0.1.2 and GenomeSTRiP version 2.00.171, and we applied the best practices recommended by authors of the tools. The annotation of structural variant was carried out using AnnTools version 1.0. 27 Only rare, exonic structural variants, which were absent in controls, were selected for further analysis. For the visualization and confirmation of structural variants, we used SAMPlot (https://github.com/ryanlayer/samplot).
We have submitted all the variants reported here to LOVD website (https://www.lovd.nl).
2.4. Genetic screening for the novel variant of IL1RL1 gene
Screening for the presence of the novel variant (2.1‐Kb deletion) of IL1RL1 gene (NM_016232, NC_000002.11:g.102967165_102969288del), identified by WGS, was performed in all 402 subjects using a tandem of 2 PCR assays (allele‐specific PCR [AS‐PCR] followed by a quantitative real‐time PCR (qRT‐PCR)]). First, samples are analysed by AS‐PCR using primers flanking a genomic region of 3 Kb encompassing the 2.1‐Kb deletion. A simultaneous amplification of a 3‐Kb fragment and a 0.9‐Kb fragment corresponds to the presence of a heterozygous deletion of exon 11 of the IL1RL1 gene, and an amplification of a 3‐Kb fragment only indicates the absence of such deletion. In order to confirm the outcome of the AS‐PCR, a qRT‐PCR was performed to quantify the copy number of the region flanking the 2.1‐Kb deletion.
Briefly, 50 ng of genomic DNA was subjected to a total of 25 μL PCR containing 200μM dNTP, 0.5 μmol/L each of forward and reverse primer and 0.5 unit Phusion® High‐Fidelity DNA polymerase (NEB), with a PCR program of 95°C for 1’30’’, followed by 35 cycles at 94°C for 25”, 65°C for 30” and 72” for 1’40” in a Veriti Thermal Cycler (Applied Biosystems). Primers were designed to amplify a 3.0‐Kb fragment encompassing the 2.1‐Kb deletion: Forward primer 5’‐ TCTCACACTCAAGCTTGTGCTG‐3’ and reverse primer 5’‐AGAGCTCTCATACACAACTGGTG‐3’. All PCR products were examined by electrophoresis on 1.5% agarose gels and photographed with a ChemiDocTM MP Imaging System (Bio‐Rad).
To confirm the outcome of the AS‐PCR, the qRT‐PCR was performed using two sets of pair of primers; one set was used to amplify a DNA fragment within the 2.1‐Kb deletion (forward primer 5’‐AGAAGCAATAGTGCCTGCTG‐3’ and reverse primer 5’‐ATTCCTGCTCCTCACACTTC‐3’), and another set to amplify, as an endogenous control, a DNA fragment upstream the 2.1‐Kb deletion (forward primer 5’‐AACGGCTCAAGAGACTTGTG‐3’ and reverse primer 5’‐TACTTCTACCTGCATGGGTG‐3’). The qRT‐PCR was performed in a total volume of 20 μL containing 15 ng genomic DNA, 10μl GoTaq® qPCR Master Mix (Promega) and 0.5 μmol/L each of forward and reverse primer using a cycling program of 2’ at 50°C, 2’ at 95°C, 40 cycles consisting of 15” at 95°C and 45” at 60°C, and a dissociation curve analysis step of 15” of a rapid ramp to 95°C, 15” at 60°C and 15” of a slow ramp to 95°C on a QuantStudio 6 Flex Real‐Time PCR system (Applied Biosystems) in Fast 96‐well plate format. qPCR for each amplicon of each patient was performed in triplicate, and AS‐PCR–verified WGS patients with and without the 2.1‐Kb deletion were included for each plate as controls. The results were analysed using the comparative CT (ΔΔCT) method.
Chi‐square test was used to compare the frequency between the two groups of patients (patients with a single or no MEFV mutation vs patients with 2 MEFV mutations), and the Phi coefficient was used to generate the effect size of this novel variant in patients.
3. RESULTS
3.1. Characterization of MEFV mutations in patients with FMF
The 184 FMF patients of the present study were randomly selected from a large cohort for which Sanger sequencing of coding sequence of MEFV gene was performed. In order to increase the chance to unveil novel pathogenic and/or modifiers genes for FMF, we purposely enriched the patient population with more patients carrying single or no mutation in MEFV gene. Out of the 184 FMF cases, 58 (31.5%) patients had biallelic variants of the MEFV gene, 57 (31.0%) patients were heterozygous, while 69 (37.5%) patients did not carry any coding mutations in MEFV gene. The mutational analysis showed that the Met694Val mutation was the most frequent mutation, followed by the Val726Ala, p.Pro158Ser/p.Pro369Ser, p.Arg197Gln/p.Arg408Gln and Met694Ile. This result is in agreement with previous studies. 28 , 29
3.2. WGS and the search of novel pathogenic or modifier genes for FMF
To investigate the potential presence of variants in novel pathogenic and/or modifiers genes in FMF patients with single or no mutated allele in MEFV gene, we analysed the WGS data of 50 patients, randomly selected from the 3 subcategories of patients, and that of 26 healthy control subjects.
We performed the identity by descent (IBD) estimation 30 in the 76 samples, which indicated that our study subjects were unrelated (Figure 1A). Principal component analysis 31 was performed on the 76 samples along with samples from the 1000 Genomes Project data set, revealing a genetic signature with proximity to that of the European ancestry (Figure 1B).
FIGURE 1.
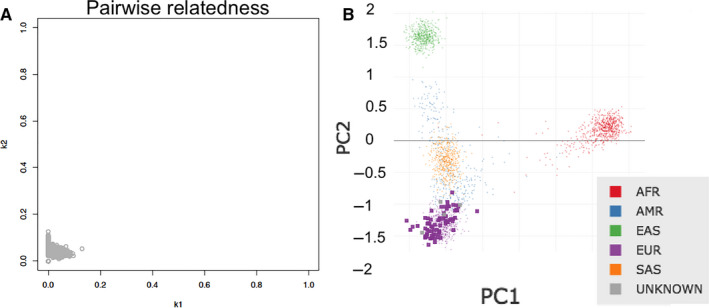
(A) Identity by descent (IBD) plot displaying un‐relatedness of the 76 samples on which whole genome sequencing was performed. (B) Principal component analysis (PCA) plot of the same samples mapped on 1000‐Genome data set; AFR = African, AMR = Ad Mixed American, EAS = East Asian, EUR = European, SAS = South Asian
The status of MEFV mutations in patients, initially defined by Sanger sequencing, was confirmed by WGS. The list of all MEFV variants, identified by WGS, found in the 50 patients with FMF is shown in Table 1. The MEFV variants were exclusively present in FMF cases and were absent in controls. In addition, WGS revealed in our patients three novel variants in the promoter region of MEFV gene: c.‐123A > G, c.‐397C > G and c.‐1309G > A (reference sequence: NC_000016.9). These heterozygous promoter variants were present in only 3 FMF cases and were predicted to cause loss of the promoter function of the gene. Beside non‐synonymous and promoter variants, two synonymous heterozygous variants (p.Pro124Pro and p.Arg290Arg/p.Arg501Arg) were found in FMF cases.
TABLE 1.
List of the MEFV variants revealed by WGS and present in patients with FMF
| Position at Chr 16 | Gene Region | Mutation description | Cases (N) | Genotype (Patient ID) | Regulatory site | Regulator | Frequency in gnomAD |
|---|---|---|---|---|---|---|---|
| 3293257 | Exon 10 | NM_000243.2:c.2230G > T (p.Ala744Ser) | 2 | Het (FMF16 & 30) | 0.0018 | ||
| 3293310 | Exon 10 | NM_000243.2:c.2177T > C (p.Val726Ala) | 10 | Het (FMF1, 5, 6,7,11, 27, 28); Homo (FMF 3, 4, 10); | 0.0020 | ||
| 3293403 | Exon 10 | NM_000243.2:c.2084A > G (p.Lys695Arg) | 2 | Het (FMF17, 26) | 0.0058 | ||
| 3293405 | Exon 10 | NM_000243.2:c.2082G > A (p. Met694Ile) | 3 | Het (FMF8, 14, 21) | 0.0001 | ||
| 3293407 | Exon 10 | NM_000243.2:c.2080A > G (p.Met694Val) | 12 | Homo (FMF2); Het (FMF1, 6, 7, 12, 13, 15, 18, 19, 23, 24, 25) | 0.0003 | ||
| 3293447 | Exon 10 | NM_000243.2:c.2040G > A (p.Met680Ile) | 2 | Het (FMF8, 11) | 0.0 000 079 | ||
| 3299468 a | Exon3 | NM_000243.2:c.1223G > A (p.Arg408Gln) | 4 | Het (FMF9, 20, 22, 29) | 0.0134 | ||
| 3299586 a | Exon3 | NM_000243.2:c.1105C > T (p.Pro369Ser) | 4 | Het (FMF9, 20, 22, 29) | 0.0147 | ||
| 3304 626 | Exon2 | NM_000243.2:c.442G > C (p.Glu148Gln) | 2 | Het (FMF5, 9) | 0.0658 | ||
| 3306710 b | Promoter | NC_000016.9:g.3306710T > C (NG_007871.1:g.4918A > G (c.‐123A > G)) | 1 | Het (FMF31) | Encode TFBS, Promoter Loss | CEBPB, HNF1B, REST, STAT3 | NA |
| 3306984 b | Promoter | NC_000016.9:g.3306984G > C (NG_007871.1:g.4644C > G (c.‐397C > G)) | 1 | Het (FMF12) | Promoter Loss | HNF1B | NA |
| 3307896 b | Promoter | NC_000016.9:g.3307896C > T (NG_007871.1:g.3732G > A (c.‐1309G > A)) | 1 | Het (FMF13) | Promoter Loss | NFIC | NA |
Abbreviations: Chr, chromosome; gnomAD, Genome Aggregation Database version 2.1.1; Het, Heterozygous variant; Homo, Homozygous variant; NA: Not available.
Arg408Gln variant is in cis with Pro369Ser.
Indicates novel variant.
3.3. Mutational Spectrum of genes associated with other AIDs in FMF patients
After filtering out variants which had high prevalence in the general population (allele frequency > 1%) or were present in controls, we first examined variants in known AID‐associated genes. More than 50 genes associated with auto‐inflammatory disorders were selected from Systemic autoinflammatory disease (SAID; http://www.autoinflammatory‐search.org/)) and Infever database. 32 The list of the novel variants of genes associated with AIDs found in the 50 FMF patients is shown in Table 2. We observed that 10 out of the 50 FMF cases had variants in genes linked with familial haemophagocytic lymphohistiocytosis (FHL), with PRF1 NM_005041.4:c.272C > T (p.Ala91Val), NM_005041.4:c.1153C > T (p.Arg385Trp) and STXBP2 NM_006949.3:c.1034C > T (p.Thr345Met) variants present in 3 patients each, while variants in other known FHL‐associated genes (RAB27A and UNC13D) were present in one FMF case each. PRF1 p.Ala91Val variant is classified as DFP (ie disease‐associated polymorphism with additional functional evidence) in the Human Gene Mutation Database (HGMD) 33 for FHL. Screening for genes associated with hereditary fever syndromes other than MEFV revealed also the presence of variants in PSTPIP1 (NM_003978.4:c.203C > A (p.Thr68Lys) and a splice site variant NC_000015.9(NM_003978.4):c.37‐10081C > G), TNFRSF11A (NM_003839.3:c.1234G > T (p.Asp412Tyr), NM_003839.3:c.1348C > T (p.Arg450Trp)) and in NLRP3 (NM_001079821.2:c.2861C > T (p.Thr954Met)). Furthermore, four different variants in NOD2 gene (NM_022162.2:c.2230C > T (p.Arg744Trp), NM_022162.2:c.2127G > A (p.Trp709*), NM_022162.2:c.676_691del (p.Arg227fs*145) and NC_000016.9(NM_022162.2):c.2883‐2A > G) were also observed in FMF cases. Other auto‐inflammatory disorder genes, which had missense substitution in our present cohort of FMF cases, were IFIH1 (NM_022168.3:c.1126G > A (p.Glu376Lys), and NM_022168.3:c.2597C > T (p.Pro866Leu)), PLCG2 (NM_002661.4:c.82A > T (p.Met28Leu)), TNFAIP3 (NM_001270508.1: c.406C > T (p.Arg136Cys)) and SH3BP2 (NM_001145856.1:c.1600C > T (p.Arg534Trp)). We observed also three predicted pathogenic variants in genes associated with Psoriasis 2 and 15 (CARD14 NM_024110.4:c.1789C > T (p.Arg597Trp) and NM_024110.4:c.1789C > T (p.Arg597Trp) and AP1S3 NM_001039569.1:c.11T > G (p.Phe4Cys)) in FMF cases, with AP1S3 (p.Phe4Cys) listed as disease‐causing mutation (DM) for psoriasis 15 in the HGMD.
TABLE 2.
List of variants of auto‐inflammatory disorders genes found in FMF patients
| Gene | Variant details | Chr: position | Type of Mutation | dbSNP ID | Cases (N) | SIFT | Polyphen‐2 | CADD Score | Frequency in gnomAD | Acronym of SAID | AID Mode of Inheritance | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRF1 | NM_005041.4:c.272C > T (p.Ala91Val) | 10:72 360 387 | Missense | rs35947132 | 3 | Damaging | Probably Damaging | 26 | 0.0292 | FHL | Autosomal recessive | |
| PRF1 | NM_005041.4:c.1153C > T (p.Arg385Trp) | 10:72 358 324 | Missense | rs72358324 | 3 | Damaging | Probably Damaging | 20.8 | 0.0002 | FHL | ||
| STXBP2 | NM_006949.3:c.1034C > T (p.Thr345Met) | 19:7 708 058 | Missense | rs117761837 | 3 | Damaging | Probably Damaging | 26.3 | 0.0106 | FHL | ||
| RAB27A | NM_004580.4:c.17A > G (p.Tyr6Cys) | 15:55 527 116 | Missense | rs145253993 | 1 | Damaging | Probably Damaging | 27 | 0.0001 | FHL | ||
| UNC13D | NM_199242.2:c.670C > T (p.His224Tyr) | 17:73 836 856 | Missense | rs145607492 | 1 | Damaging | Probably Damaging | 25.5 | 0.0002 | FHL | ||
| UNC13D | NM_199242.2:c.610A > G (p.Met204Val) | 17:73 837 042 | Missense | rs144722609 | 1 | Tolerated | Benign | 22.6 | 0.0007 | FHL | ||
| TNFAIP3 | NM_001270508.1:c.406C > T (p.Arg136Cys) | 6:138 196 092 | Missense | rs200740561 | 1 | Damaging | Probably Damaging | 35 | 0.0001 | AISBL | Autosomal dominant | |
| PSTPIP1 | NM_003978.4:c.203C > A (p.Thr68Lys) | 15:77 310 863 | Missense | NA | 1 | Damaging | Possibly Damaging | 25.7 | NA | PAPA | Autosomal dominant | |
| PSTPIP1 | NC_000015.9(NM_003978.4):c.37‐10081C > G | 15:77 300 408 | Splice site a | rs1020233393 | 1 | NA | NA | <10 | NA | PAPA | ||
| NOD2 | NM_022162.2:c.2230C > T (p.Arg744Trp) | 16:50 746 052 | Missense | rs140876663 | 1 | Damaging | Probably Damaging | 26.8 | 0.0001 | Blau syndrome | Autosomal dominant | |
| NOD2 | NM_022162.2:c.2127G > A (p.Trp709*) | 16:50 745 949 | Stop gain | rs776701942 | 1 | NA | NA | 35 | 0.000008 | Blau syndrome | ||
| NOD2 | NM_022162.2:c.679_694del (p.Arg227fs*145) | 16:50 744 498 | Frameshift | NA | 1 | NA | NA | NA | Blau syndrome | |||
| NOD2 | NC_000016.9(NM_022162.2):c.2883‐2A > G | 16:50 759 398 | Splice Site | rs564226539 | 1 | NA | NA | 24.8 | 0.00002 | Blau syndrome | ||
| TNFRSF11A | NM_003839.3:c.1234G > T (p.Asp412Tyr) | 18:60 036 384 | Missense | NA | 1 | Damaging | Probably Damaging | 24.5 | NA | TRAPS11 | Autosomal dominant | |
| TNFRSF11A | NM_003839.3:c.1348C > T (p.Arg450Trp) | 18:60 036 498 | Missense | rs34945627 | 1 | Damaging | Probably Damaging | 22.9 | 0.0009 | TRAPS11 | ||
| NLRP3 | NM_001079821.2:c.2861C > T (p.Thr954Met) | 1:247 607 973 | Missense | rs139814109 | 1 | Damaging | Probably Damaging | 33 | 0.0012 | CAPS | Autosomal dominant. | |
| IFIH1 | NM_022168.3:c.1126G > A (p.Glu376Lys) | 2:163 139 056 | Missense | 1 | Damaging | Probably Damaging | 33 | NA | AGS7 | Autosomal recessive | ||
| IFIH1 | NM_022168.3:c.2597C > T (p.Pro866Leu) | 2:163 128 755 | Missense | rs200833729 | 1 | Tolerated | Possibly Damaging | 23.0 | 0.0004 | AGS7 | ||
| PLCG2 | NM_002661.4:c.82A > T (p.Met28Leu) | 16:81 819 676 | Missense | rs61749044 | 1 | Tolerated | Possibly Damaging | 24.0 | 0.0106 | APLAID | Autosomal dominant | |
| SH3BP2 | NM_001145856.1:c.1600C > T (p.Arg534Trp) | 4:2 834 080 | Missense | rs14876133 | 2 | Damaging | Probably Damaging | 32 | 0.0043 | Cherubism | Autosomal dominant | |
| CARD14 | NM_024110.4:c.1789C > T (p.Arg597Trp) | 17:78 172 328 | Missense | NA | 1 | Damaging | Probably Damaging | 34 | 0.0037 | PSORS2 | Autosomal dominant | |
| CARD14 | NM_024110.4:c.239G > A (p.Arg80Gln) | 17:78 156 479 | Missense | NA | 1 | Tolerated | Probably Damaging | 25 | NA | PSORS2 | ||
| AP1S3 | NM_001039569.1:c.11T > G (p.Phe4Cys) | 2:224 642 579 | Missense | rs116107386 | 1 | Damaging | Probably Damaging | 27.2 | 0.0079 | PSOR15 | Autosomal dominant | |
Abbreviations: AGS7, Aicardi‐Goutieres syndrome 7; AIBSL, autoinflammatory syndrome, familial, Behcet‐like; APLAID, auto‐inflammation and PLCG2‐associated antibody deficiency and immune dysregulation; CAPS, cryopyrin‐associated periodic syndromes; Chr, chromosome; FHL, familial haemophagocytic lymphohistiocytosis; gnomAD, Genome Aggregation Database version 2.1; NA, not available; PAPA, pyogenic sterile arthritis, pyoderma gangrenosum and acne syndrome; PSOR15, pustular psoriasis; PSOR2, familial psoriasis; TRAPS11, TNFRSF11A‐associated hereditary fever disease.
All the listed variants were present in heterozygous state in FMF cases and were absent in controls; software version: SIFT version 5.1.1, PolyPhen‐2 version 2.2.2r398, CADD version 1.3.
Results in splice site Loss.
3.4. Identification of novel variants in inflammatory genes in FMF patients
Variants in known AID‐associated genes identified in our cohort were not sufficient to completely draw the genetic variation pattern in our FMF patients. We further looked for the predicted pathogenic variants in inflammatory genes either interacting with known genes associated to AIDs or involved in auto‐inflammation processes, using knowledge base of Ingenuity variant analysis. The list of variants in inflammatory genes found in the 50 FMF patients is shown in Table 3. We observed that IFNAR2 (NM_207585.2:c.611C > G: (p.Thr204Arg)) was the most common variant among FMF cases, and it was present in 7 out of 50 FMF cases (from the three FMF subgroups). IFNAR2 associates with IFNAR1 to form a receptor for interferons alpha (IFNA1) and beta (IFNB1). In the present study, FMF cases also had variants in IFNAR1 (NM_000629.2:c.954G > C (p.Trp318Cys)) and in IFNB1 (NM_002176.3:c.498A > G (p.Ile166Met)), which were present in two FMF cases and one FMF case, respectively. A more comprehensive screening from the list of inflammatory genes identified from Ingenuity revealed that our FMF patients had many variants in genes of the superfamily of TNF and its receptors. A stop gain variant in TNFRSF4 (NM_003327.3:c.384C > A (p.Cys128*)) was present in two FMF cases, whereas missense variants in TNFRSF8 (NM_001243.4:c.1511G > A (p.Arg504Gln)) and TNFRSF9 (NM_003811.3:c.716G > A (p.Arg239Gln)) were present in single FMF case each. We also identified two variants in genes involved in TLR pathway: TLR1 (NM_003263.3:c.1013T > C (p.Met338Thr)) and TRAFD1 (NM_001143906.1:c.908A > C (p.Glu303Ala). Many interleukins and their receptors sequences were also found to be altered in FMF patients like IL17RB (NM_018725.3:c.529G > A (p.Gly177Arg)), IL17RD (NM_017563.4:c.1696C > T (p.Pro566Ser)), IL1R2 (NM_004633.3:c.932T > C (p.Ile311Thr)), IL20 (NC_000001.10(NM_018724.3):c.225 + 1G>T), IL12A (NM_000882.3:c.631G > A (p.Val211Met)) and IL1A (NM_000575.4:c.526G > C (p.Asp176His)), with IL17RB (NM_018725.3:c.529G > A (p.Gly177Arg)) and IL1R2 (NM_004633.3:c.932T > C (p.Ile311Thr)) variants present in three patients each, and the remaining other variants present in one case each. Among NLR family of genes, NLRC3 NM_178844.3:c.2401G > A: (p.Ala801Thr), NLRP2 NM_017852.4:c.2672G > T (p.Gly891Val) and NLRX1 NM_024618.3:c.1480G > A (p.Val494Met) were present in one FMF case each. A missense variant in CASP14 gene (NM_012114.2:c.418G > A (p.Gly140Ser)) was observed in one FMF case. Endoplasmic reticulum aminopeptidases genes, ERAP1 and ERAP2, which encode proteins involved in peptide trimming for HLA class I molecules, 34 were altered in four and one FMF cases, respectively. Some other predicted pathogenic variants in FMF cases were LILRB1 NM_006669.6:c.997G > T (p.Gly333Cys), RAB27B NM_004163.4:c.274G > A (p.Ala92Thr) and ICAM1 NM_000201.2:c.1099C > T (p.Arg367Cys).
TABLE 3.
List of variants in inflammatory genes in FMF patients
| Gene | Variant details | Chr: position | Type of variant | dbSNP ID | Cases (N) | SIFT | Polyphen‐2 | CADD Score | Frequency in gnomAD |
|---|---|---|---|---|---|---|---|---|---|
| IFNAR2 |
NM_207585.2:c.611C > G (p.Thr204Arg) |
21:34 625 037 | Missense | rs147496374 | 7 | Damaging | Probably Damaging | 29.3 | 0.0046 |
| IFNAR1 | NM_000629.2:c.954G > C (p.Trp318Cys) | 21:34 721 562 | Missense | rs578193831 | 2 | Damaging | Probably Damaging | 28.7 | 0.00 002 |
| IFNB1 | NM_002176.3:c.498A > G (p.Ile166Met) | 9:21 077 371 | Missense | rs141894933 | 1 | Damaging | Probably Damaging | 15.94 | 0.0016 |
| TNFRSF4 | NM_003327.3:c.384C > A (p.Cys128 a ) | 1:1 148 071 | Stop gain | NA | 2 | NA | NA | 36 | NA |
| TNFRSF8 | NM_001243.4:c.1511G > A (p.Arg504Gln) | 1:12 198 461 | Missense | rs2230627 | 1 | Tolerated | Probably Damaging | 28.8 | 0.0002 |
| TNFSF9 | NM_003811.3:c.716G > A (p.Arg239Gln) | 19:6 535 028 | Missense | rs755292822 | 1 | Tolerated | Probably Damaging | 23.3 | NA |
| TRAFD1 | NM_001143906.1:c.908A > C (p.Glu303Ala) | 12:112 583 447 | Missense | rs79680080 | 1 | Damaging | Probably Damaging | 26 | 0.0158 |
| TLR1 | NM_003263.3:c.1013T > C (p.Met338Thr) | 4:38 799 440 | Missense | rs990267834 | 1 | Damaging | Possibly Damaging | 23.5 | NA |
| IL1R2 | NM_004633.3:c.932T > C (p.Ile311Thr) | 2:102 642 617 | Missense | rs144482163 | 3 | Damaging | Probably Damaging | 26.1 | 0.0023 |
| IL1A | NM_000575.4:c.526G > C p.(Asp176His) | 2:113 535 653 | Missense | rs1801715 | 1 | Damaging | Probably Damaging | 23.3 | 0.00 006 |
| IL12A | NM_000882.3:c.631G > A (p.Val211Met) | 3:159 713 215 | Missense | rs35990253 | 1 | Tolerated | Probably Damaging | 18.74 | 0.0040 |
| IL17RB | NM_018725.3:c.529G > A (p.Gly177Arg) | 3:53 889 368 | Missense | rs2232337 | 3 | Damaging | Probably Damaging | 29.7 | 0.0044 |
| IL17RD | NM_017563.4:c.1696C > T (p.Pro566Ser) | 3:57 132 035 | Missense | rs61742267 | 1 | Tolerated | Probably Damaging | 23.6 | 0.0142 |
| IL20 | NC_000001.10(NM_018724.3):c.225 + 1G>T | 1:207 039 710 | Splice site a | rs138566326 | 1 | NA | NA | 23.5 | 0.0006 |
| NLRP2 | NM_017852.4:c.2672G > T (p.Gly891Val) | 19:55 502 004 | Missense | NA | 1 | Tolerated | Probably Damaging | 22.1 | NA |
| NLRC3 | NM_178844.3:c.2401G > A: (p.Ala801Thr) | 16:3 600 448 | Missense | rs767176921 | 1 | Damaging | Possibly Damaging | 24.4 | 0.00 001 |
| NLRX1 | NM_024618.3:c.1480G > A (p.Val494Met) | 11:119 045 792 | Missense | rs780397677 | 1 | Tolerated | Probably Damaging | 23.7 | 0.0000 |
| CASP14 | NM_012114.2:c.418G > A (p.Gly140Ser) | 19:15 165 983 | Missense | rs761542772 | 1 | Damaging | Probably Damaging | 25.2 | 0.00 004 |
| LILRB1 | NM_006669.6:c.997G > T (p.Gly333Cys) | 19:55 144 505 | Missense | rs201421803 | 1 | Damaging | Probably Damaging | 22.7 | 0.0006 |
| RAB27B | NM_004163.4:c.274G > A (p.Ala92Thr) | 18:52 551 598 | Missense | rs9962265 | 1 | Damaging | Probably Damaging | 34.0 | 0.000 003 |
| ICAM1 | NM_000201.2:c.1099C > T (p.Arg367Cys) | 19:10 395 252 | Missense | rs139178890 | 1 | Damaging | Probably Damaging | 32 | 0.0005 |
| ERAP2 | NM_022350.4:c.1040C > T (p.Thr347Met) | 5:96 228 072 | Missense | rs75263594 | 4 | Damaging | Probably Damaging | 26.8 | 0.02 155 |
| ERAP1 | NM_016442.4:c.1378G > C (p.Gly460Arg) | 5:96 126 289 | Missense | rs771994807 | 1 | Damaging | Probably Damaging | 27.8 | 0.000 015 |
Abbreviations: Chr, Chromosome; NA, Not available.
Note: All the listed variants were present in heterozygous state in FMF cases and were absent in controls.
Software version: SIFT version 5.1.1, PolyPhen‐2 version 2.2.2r398, CADD version 1.3.
Splice site loss
3.5. Copy number variant (CNV) analysis in FMF
Beside point mutations and small indels, we also looked for the structural variants in the whole genome of the 50 FMF cases. Variant calling was done using Delly version 0.7.8, GenomeSTRiP version 2.00.17.1 and Speedseq version 0.1.2 using best practices recommended by authors of the tools. Later, final output from these 3 tools annotated with Anntools version 1.0. After removing variants, which either were present in controls or were located in non‐coding regions, 164 deletions were identified by GenomeSTRiP version 2.00.17.1, 704 variants (358 deletions, 334 duplications, 12 inversions) were found by Speedseq version 0.1.2 and 1178 variants (338 duplications, 398 deletions, 442 inversions) were identified by Delly version 0.7.8. For genotyping structural variants, we used their respective genotyper modules or tools such as SVTyper for speedseq, SVGenotyper module of GenomeSTRiP and integrated genotyper of Delly. We performed manual inspection of all these variants and found a deletion in IL1RL1 gene, which was consistently detected by all three software. This heterozygous deletion in exon 11 of IL1RL1 gene (NM_016232, NC_000002.11:g. 102967165_102969288 del) was around 2.1 Kb in size and was present in 9 FMF cases carrying one mutated allele of the MEFV gene and reported by three software on same subjects. For the visualization and confirmation of structural variants, we used SAMPlot. The representative figure of IL1RL1 deletion in FMF cases and controls is shown in Figure S1. The search of the identified IL1RL1 variant in the 1000G phase 3 data set showed the presence of a larger deletion of 3.1 Kb (Variant: esv3591789; http://dgv.tcag.ca/dgv/app/variant?id=esv3591789&ref=hg19), overlapping with the 2.1‐Kb IL1RL1 deletion, in only one subject among 2504.
Table S1 shows the summary of all the variants from WGS (listed in Tables 1, 2, 3 including IL1RL1 deletion variant) per patient to demonstrate the genotype of all FMF patients at these loci. There is no distinct pattern of distribution of AID‐associated variants and inflammatory gene variants among three subgroup of FMF patients (with zero, single and double MEFV variants) IL1RL1 deletion variant was particularly enriched in FMF patient with single MEFV variant in WGS cohort. Few FMF patients had burden of several rare variants of AID and/or inflammatory genes.
3.6. The IL1RL1 gene deletion in familial Mediterranean fever patients
To validate the finding revealed by WGS and CNV analysis, a search of the 2.1‐Kb deletion detected in the IL1RL1 gene was performed in all 402 study subjects using allele‐specific PCR (AS‐PCR) followed by quantitative real‐time PCR (qRT‐PCR). No discrepancies in IL1RL1 variant genotyping were found between AS‐PCR and qRT‐PCR. Figure 2 shows both the gel electrophoresis of the AS‐PCR products of samples with or without the 2.1‐Kb deletion of the IL1RL1 gene and the quantification by qRT‐PCR of the copy number of the region flanking the 2.1‐Kb deletion. The distribution of the IL1RL1 deletion in FMF according to the number of the mutated MEFV alleles is shown in Table 4. This novel variant in IL1RL1 was found in FMF patients only. More than 19% of FMF patients are carriers of the IL1RL1 deletion. The frequency of IL1RL1 variant was found higher in patients with a single or no mutation in MEFV gene compared to that in patients carrying 2 MEFV mutations (0.222 vs 0.120, P = .05) with an effect size of 0.12. No control subject was found to be a carrier of this variant.
FIGURE 2.
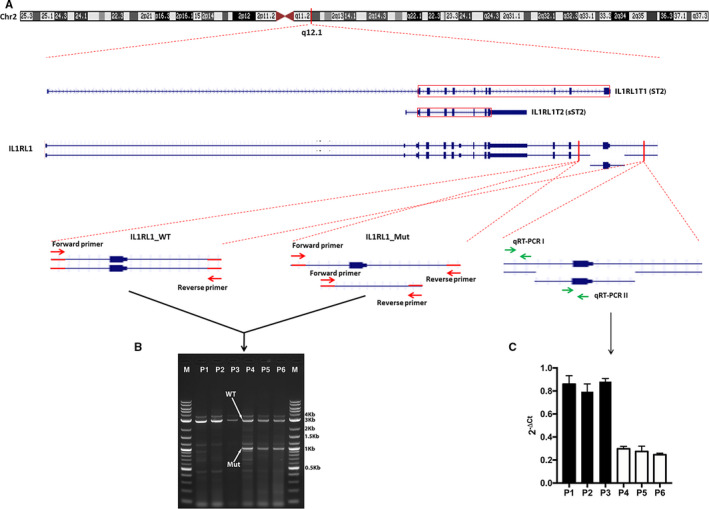
Screening of the 2.1‐Kb deletion of the IL1RL1 gene using AS‐PCR and qRT‐PCR. (A) Schematic representation of IL1RL1 transcripts that encode ST2 and sST2, respectively, and of the IL1RL1 heterozygous deletion containing exon 11 and experimental design to confirm the presence of the deletion using AS‐PCR and qRT‐PCR. The red box represents the coding sequence of the transcript. Hom: homozygous; Het: Heterozygous; WT: wild type; Mut: mutant. (B) DNA gel of AS‐PCR products of 6 FMF patients carrying (P4, P5 and P6) or not (P1, P2 and P3) the 2.1‐Kb deletion of exon 11 of the IL1RL1 gene. A simultaneous amplification of a 3 Kb fragment and a 0.9 Kb fragment corresponds to the presence of the heterozygous deletion, and an amplification of a 3 Kb fragment only indicates the absence of such deletion. M: 1Kb plus DNA marker (New England Biolabs, US). (C) qRT‐PCR results of the IL1RL1 deletion region containing exon 11 compared to its 5’ wild‐type region among 6 FMF patients. ΔCt = Ct(RT‐PCR II)‐Ct(RT‐PCR I)
TABLE 4.
Distribution of the novel variant (2.1‐Kb deletion) of IL1RL1 gene (NC_000002.11:g. 102967165_102969288 del) in FMF patients and in controls
| Subjects (N = 402) | IL1RL1 deletion |
|---|---|
|
Total FMF N = 184 |
35 (19.02%) |
| MEFV | |
|
2 mutations N = 58 |
7 (12.06%) |
|
1 mutation N = 57 |
12 (21.05%) |
|
0 mutation N = 69 |
16 (23.18%) |
|
Controls N = 218 |
0 (0%) |
4. DISCUSSION
In the present report, we showed significant genetic heterogeneity in FMF patients having single or no mutated allele of MEFV gene, with several patients carrying a burden of rare variants in auto‐inflammatory genes.
We first performed Sanger sequencing of coding region of MEFV gene in FMF cases to characterize MEFV mutations and to stratify patients based on the number of mutated alleles of MEFV. The most common MEFV mutation in our patient group was pMet694Val followed by p.Val726Ala, which is similar to other published reports in Lebanese and Middle Eastern populations. 28 , 29
As some recent familial and non‐familial studies on FMF have identified the role of selected auto‐inflammatory genes like NLRP3, TNFRSF1A and MVK, 35 , 36 we decided to screen our patients for the possibility of having rare/pathogenic mutations in other known auto‐inflammatory genes. Six broad categories of AID have been proposed based on the genetic defect in different component of the immune system: (a) IL‐1beta activation disorders (inflammasomopathies), (b) NF‐kB activation syndromes, (c) protein misfolding disorders, (d) complement regulatory diseases, (e) disturbances in cytokine signalling and (f) macrophage activation syndromes. 37 We filtered our WGS data for the variants in more than 50 genes associated to AID belonging to one or another of the above‐mentioned AID categories and investigated for potential pathogenic variants common to FMF cases and absent in controls. Although no single variant in an AID‐associated gene seemed frequent in FMF cases, we found that six different variants in four known genes (PRF1, STXBP2, RAB27A and UNC13D) associated with familial haemophagocytic lymphohistiocytosis (FHL) were present in about 20% of our FMF patients. Genes associated with FHL are known to encode cytotoxic proteins: PRF1 encodes perforin, which permeabilizes the target cell membrane, UNC13D encodes Munc13‐4 protein that causes cytolytic granule fusion with the cell membrane during degranulation, RAB27A encodes small Rab GTPase, which plays a role in exocytosis of cytotoxic vesicles, while STXBP2 is involved in the release of cytotoxic granules by natural killer cells. 38 Mutations in these genes are supposed to impair their normal function and could lead to increased macrophages activation and cytokine production. 39 Other AID‐associated gene variants were identified in our patients, but they were not frequent and were present only in one or two cases each.
We further investigated variations in novel genes, which are reported to interact with known auto‐inflammatory genes or which may have a role in auto‐inflammation process. The top candidate variant identified in this analysis was IFNAR2 NM_207585.2:c.611C > G: (p.Thr204Arg), which was present in 14% of FMF cases from all subcategories (with 0, 1 and 2 MEFV mutations) and which is involved in type 1 interferon signalling.
Our initial search for rare structural variants in exonic regions performed on the 76 WGS (50 FMF cases and 26 controls) led to the discovery of a novel (2.1‐Kb deletion) variant in interleukin‐1 receptor‐like 1 (IL1RL1) gene. This deletion initially revealed by WGS was present in 9 FMF patients with a single mutated allele of the MEFV gene. The high frequency of this genetic alteration in our patients compared to controls and its relevance to the pathophysiology of inflammatory diseases stimulated the search of its presence in all 402 study subjects. Interestingly, the IL1RL1 variant, absent in controls, was confirmed in more than 19% of FMF patients belonging to the different MEFV subgroups. The IL1RL1 variant was found even higher in FMF patients carrying a single or no mutation in MEFV gene.
The IL1RL1 gene product, which has been given the alias ST2, is defined as the IL‐33 receptor. 40 , 41 ST2 is a member of the IL‐1 receptor family. There are two main isoforms: a membrane‐bound form (ST2), which promotes NF‐κB signalling, and a soluble receptor (sST2) which prevents its signalling. ST2/IL‐33 pathway has been implicated in a wide range of disease settings, in anti‐inflammatory responses and homeostasis, and thus, signalling must be strictly regulated. 42 Dysregulation of ST2/IL‐33 signalling and sST2 production have been implicated in a variety of inflammatory diseases. 43 , 44 ST2 contains an extracellular domain, which binds IL‐33, a transmembrane domain, and an intracellular domain called a Toll/interleukin 1 receptor (TIR) domain. The novel variant (2‐Kb deletion) of IL1RL1 gene, reported in the present study, covers the totality of exon 11 encoding the TIR domain. Therefore, this deletion could lead to the disruption of the IL‐33/ST2 signalling.
Although this current study, showing the presence of many rare variants in genes associated with auto‐inflammatory disorders and a novel variant (a 2.1‐Kb deletion) in exon 11 of IL1RL1 gene (NM_016232) in atypical FMF patients (carrying a single or no mutation in MEFV), supports the multigenic inheritance model of FMF, a large‐scale typing in Lebanese FMF patients is needed. The small number of healthy control subjects included in the Genome sequencing analysis constitutes a potential limitation of our study. Replication of the present findings in other populations will be useful to determine whether the association between these genetic markers and FMF can be generalized. We believe that our findings could have potential implications in the diagnostic and disease management of FMF. The extreme variability of clinical presentation and disease severity of FMF constitute a significant challenge for clinicians. As pointed out by Gangemi et al, 45 although the MEFV genotype‐phenotype correlation in FMF patients has been intensively investigated, a clear consensus has not yet been reached. Several hypotheses have been proposed to explain the clinical heterogeneity of FMF but the clinical and diagnostic dilemma remain unsolved. While the current study showed that FMF patients carried a large spectrum of variants in several inflammatory genes, certain variants seem to be quite prevalent in patients carrying a single or no mutation in MEFV gene including variants in the 4 genes (PRF1, STXBP2, RAB27A and UNC13D) associated with FHL and the novel variant that we have discovered in the IL1RL1 gene. A more holistic approach integrating clinical data and comprehensive genetic investigations, not limited to MEFV, could constitute the most effective diagnostic process to confirm or refute the diagnosis of FMF. A large phenotype‐genotype study will be undertaken to identify potential associations between the numerous genetic variants herein reported and specific clinical features of FMF.
In conclusion, this study provides novel evidence supporting a multigenic model of inheritance in FMF. The novel IL1RL1 gene variant that we have identified in a significant proportion of our patients qualifies as an additional genetic marker for FMF. These findings pave the way for future studies that would provide more insight into the molecular mechanisms underlying FMF and for the design of new and more effective genetic tests for the diagnosis of FMF.
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTION
Meenakshi Umar: Data curation (equal); Investigation (equal); Methodology (equal); Writing‐original draft (lead); Writing‐review & editing (supporting). Andre Megarbane: Conceptualization (supporting); Data curation (equal); Investigation (equal); Resources (lead). Jingxuan Shan: Investigation (equal); Methodology (supporting); Writing‐original draft (supporting). Najeeb Syed: Data curation (equal); Formal analysis (equal). Eliane Chouery: Data curation (equal); Formal analysis (equal). Elbay Aliyev: Data curation (equal); Formal analysis (equal). Puthen Jithesh: Data curation (supporting); Formal analysis (equal). Ramzi Temanni: Data curation (supporting); Formal analysis (equal). Issam Mansour: Data curation (supporting); Resources (supporting). Lotfi Chouchane: Conceptualization (equal); Project administration (supporting); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Aouatef Ismail Chouchane: Conceptualization (equal); Funding acquisition (lead); Project administration (lead); Supervision (lead); Writing‐original draft (equal); Writing‐review & editing (equal).
Supporting information
Fig S1
Table S1
ACKNOWLEDGEMENTS
This work was supported by Sidra Medicine, Qatar and Open Access funding provided by the Qatar National Library.
Umar M, Megarbane A, Shan J, et al. Genome sequencing unveils mutational landscape of the familial Mediterranean fever: Potential implications of IL33/ST2 signalling. J Cell Mol Med. 2020;24:11294–11306. 10.1111/jcmm.15701
Meenakshi Umar, Andre Megarbane and Jingxuan Shan contributed equally to this manuscript.
DATA AVAILABILITY STATEMENT
All the variants reported here have been submitted to LOVD website (https://www.lovd.nl).
REFERENCES
- 1. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. 2017;18:832‐842. [DOI] [PubMed] [Google Scholar]
- 2. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next‐of‐kin. Nat Rev Rheumatol. 2014;10:135‐147. [DOI] [PubMed] [Google Scholar]
- 3. Jamilloux Y, Belot A, Magnotti F, et al. Geoepidemiology and immunologic features of autoinflammatory diseases: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:454‐479. [DOI] [PubMed] [Google Scholar]
- 4. Onen F. Familial Mediterranean fever. Rheumatol int. 2006;26:489‐496. [DOI] [PubMed] [Google Scholar]
- 5. Bernot A, Clepet C, Dasilva C, et al. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25. [DOI] [PubMed] [Google Scholar]
- 6. International FMF Consortium . Ancient missense pathogenic variants in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797‐807. [DOI] [PubMed] [Google Scholar]
- 7. Centola M, Wood G, Frucht DM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223‐3231. [PubMed] [Google Scholar]
- 8. Diaz A, Hu C, Kastner DL, et al. Lipopolysaccharide‐induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C‐terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum. 2004;50:3679‐3689. [DOI] [PubMed] [Google Scholar]
- 9. Ben‐Chetrit E, Levy M. Familial mediterranean fever. Lancet. 1998;351:659‐664. [DOI] [PubMed] [Google Scholar]
- 10. Aksentijevich I, Torosyan Y, Samuels J, et al. Pathogenic variant and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Ashkenazi Jewish population. Am J Hum Genet. 1999;64:949‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarrabay G, Touitou I. Dominant familial Mediterranean fever. Rheumatology (Oxford). 2017;56:173‐175. [DOI] [PubMed] [Google Scholar]
- 12. Booty MG, Chae JJ, Masters SL, et al. Familial Mediterranean fever with a single MEFV pathogenic variant: where is the second hit? Arthritis Rheum. 2009;60:1851‐1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ben‐Zvi I, Herskovizh C, Kukuy O, et al. Familial Mediterranean fever without MEFV pathogenic variants: a case‐control study. Orphanet J Rare Dis. 2015;10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turkcapar N, Tuncalı T, Kutlay S, et al. The contribution of genotypes at the MICA gene triplet repeat polymorphisms and MEFV pathogenic variants to amyloidosis and course of the disease in the patients with familial Mediterranean fever. Rheumatol Int. 2007;27:545‐551. [DOI] [PubMed] [Google Scholar]
- 15. Cazeneuve C, Ajrapetyan H, Papin S, et al. Identification of MEFV‐independent modifying genetic factors for familial Mediterranean fever. Am J Hum Genet. 2000;67:1136‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879‐1885. [DOI] [PubMed] [Google Scholar]
- 17. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 2012;6:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Genomes Project Consortium . A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense pathogenic variants. Nat Methods. 2010;7:248‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073‐1081. [DOI] [PubMed] [Google Scholar]
- 26. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Makarov V, O'Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics. 2012;28:724‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Medlej‐Hashim M, Chouery E, Salem N, et al. Familial Mediterranean fever in a large Lebanese family: multiple MEFV pathogenic variants and evidence for a Founder effect of the p.[M694I] pathogenic variant. Eur J Med Genet. 2011;54:50‐54. [DOI] [PubMed] [Google Scholar]
- 29. Jalkh N, Génin E, Chouery E, et al. Familial Mediterranean fever in Lebanon: founder effects for different MEFV pathogenic variants. Ann Hum Genet. 2008;72:41‐47. [DOI] [PubMed] [Google Scholar]
- 30. Chang CC, Chow CC, Tellier LC, et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pedersen BS, Quinlan AR. Who’s Who? Detecting and resolving sample anomalies in human DNA sequencing studies with Peddy. Am J Hum Genet. 2017;100:406‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milhavet F, Cuisset L, Hoffman HM, et al. The infevers autoinflammatory pathogenic variant online registry: update with new genes and functions. Hum Mutat. 2008;29:803‐808. [DOI] [PubMed] [Google Scholar]
- 33. Stenson PD, Ball EV, Mort M, et al. Human Gene Pathogenic variant Database (HGMD). Hum Mutat. 2003;6:577‐581. [DOI] [PubMed] [Google Scholar]
- 34. Saveanu L, Carroll O, Lindo V, et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol. 2005;6:689‐697. [DOI] [PubMed] [Google Scholar]
- 35. Karacan İ, Uğurlu S, Tolun A, et al. Other autoinflammatory disease genes in an FMF‐prevalent population: a homozygous MVK pathogenic variant and a novel heterozygous TNFRSF1A pathogenic variant in two different Turkish families with clinical FMF. Clin Exp Rheumatol. 2017;35(Suppl 108):75‐81. [PubMed] [Google Scholar]
- 36. Neocleous V, Byrou S, Toumba M, et al. Evidence of digenic inheritance in autoinflammation‐associated genes. J Genet. 2016;95:761‐766. [DOI] [PubMed] [Google Scholar]
- 37. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Filipovich AH, Chandrakasan S. Pathogenesis of Hemophagocytic Lymphohistiocytosis Filipovich AH. Hematol Oncol Clin North Am. 2015;29:895‐902. [DOI] [PubMed] [Google Scholar]
- 39. de Jesus AA, Canna SW, Liu Y, Goldbach‐Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. 2015;33:823‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Werenskiold AK, Hoffmann S, Klemenz R. Induction of a mitogen‐responsive gene after expression of the Ha‐ras oncogene in NIH 3T3 fibroblasts. Mol Cell Biol. 1989;9:5207‐5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tominaga S. A putative protein of a growth specific cDNA from BALB/c‐3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. 1989;258:301‐304. [DOI] [PubMed] [Google Scholar]
- 42. Griesenauer B, Paczesny S. The ST2/IL‐33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Díaz‐Jiménez D, De la Fuente M, Dubois‐Camacho K, et al. Soluble ST2 is a sensitive clinical marker of ulcerative colitis evolution. BMC Gastroenterol. 2016;16:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caselli C. Inflammation in cardiac disease: focus on Interleukin‐33/ST2 pathway. Inflamm Cell Signal. 2014;1:e149. [Google Scholar]
- 45. Gangemi S, Manti S, Procopio V, et al. Lack of clear and univocal genotype‐phenotype correlation in familial Mediterranean fever patients: A systemic review. Clin Genet. 2018;94:81‐94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Data Availability Statement
All the variants reported here have been submitted to LOVD website (https://www.lovd.nl).