

Summary
Misfolded or unfolded proteins in the ER are said to be degraded only after translocation or isolation from the ER. Here, we describe a mechanism by which mutant proteins are degraded within the ER. Aggregates of mutant arginine vasopressin (AVP) precursor were confined to ER-associated compartments (ERACs) connected to the ER in AVP neurons of a mouse model of familial neurohypophysial diabetes insipidus. The ERACs were enclosed by membranes, an ER chaperone and marker protein of phagophores and autophagosomes were expressed around the aggregates, and lysosomes fused with the ERACs. Moreover, lysosome-related molecules were present within the ERACs, and aggregate degradation within the ERACs was dependent on autophagic-lysosomal activity. Thus, we demonstrate that protein aggregates can be degraded by autophagic-lysosomal machinery within specialized compartments of the ER.
Subject Areas: neuroscience, cell biology, technical aspects of cell biology
Graphical Abstract
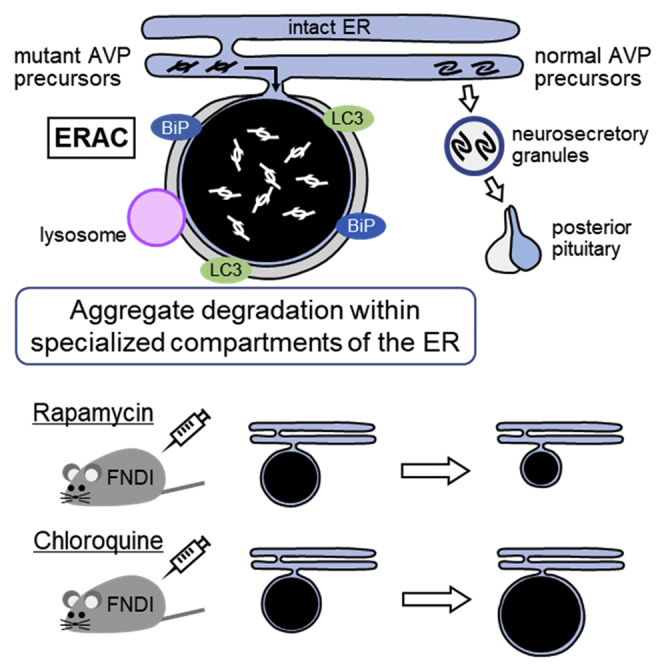
Highlights
-
•
Mutant AVP precursors are confined to ERACs connected to the ER of FNDI AVP neurons
-
•
Lysosomes fuse with ERACs surrounded by phagophore-like membranes
-
•
Lysosome-related molecules are localized within ERACs
-
•
Rapamycin reduces and chloroquine increases protein aggregate accumulation in ERACs
neuroscience; cell biology; technical aspects of cell biology
Introduction
The ER is an organelle mainly responsible for the synthesis, folding, assembly, and transport of proteins (Kaufman, 1999). While properly folded proteins are packed into secretory granules as secretory proteins or transported to the cellular membrane as membrane proteins through the Golgi apparatus (Braakman and Bulleid, 2011; Gidalevitz et al., 2013), misfolded or unfolded proteins accumulate in the ER causing ER stress (Hetz, 2012; Schroder and Kaufman, 2005; Wang and Kaufman, 2012). The unfolded protein response, including ER-associated degradation (ERAD), is a cellular mechanism by which ER stress is reduced. Through the ERAD machinery, misfolded or unfolded proteins are translocated from the ER to the cytosol and degraded by the ubiquitin-proteasome system (UPS) (Guerriero and Brodsky, 2012; Qi et al., 2017; Smith et al., 2011). In addition, a growing body of evidence has accumulated regarding ER-phagy machinery targeting protein aggregates in the ER lumen that cannot be degraded by ERAD (Fregno and Molinari, 2018; Smith and Wilkinson, 2017; Song et al., 2018; Wilkinson, 2019). In macro-ER-phagy, an isolation membrane called a phagophore sequesters a portion of the ER containing the aggregates to form an autophagosome, which fuses with a lysosome to degrade the contents (Cunningham et al., 2019; Forrester et al., 2019; Schultz et al., 2018). Recent studies have also reported another type of ER-phagy, micro-ER-phagy, in which a lysosome engulfs or fuses with aggregate-containing ER buds independent of autophagosome biogenesis (Fregno et al., 2018; Omari et al., 2018). In either case of ERAD or ER-phagy, aggregates in the ER are degraded only after translocation or isolation from the ER.
Arginine vasopressin (AVP), an antidiuretic hormone, is synthesized in magnocellular neurons of the supraoptic nuclei (SON) and paraventricular nuclei (PVN) in the hypothalamus (Bisset and Chowdrey, 1988). The AVP gene encodes a signal peptide, AVP, the AVP carrier protein neurophysin II (NPII), and a glycoprotein, also referred to as copeptin (Sausville et al., 1985). Upon removal of the signal peptide, prepro-AVP is truncated to pro-AVP, which is folded into its native conformation in the ER and then is packed into secretory granules. AVP, NPII, and the glycoprotein are cleaved from pro-AVP in the vesicle during transport to the posterior pituitary, from which AVP is released into systemic circulation in response to changes in plasma osmolality and blood pressure (Brownstein et al., 1980; Burbach et al., 2001). AVP is also known to be released from dendrites and to modulate the phasic activity of AVP neurons by autocrine regulation in order to maximize AVP secretion efficiency (Ludwig and Leng, 2006; Pow and Morris, 1989).
Familial neurohypophysial diabetes insipidus (FNDI) is an autosomal dominant disease caused by mutations in the AVP gene locus, predominantly in the region encoding NPII (Arima et al., 2016; Babey et al., 2011; Christensen and Rittig, 2006). We previously generated FNDI model mice by introducing an NPII mutation (that causes FNDI in humans) into the AVP gene locus, and the resulting heterozygous mice recapitulated the phenotypes of patients with FNDI. Owing to AVP deficiency, urine volumes and water intake were significantly increased, and urine osmolality was significantly reduced in FNDI mice compared with wild-type mice (Hayashi et al., 2009). In the AVP neurons of FNDI mice, inclusion bodies were present and increased in size as the mice aged in proportion to the increase in urine volumes (Hayashi et al., 2009) and decreased in size when the FNDI mice were treated with desmopressin (Hiroi et al., 2010), an AVP agonist (Edwards et al., 1973). Electron microscopic analyses of AVP neurons in FNDI mice revealed that aggregates were confined to a specific compartment of the rough ER, termed the ERAC (ER-associated compartment) (Hagiwara et al., 2014). Despite the presence of massive aggregates in the ER of AVP neurons in FNDI mice, there was no significant difference in the expression levels of an ER chaperone immunoglobulin heavy chain binding protein (BiP) in AVP neurons between wild-type and FNDI mice at 3 months of age. This suggests that the pathophysiological significance of ERACs is their ability to maintain function in the remainder of the ER by sequestering and confining aberrant proteins to the ERAC (Hagiwara et al., 2014). However, it remains to be elucidated whether ERACs are connected to the intact ER lumen or if there are any mechanisms by which aggregates are degraded within the ERACs.
In the present study, we hypothesized that aggregates are degraded by lysosomes within the ERACs which maintain connection to the intact ER lumen. To test this hypothesis, we investigated the following: (1) the structural relationships between ERACs, ER membranes, and lysosomes by serial block-face scanning electron microscopy (SBF-SEM), (2) the localization of several molecules involved in autophagic-lysosomal degradation, as well as ER chaperones by immunoelectron microscopy, and (3) the pharmacological effects of inducing or inhibiting the autophagic-lysosomal degradation system on ERAC formation in AVP neurons of FNDI mice.
Results
ERACs Are Connected to the Intact ER Lumen
In order to elucidate the detailed structural relationships between organelles, three-dimensional electron microscopic analyses were performed by acquiring several sets of serial electron microscopic images from the SON of FNDI mice using SBF-SEM. Each dataset spanned volumes of approximately 30–50 μm x 30–40 μm x 16–20 μm at subcellular resolution (Figure 1A). These datasets included SON neurons which possessed electron-dense ERACs located in their cytosol and surrounded by various organelles including the rough ER (Figure 1B). Detailed analyses of these serial images showed that the ERACs were enclosed by membranes of high electron density surrounded by an electron-lucent area and that they frequently had small protrusions (Figure 2A). Careful tracing revealed that these ERAC-derived protrusions were often connected to the intact ER lumen (Figures 2A3, 2B1-4, and 2C, and Video S1). A montage image of two angularly connected electron microscopic images reconstructed from the original serial images also showed the connection between the ERAC and intact ER lumen (Figure S1). In addition, another protrusion originating from the ER-connected ERAC was also found connected to electron-dense lysosomes in the cytosol (Figures 2D and 2E, and Video S2). ERACs containing a relatively small amount of aggregates were also fused with lysosomes but not via the protrusions (Figure S2, and Videos S3 and S4). These results suggest that ERACs have direct connections with both the ER lumen and lysosomes in the cytosolic compartment of AVP neurons and that mutant aggregates in the ER could undergo degradation by lysosomes from relatively early stages of ERAC formation.
Figure 1.

SBF-SEM Analysis of the SON from FNDI Mice
(A) Three-dimensional reconstruction of a brain volume obtained from serial electron microscopic images of the SON from FNDI mice. An AVP neuron (green) is observed on the surface of the block. Scale bars: 5 μm.
(B) Two adjacent AVP neurons (blue and light blue) in the SBF-SEM image which have large electron-dense ERACs (asterisks, colored red in inset) and surrounding intact rough ER (inset, arrowheads). The boxed region is shown at higher magnification (inset). Scale bars: 5 μm.
Figure 2.

ERACs Are Connected to the Intact ER Lumen and Lysosomes
(A–E) Serial images of an ERAC (A, red) with multiple small protrusions (A, arrows). The boxed regions in A1-3 are magnified in the inset (A2) or in other panels (B4 and D3). Magnified serial images of one ERAC protrusion connected to the intact ER lumen (B1-4, B1′-4′, arrows). The compartments associated with the connection are colored red (B1′-4′). Three-dimensional reconstruction of an ERAC (C, red) and intact ER (C, blue). Magnified serial electron microscopic images (D1-12) and 3D reconstruction (E) of one protrusion (D2-9, arrowheads, colored red) connected to an ERAC (partly colored red in D1-3) which is shown to be connected to the ER in B and C. The tip of the protrusion (D10-12, white arrowheads) exposed to the cytosol is connected to a lysosome (D9-12, colored purple). A three-dimensional reconstruction of an ERAC (E, red) and a lysosome (E, purple) is shown. The numbers in the upper-right corners indicate the respective slice within the electron microscopic image stack. Scale bars: 1 μm (A1) and 500 nm (B1, C, D1, and E). See also Figures S1 and S2 and Videos S1 and S2.
Red and blue represent an ERAC and the intact ER, respectively.
Red and purple represent an ERAC and a lysosome, respectively.
Red, blue, and purple represent an ERAC, the intact ER, and lysosomes, respectively.
Red and purple represent an ERAC and lysosomes, respectively.
Mutant NPII Is Confined within ERACs of AVP Neurons in FNDI Mice
To distinguish normal from mutant NPII, we used two types of antibodies, as described previously (Ben-Barak et al., 1984, 1985; Hayashi et al., 2009). In AVP neurons of FNDI mice, immunofluorescence staining showed that mutant NPII was expressed in round structures; in contrast, there were no normal NPII signals within these structures (Figures 3A–3C). Immunoelectron microscopy demonstrated that mutant NPII was expressed in ERACs (Figures 3D and 3E) and in aggregates in the ER lumen (Figure 3G), suggesting that immunohistochemical staining of mutant NPII in areas other than the round structures represents regions in the ER where mutant NPII is also present. Mutant NPII was not expressed in the electron-lucent area around the aggregates (Figure 3D). While our previous study clearly demonstrated that aggregates were surrounded by membranous structures studded with ribosomes (Morishita et al., 2011), this is less clear in the immunoelectron microscopic analyses in which the membranous structures may be difficult to maintain. Normal NPII was found in neurosecretory granules of the cell bodies in AVP neurons and in the nerve terminals of the posterior pituitary, but not in the ER, in both FNDI (Figures 3F–3I) and wild-type mice (Figure S3). In contrast, mutant NPII was not observed in either neurosecretory granules or the nerve terminals of the posterior pituitary in FNDI mice (Figures 3D–3I). These data indicate that mutant AVP precursors are confined to ERACs and not subjected to proper cellular trafficking out of the ER.
Figure 3.

Mutant NPII Is Confined within ERACs in AVP Neurons of FNDI Mice
(A–C) Immunofluorescence staining for mutant (green) and normal (magenta) NPII, as well as DAPI (blue) in the SON of FNDI mice. Higher magnification images of the boxed areas in the left panels are shown at right. Scale bars: 50 μm (left panels) and 10 μm (right panels).
(D–I) Immunoelectron microscopic analysis of mutant NPII (10 nm gold particles) and normal NPII (15 nm gold particles) in AVP neurons from the SON (D–G) and posterior pituitary (H and I) of FNDI mice. (D) An ERAC and a vesicle containing neurosecretory granules are shown. (G) Aggregates in the ER lumen. Higher magnification images of the boxed areas in D and H are shown in E, F, and I, respectively. The white arrowheads in F, G, and I indicate neurosecretory granules containing AVP. m: mitochondria. Scale bars: 100 nm (D, E, F, and I) and 500 nm (G and H).
See also Figure S3.
ERACs Are Surrounded by Phagophore-like Membranes Derived from the ER
Immunofluorescent signals for BiP appeared to surround the round structures that were positive for mutant NPII antibodies in AVP neurons of FNDI mice (Figures 4A–4C). Immunoelectron microscopic analyses revealed that BiP was expressed in aggregate-surrounding areas including the electron-lucent regions (Figures 4D and 4E). Our data, derived using immunoelectron microscopy, also showed that both mutant NPII (Figure 3G) and BiP (Figure S4) were expressed in the ER of AVP neurons of FNDI mice, suggesting that the overlapping areas between mutant NPII and BiP immunostaining in areas other than the round structures likely correspond to the ER. In wild-type mice, BiP was expressed in the ER of AVP neurons (Figures S3A–S3C). Green fluorescent protein (GFP)–labeled microtubule-associated protein 1 light chain 3 (LC3) was also observed surrounding these rounded structures that were positively stained with mutant NPII antibodies in the AVP neurons of FNDI/GFP-LC3 mice (Figures 4F–4H) and was also detected around aggregates based on immunoelectron microscopy (Figure 4I). In contrast, immunofluorescent signals for GFP-LC3 were not visible in AVP neurons of GFP-LC3 mice (Figure S5). These findings suggest that ERACs are surrounded by membranes characteristic of phagophores derived from the ER.
Figure 4.

ERACs Are Surrounded by Membranes that Express BiP and LC3
(A–C) Immunofluorescence staining for mutant NPII (green) and BiP (magenta) in the SON of FNDI mice. Higher magnification images of the boxed areas in the left panels are shown at right. Scale bars: 50 μm (left panels) and 10 μm (right panels).
(D and E) Immunoelectron microscopic analysis for BiP (10 nm gold particles) and normal NPII (15 nm gold particles) in AVP neurons from the SON of FNDI mice. Higher magnification image of the boxed area in D is shown in E. m: mitochondria, Gol: Golgi apparatus. Scale bars: 500 nm (D) and 100 nm (E).
(F–H) Immunofluorescence staining for GFP-LC3 (green) and mutant NPII (magenta) in the SON of FNDI/GFP-LC3 mice. Higher magnification images of the boxed areas in the left panels are shown at right. Scale bars: 50 μm (left panels) and 10 μm (right panels).
(I) Immunoelectron microscopic analysis of GFP-LC3 (12 nm gold particles, white arrowheads) in AVP neurons from the SON of FNDI/GFP-LC3 mice. A small portion of an ERAC is shown. Scale bar: 100 nm.
See also Figures S3–S5.
Lysosome-Related Molecules Are Expressed within ERACs
Immunofluorescence images demonstrated that lysosome-associated membrane protein 2 (LAMP2) (Figures 5A–5C) and cathepsin D (Figures 5D–5F) were localized within the round mutant NPII-positive structures in AVP neurons of FNDI mice, while these signals were not observed in the AVP neurons of wild-type mice (Figure S6). Further analyses using immunoelectron microscopy revealed that LAMP2 and cathepsin D were present inside the ERACs (Figures 5G and 5H) but not in the electron-lucent areas around the aggregates, suggesting that lysosomes were incorporated into ERACs. Furthermore, the overlap between mutant NPII and LAMP2 immunostaining in areas other than the round structures suggests that lysosomes also fuse to the ER where relatively small aggregates are found, as shown in Figure S2. On the other hand, the finding that cathepsin D was preferentially located in the aggregates within ERACs suggests the possibility that lysosomal acid hydrolases including cathepsin D are confined to ERACs and do not spread into the intact ER lumen.
Figure 5.

Lysosome-Related Molecules Are Expressed within ERACs
(A–F) Immunofluorescence staining for mutant NPII (green) and LAMP2 or cathepsin D (magenta) in the SON of FNDI mice. Higher magnification images of the boxed areas in the left panels are shown at right. Scale bars: 50 μm (left panels) and 10 μm (right panels).
(G and H) Immunoelectron microscopic analysis of LAMP2 (G, 10 nm gold particles, white arrowheads) or cathepsin D (H, 10 nm gold particles, white arrowheads) in AVP neurons from the SON of FNDI mice. The large images show a small portion of an ERAC; higher magnification images of the boxed areas are shown in the insets at lower left. Scale bars: 500 nm.
See also Figure S6.
The Accumulation of Aggregates within ERACs Is reduced by the Autophagy Inducer Rapamycin and Increased by the Lysosome Inhibitor Chloroquine
To examine whether peripheral injection of rapamycin and chloroquine could act on the hypothalamus, LC3 conversion (LC3-I to LC3-II) in the hypothalamus of wild-type mice injected with rapamycin or chloroquine was analyzed by immunoblotting. The LC3-II/LC3-I ratio was increased in the rapamycin group compared with the control (Figure S7A), suggesting that autophagic flux was increased in the hypothalamus of mice treated with the autophagy inducer rapamycin. The LC3-II/LC3-I ratio was also increased in the chloroquine group compared with the control (Figure S7B). This is consistent with previous studies showing that LC3-II, a substitute of autophagic degradation, was increased relative to LC3-I when autophagic degradation was inhibited by the lysosome inhibitor chloroquine (Han et al., 2019).
In FNDI mice, the number of inclusion bodies over 4.5 μm in diameter [the mean size in 3-month-old FNDI mice (Hagiwara et al., 2014)] was significantly decreased by rapamycin treatment (Figure 6A), while the number of inclusion bodies was significantly increased by chloroquine administration (Figure 6B).
Figure 6.

Accumulation of Aggregates within ERACs Is Reduced by the Autophagy Inducer Rapamycin and Increased by the Lysosome Inhibitor Chloroquine
(A–D) Representative images of immunohistochemical staining for mutant NPII in the SON and the number of inclusion bodies with a diameter >4.5 μm in the SON of 3-month-old FNDI mice in the control and rapamycin (Rapa, A and B) or chloroquine (CQ, C and D) or groups. Results are expressed as means ± SE; n = 5–7 animals per group. White arrowheads indicate the inclusion bodies. Scale bars: 50 μm.
(E) Possible mechanisms of ERAC formation and mutant NPII degradation in AVP neurons of FNDI mice. Mutant NPII is confined to the ERACs of AVP neurons in FNDI mice. The ER chaperone BiP was localized around the ERACs, indicating that ERACs are connected to the ER and that BiP might be associated with ERAC formation. Furthermore, LAMP2 and cathepsin D were expressed in ERACs surrounded by membranes with LC3, suggesting that lysosomes degrade mutant NPII within ERACs which are connected to the ER. mNPII: mutant NPII.
See also Figure S7.
Discussion
In the present study, we showed that ERACs, in which mutant NPII was accumulated, were connected to the intact ER and enclosed by membranes. We also showed that BiP and LC3 were expressed around the aggregates and that LAMP2 and cathepsin D were present within the ERACs. Furthermore, our data showed that the number of ERACs was decreased or increased by rapamycin or chloroquine treatment, respectively. Figure 6E summarizes the findings of our present study.
The formation of ERAC-like structures has been reported not only in FNDI but also in other diseases, such as α1-antitrypsin deficiency (Granell et al., 2008), familial encephalopathy with neuroserpin inclusion bodies (Hagen et al., 2011), seipinopathy (Ito et al., 2012), and autosomal dominant retinitis pigmentosa (Chiang et al., 2012; Saliba et al., 2002). While previous studies including ours suggested that aggregates were confined to a subcompartment of the ER, it has been unclear whether there is any communication between the ER and these compartments. In the present study, we demonstrated that ERACs were connected to the intact ER lumen using SBF-SEM. Furthermore, our data showed that membranes enclosed the aggregates, around which an ER chaperone (BiP) was expressed, suggesting that the ERAC membranes were derived from the ER. As ERAC formation reportedly mitigates ER stress and improves cellular viability not only in FNDI (Hagiwara et al., 2014) but also in α1-antitrypsin deficiency (Granell et al., 2008), it could be a common unfolded protein response shared by several cell types for coping with ER stress. Whether aggregates within ERACs are degraded by autophagic-lysosomal machinery in other diseases remains to be elucidated.
The finding that mutant NPII was not observed in the neurosecretory granules but was exclusively within ERACs and small aggregates in the ER lumen is consistent with previous in vivo and in vitro studies showing that mutant AVP precursors, resulting from various AVP gene mutations, also accumulated in the ER (Birk et al., 2009; Ito and Jameson, 1997; Ito et al., 1999; Si-Hoe et al., 2000). In contrast, normal NPII was undetectable in ERACs of FNDI mice. This finding does not exclude the possibility that normal NPII exists in ERACs, given that the anti-normal NPII antibody PS41 can only detect the normal AVP precursor where it is sufficiently concentrated. This is supported by the findings in wild-type mice in which PS41 could detect normal NPII in the dense-core neurosecretory granules but not in the ER.
In this study, we showed that lysosomes were fused to the ERAC membrane and that LAMP2 as well as cathepsin D were found within ERACs. While previous studies also suggested that a lysosome might be involved in the degradation of mutant AVP precursor proteins (Castino et al., 2005; Davies and Murphy, 2002), our data suggest that mutant protein aggregates are degraded within the ER by autophagic-lysosomal machinery. A clear difference between macro-ER-phagy and ERAC degradation, as shown in the present study, is that ERAC degradation occurs in a compartment derived from the ER that maintains connection to the ER, whereas in macro-ER-phagy, the ER contents are degraded together with the ER itself following the complete sequestration by phagophores. We showed that aggregates were surrounded by membranes of high electron density based on SEM. Similarly, a previous study reported that the membranes of phagophores showed high electron density when observed by SBF-SEM, although the composition of this high density material has yet to be determined (Arai and Waguri, 2019). Our data also revealed that LC3 was expressed around the aggregates and that rapamycin, which is known to accelerate the formation of phagophores (Vakifahmetoglu-Norberg et al., 2015), increased the degradation of aggregates surrounded by the membranes. Combined, these data suggest that the membranes possess characteristics of phagophores (Kabeya et al., 2000; Mizushima and Komatsu, 2011; Mizushima et al., 2004) and indicate that ERAC degradation is different to micro-ER-phagy. To the best of our knowledge, this is the first report showing that accumulated aggregates are degraded within specialized compartments of the ER. While previous in vitro studies suggested that phagophores originate from the ER (Axe et al., 2008; Graef et al., 2013; Hamasaki et al., 2013; Hayashi-Nishino et al., 2009; Uemura et al., 2014), our data clearly show that this is also the case for the phagophore-like membrane surrounding ERACs to which the lysosomes fuse to form the autolysosome-like structures.
SBF-SEM analyses suggested that lysosomes were fused to the ERACs via ERAC protrusions in some AVP neurons. Given that not only protein aggregates but also the lysosomal acid hydrolase cathepsin D is localized exclusively in ERACs, some mechanisms should exist by which protein aggregates and lysosome-related molecules are confined to ERACs in order to protect the remainder of the intact ER lumen. The electron-lucent areas shown in the electron microscopic analyses have also been observed in our previous studies (Hayashi et al., 2009). Here, we showed that BiP is expressed in these electron-lucent areas. Thus, it is possible that molecules related to ERAC formation and confinement of aggregates as well as lysosomal acid hydrolases to the ERACs might be present in this region, although further studies are required to clarify the underlying mechanisms.
We previously reported that macroautophagy or cell death was not observed as long as mutant NPII was confined to the ERACs in FNDI mice with free access to water (Hagiwara et al., 2014). Conversely, ERAC formation was disrupted and mutant NPII aggregates were spread throughout the ER lumen after intermittent water deprivation, leading to autophagy-associated cell death of AVP neurons in FNDI mice (Hagiwara et al., 2014, 2019). Taken together, it seems that ERAC formation as well as degradation of mutant proteins within ERACs is essential for protecting AVP neurons from cell death in FNDI.
In general, misfolded or unfolded proteins in the ER are targeted for ERAD, in which substrates are translocated from the ER to the cytosol and degraded by UPS (Guerriero and Brodsky, 2012; Qi et al., 2017; Smith et al., 2011). In AVP neurons, wild-type AVP precursors are reported to undergo proteasomal degradation (Friberg et al., 2004). Furthermore, a recent study demonstrated that deficiency of the Sel1L-Hrd1 protein complex, a principal ER-resident E3 ligase in mammalian ERAD, caused marked retention and aggregation of wild-type AVP precursors in the ER, resulting in polyuria due to AVP deficiency (Shi et al., 2017). While these findings indicate that ERAD is essential to the cellular function of AVP neurons, our present data showed that, in addition to ERAD, there exists another mechanism by which protein aggregates could be degraded without translocation from the ER to the cytosol in AVP neurons.
In conclusion, our data demonstrate that mutant proteins undergo autophagic-lysosomal degradation within ERACs, without isolation or translocation from the ER, in AVP neurons of FNDI mice.
Limitations of the Study
It is unclear from this study how aggregates and lysosomal acid hydrolases are confined to the ERACs. Furthermore, the roles of the protrusions in the ERACs and the electron-lucent areas around the aggregates also remain to be determined.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hiroshi Arima (arima105@med.nagoya-u.ac.jp).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets or code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Dr. Noboru Mizushima (Tokyo University) for providing the GFP-LC3 transgenic mice and Atsuko Imai, Nobuko Hattori (National Institute for Physiological Sciences), and Michiko Yamada for their helpful technical assistance. This work was supported by JSPS KAKENHI Grant Number JP15K19530 (to D.H.); JP16H06280 (to D.H. and H.S.), Grant-in-Aid for Scientific Research on Innovative Areas — Platforms for Advanced Technologies and Research Resources “Advanced Bioimaging Support”; Alexander von Humboldt Foundation Research Fellowship (to D.H.); the Acceleration Program for Intractable Diseases Research utilizing Disease-specific iPS cells (to H.S.) of the Research Center Network for Realization of Regenerative Medicine from the Japanese Agency for Medical Research and Development (AMED); Suzuken Memorial Foundation (to H.A.); and Cooperative Study Programs of National Institute for Physiological Sciences (to H.A. and N.O.).
Author Contributions
T. Miyata, D.H., H. Sakamoto, N.O., and H.A. designed the studies and wrote the manuscript. T. Miyata, Y.H., T. Miwata, Y.K., J.K., H.O., K.M., H.T., H. Suga, T.K., M.S., T.O., Y.I., S.I., and R.B. performed most of the experiments. N.K. and H. Sakamoto performed the immunoelectron microscopy experiments. M.M. and N.O. performed the SBF-SEM analysis. All authors discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101648.
Contributor Information
Daisuke Hagiwara, Email: d-hagiwara@med.nagoya-u.ac.jp.
Hiroshi Arima, Email: arima105@med.nagoya-u.ac.jp.
Supplemental Information
References
- Arai R., Waguri S. Improved electron microscopy fixation methods for tracking autophagy-associated membranes in cultured mammalian cells. Methods Mol. Biol. 2019;1880:211–221. doi: 10.1007/978-1-4939-8873-0_13. [DOI] [PubMed] [Google Scholar]
- Arima H., Azuma Y., Morishita Y., Hagiwara D. Central diabetes insipidus. Nagoya J. Med. Sci. 2016;78:349–358. doi: 10.18999/nagjms.78.4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babey M., Kopp P., Robertson G.L. Familial forms of diabetes insipidus: clinical and molecular characteristics. Nat. Rev. Endocrinol. 2011;7:701–714. doi: 10.1038/nrendo.2011.100. [DOI] [PubMed] [Google Scholar]
- Ben-Barak Y., Russell J.T., Whitnall M., Ozato K., Gainer H. Phylogenetic cross-reactivities of monoclonal antibodies produced against rat neurophysin. Cell Mol. Neurobiol. 1984;4:339–349. doi: 10.1007/BF00733596. [DOI] [PubMed] [Google Scholar]
- Ben-Barak Y., Russell J.T., Whitnall M.H., Ozato K., Gainer H. Neurophysin in the hypothalamo-neurohypophysial system. I. Production and characterization of monoclonal antibodies. J. Neurosci. 1985;5:81–97. doi: 10.1523/JNEUROSCI.05-01-00081.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk J., Friberg M.A., Prescianotto-Baschong C., Spiess M., Rutishauser J. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J. Cell Sci. 2009;122:3994–4002. doi: 10.1242/jcs.051136. [DOI] [PubMed] [Google Scholar]
- Bisset G.W., Chowdrey H.S. Control of release of vasopressin by neuroendocrine reflexes. Q. J. Exp. Physiol. 1988;73:811–872. doi: 10.1113/expphysiol.1988.sp003223. [DOI] [PubMed] [Google Scholar]
- Braakman I., Bulleid N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Brownstein M.J., Russell J.T., Gainer H. Synthesis, transport, and release of posterior pituitary hormones. Science. 1980;207:373–378. doi: 10.1126/science.6153132. [DOI] [PubMed] [Google Scholar]
- Burbach J.P., Luckman S.M., Murphy D., Gainer H. Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol. Rev. 2001;81:1197–1267. doi: 10.1152/physrev.2001.81.3.1197. [DOI] [PubMed] [Google Scholar]
- Castino R., Davies J., Beaucourt S., Isidoro C., Murphy D. Autophagy is a prosurvival mechanism in cells expressing an autosomal dominant familial neurohypophyseal diabetes insipidus mutant vasopressin transgene. FASEB J. 2005;19:1021–1023. doi: 10.1096/fj.04-3162fje. [DOI] [PubMed] [Google Scholar]
- Chiang W.C., Messah C., Lin J.H. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol. Biol. Cell. 2012;23:758–770. doi: 10.1091/mbc.E11-08-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J.H., Rittig S. Familial neurohypophyseal diabetes insipidus--an update. Semin. Nephrol. 2006;26:209–223. doi: 10.1016/j.semnephrol.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Cunningham C.N., Williams J.M., Knupp J., Arunagiri A., Arvan P., Tsai B. Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol. Cell. 2019;75:442–456.e4. doi: 10.1016/j.molcel.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J., Murphy D. Autophagy in hypothalamic neurones of rats expressing a familial neurohypophysial diabetes insipidus transgene. J. Neuroendocrinol. 2002;14:629–637. doi: 10.1046/j.1365-2826.2002.00822.x. [DOI] [PubMed] [Google Scholar]
- Edwards C.R., Kitau M.J., Chard T., Besser G.M. Vasopressin analogue DDAVP in diabetes insipidus: clinical and laboratory studies. Br. Med. J. 1973;3:375–378. doi: 10.1136/bmj.3.5876.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester A., De Leonibus C., Grumati P., Fasana E., Piemontese M., Staiano L., Fregno I., Raimondi A., Marazza A., Bruno G. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019;38:e99847. doi: 10.15252/embj.201899847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I., Fasana E., Bergmann T.J., Raimondi A., Loi M., Solda T., Galli C., D'Antuono R., Morone D., Danieli A. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018;37:e99259. doi: 10.15252/embj.201899259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I., Molinari M. Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance. F1000Res. 2018;7:454. doi: 10.12688/f1000research.13968.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg M.A., Spiess M., Rutishauser J. Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J. Biol. Chem. 2004;279:19441–19447. doi: 10.1074/jbc.M310249200. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T., Stevens F., Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta. 2013;1833:2410–2424. doi: 10.1016/j.bbamcr.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef M., Friedman J.R., Graham C., Babu M., Nunnari J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell. 2013;24:2918–2931. doi: 10.1091/mbc.E13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granell S., Baldini G., Mohammad S., Nicolin V., Narducci P., Storrie B. Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function. Mol. Biol. Cell. 2008;19:572–586. doi: 10.1091/mbc.E07-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero C.J., Brodsky J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen M.C., Murrell J.R., Delisle M.B., Andermann E., Andermann F., Guiot M.C., Ghetti B. Encephalopathy with neuroserpin inclusion bodies presenting as progressive myoclonus epilepsy and associated with a novel mutation in the Proteinase Inhibitor 12 gene. Brain Pathol. 2011;21:575–582. doi: 10.1111/j.1750-3639.2011.00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D., Arima H., Morishita Y., Wenjun L., Azuma Y., Ito Y., Suga H., Goto M., Banno R., Sugimura Y. Arginine vasopressin neuronal loss results from autophagy-associated cell death in a mouse model for familial neurohypophysial diabetes insipidus. Cell Death Dis. 2014;5:e1148. doi: 10.1038/cddis.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D., Grinevich V., Arima H. A novel mechanism of autophagy-associated cell death of vasopressin neurons in familial neurohypophysial diabetes insipidus. Cell Tissue Res. 2019;375:259–266. doi: 10.1007/s00441-018-2872-4. [DOI] [PubMed] [Google Scholar]
- Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Han Y., Wang S., Wang Y., Zeng S. IGF-1 inhibits apoptosis of porcine primary granulosa cell by targeting degradation of Bim(EL) Int. J. Mol. Sci. 2019;20:5356. doi: 10.3390/ijms20215356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M., Arima H., Ozaki N., Morishita Y., Hiroi M., Nagasaki H., Kinoshita N., Ueda M., Shiota A., Oiso Y. Progressive polyuria without vasopressin neuron loss in a mouse model for familial neurohypophysial diabetes insipidus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;296:R1641–R1649. doi: 10.1152/ajpregu.00034.2009. [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M., Fujita N., Noda T., Yamaguchi A., Yoshimori T., Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hiroi M., Morishita Y., Hayashi M., Ozaki N., Sugimura Y., Nagasaki H., Shiota A., Oiso Y., Arima H. Activation of vasopressin neurons leads to phenotype progression in a mouse model for familial neurohypophysial diabetes insipidus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;298:R486–R493. doi: 10.1152/ajpregu.00529.2009. [DOI] [PubMed] [Google Scholar]
- Ito D., Yagi T., Ikawa M., Suzuki N. Characterization of inclusion bodies with cytoprotective properties formed by seipinopathy-linked mutant seipin. Hum. Mol. Genet. 2012;21:635–646. doi: 10.1093/hmg/ddr497. [DOI] [PubMed] [Google Scholar]
- Ito M., Jameson J.L. Molecular basis of autosomal dominant neurohypophyseal diabetes insipidus. Cellular toxicity caused by the accumulation of mutant vasopressin precursors within the endoplasmic reticulum. J. Clin. Invest. 1997;99:1897–1905. doi: 10.1172/JCI119357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Yu R.N., Jameson J.L. Mutant vasopressin precursors that cause autosomal dominant neurohypophyseal diabetes insipidus retain dimerization and impair the secretion of wild-type proteins. J. Biol. Chem. 1999;274:9029–9037. doi: 10.1074/jbc.274.13.9029. [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman R.J. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Ludwig M., Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat. Rev. Neurosci. 2006;7:126–136. doi: 10.1038/nrn1845. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita Y., Arima H., Hiroi M., Hayashi M., Hagiwara D., Asai N., Ozaki N., Sugimura Y., Nagasaki H., Shiota A. Poly(A) tail length of neurohypophysial hormones is shortened under endoplasmic reticulum stress. Endocrinology. 2011;152:4846–4855. doi: 10.1210/en.2011-1415. [DOI] [PubMed] [Google Scholar]
- Omari S., Makareeva E., Roberts-Pilgrim A., Mirigian L., Jarnik M., Ott C., Lippincott-Schwartz J., Leikin S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. U S A. 2018;115 doi: 10.1073/pnas.1814552115. E10099–e10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow D.V., Morris J.F. Dendrites of hypothalamic magnocellular neurons release neurohypophysial peptides by exocytosis. Neuroscience. 1989;32:435–439. doi: 10.1016/0306-4522(89)90091-2. [DOI] [PubMed] [Google Scholar]
- Qi L., Tsai B., Arvan P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 2017;27:430–440. doi: 10.1016/j.tcb.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba R.S., Munro P.M., Luthert P.J., Cheetham M.E. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- Sausville E., Carney D., Battey J. The human vasopressin gene is linked to the oxytocin gene and is selectively expressed in a cultured lung cancer cell line. J. Biol. Chem. 1985;260:10236–10241. [PubMed] [Google Scholar]
- Schroder M., Kaufman R.J. ER stress and the unfolded protein response. Mutat. Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Schultz M.L., Krus K.L., Kaushik S., Dang D., Chopra R., Qi L., Shakkottai V.G., Cuervo A.M., Lieberman A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018;9:3671. doi: 10.1038/s41467-018-06115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Somlo D.R.M., Kim G.H., Prescianotto-Baschong C., Sun S., Beuret N., Long Q., Rutishauser J., Arvan P., Spiess M. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Invest. 2017;127:3897–3912. doi: 10.1172/JCI94771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Hoe S.L., De Bree F.M., Nijenhuis M., Davies J.E., Howell L.M., Tinley H., Waller S.J., Zeng Q., Zalm R., Sonnemans M. Endoplasmic reticulum derangement in hypothalamic neurons of rats expressing a familial neurohypophyseal diabetes insipidus mutant vasopressin transgene. FASEB J. 2000;14:1680–1684. doi: 10.1096/fj.99-0892fje. [DOI] [PubMed] [Google Scholar]
- Smith M., Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61:625–635. doi: 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.H., Ploegh H.L., Weissman J.S. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Tan J., Miao Y., Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018;233:3867–3874. doi: 10.1002/jcp.26137. [DOI] [PubMed] [Google Scholar]
- Uemura T., Yamamoto M., Kametaka A., Sou Y.S., Yabashi A., Yamada A., Annoh H., Kametaka S., Komatsu M., Waguri S. A cluster of thin tubular structures mediates transformation of the endoplasmic reticulum to autophagic isolation membrane. Mol. Cell Biol. 2014;34:1695–1706. doi: 10.1128/MCB.01327-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H., Xia H.G., Yuan J. Pharmacologic agents targeting autophagy. J. Clin. Invest. 2015;125:5–13. doi: 10.1172/JCI73937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Kaufman R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. ER-phagy: shaping up and destressing the endoplasmic reticulum. FEBS J. 2019;286:2645–2663. doi: 10.1111/febs.14932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Red and blue represent an ERAC and the intact ER, respectively.
Red and purple represent an ERAC and a lysosome, respectively.
Red, blue, and purple represent an ERAC, the intact ER, and lysosomes, respectively.
Red and purple represent an ERAC and lysosomes, respectively.
Data Availability Statement
This study did not generate datasets or code.