

Abstract
Rhamnan and rhamnan sulfate are naturally occurring carbohydrates that have important biological functions and possible therapeutic applications, but studies are limited to the microheterogeneous mixtures from natural sources. This work reports the first synthesis of any sulfated rhamnan fragments and successful automation of the process with a recently developed automated solution-phase approach using N-iodosuccinimide/trimethylsilyl triflate (NIS/TMSOTf) promotor and levulinoyl ester deprotection conditions. The automated solution-phase activation/deprotection approach was initially able to create alpha 1→2, 1→3 type rhamnan di- and trisaccharide in moderate yields. Once these targets were achieved, a process to use SO3•pyridine complex in DMF for sulfation compatible with an automated solution-phase liquid handling system was developed and successfully applied to carbohydrate sulfation to create two rhamnan sulfate fragments with differing monosulfation patterns.
Keywords: Rhamnan, Rhamnan Sulfate, Solution-Phase Automated Synthesis, Sulfation
Graphical Abstract
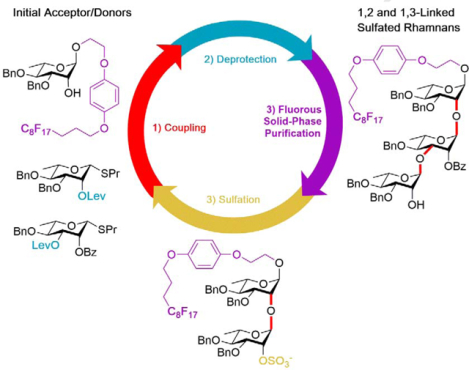
1. Introduction
The automated syntheses of peptide and nucleic acid biopolymers have become commonplace within the organic chemistry field. The ability to efficiently and rapidly access these biopolymers has been instrumental in expanding our understanding of their biological functions. It has also allowed for the exploration of these molecules in non-natural applications such as biomaterials and drug candidates. [1, 2] Unfortunately, the use of automated technology to create other types of organic molecules is still in its infancy and presents unique challenges compared to peptides or nucleic acids. Specifically, the development of automated syntheses for carbohydrates has experienced difficulties from both a chemical and practical standpoint. The extensive number of conceivable linear and branched structures significantly contributes to the complexity of carbohydrate syntheses.[3] Many of these linkages are difficult to form and require non-commercially available building blocks for precise chemical control to obtain appropriate regio- and chemoselectivity. Also, once the desired carbohydrate is formed, purification and isolation of a single structure is a time-consuming affair.
Despite these practical and chemical challenges, several automated methods have been developed for the synthesis of carbohydrates. Many automated oligosaccharide syntheses have been modeled after solid-phase peptide synthesis, which uses a polymeric or gold-based support for chain extension.[4–7] Several solution-phase automated methods have also emerged alongside the solid-phase methods. These methods include the use of electrochemical glycosylation, chemoenzymatic strategies, and the use of fluorous tags to facilitate intermediate purification.[7–16] The fluorous-tagged strategy has been successful in providing a number desired oligosaccharide targets both manually and on an automated system with efficient usage of building block equivalents.[17–20] Most recently, this fluorous-tag-based approach was used to create an efficient automated process for the synthesis of an α−1→2 rhamnan di- and trisaccharide. The developed process focused on adapting a commonly used N-iodosuccinimide/trimethylsilyltriflate (NIS/TMSOTf) promoter system for thioglycoside activation for use on a solution-phase handling platform.[21] In addition, it was previously shown that it was possible to selectively deprotect the levulinoyl ester from the formed di- and trisaccharide rhamnan structures in the presence of glycosidic byproducts. This deprotection strategy eliminates the need for a purification event after glycosylation and improves the overall efficiency of this synthetic strategy.
With the success of the developed automation approach, we next wanted to see if we could extend the scope of this automated NIS/TMSOTf-promoted glycosylation and deprotection strategy to the creation of rhamnan fragments with alpha 1→2 and 1→3 linkage type patterns as well as site-selectively sulfated versions of these types of oligomers. Naturally occurring rhamnans contain both α−1→2 and α−1→3 linkages and have been found modified with sulfates at various positions on the rhamnose ring [22–25] (Figure 1); however, such sulfated rhamnan structures have not yet made by chemical or enzymatic synthesis. Rhamnan sulfate structures have been linked to a variety of possible therapeutic applications as anti-coagulant, antiviral, anti-obesity, and anti-tumor agents.[26–29] The unmodified rhamnan structures are frequently found in virulent strains of both Gram-positive and negative bacteria.[22, 23] It has also been shown that an α−1→3 linked rhamnan disaccharide on a multivalent scaffold reduces the binding activity of a horseshoe crab lectin to P. aeruginosa PA01, a virulent Gram-negative bacteria that contains rhamnan structures on its outer cellular membrane.[30] Based on these findings, smaller rhamnan fragments displayed on a multivalent scaffold could serve as diagnostic or therapeutic tool. In an effort to provide alternative, well-defined chemical structures for screening, a variety of synthetic di- and trisaccharides with varying linkages and sulfation patterns are needed.
Figure 1.

Rhamnan and rhamnan sulfate fragment structures.
2. Results and Discussion
2.1. Building Block Syntheses
Even with automation of the oligosaccharide assembly itself, half or more of the chemical steps remain in the syntheses of properly protected building blocks required for the various automation platforms. In order to create rhamnan libraries, building blocks amenable to the formation of both α 1→2 and 1→3 linkages are needed. The initial acceptor and donor for α 1→2 linkage syntheses were reported in a prior study using established methods.[21, 31] However, we envisioned obtaining all of the necessary building blocks for these rhamnan structures in a divergent manner from a common intermediate (1) found in the previously reported synthesis for donor 2 (Scheme 1). To that end, the initial acceptor 3 was easily obtained in two steps from donor 2 in high yield (86%). This approach eliminates the need to use a separate pathway as shown in prior work. The other donor 6 could also be effectively reached from the advanced diol intermediate 1 in the α 1→2 donor pathway (Scheme 1). In lieu of a selective benzylation at the 3-OH, a selective p-methoxybenzyl (PMB) protection with dibutyltin oxide was performed to allow for subsequent selective deprotection.
Scheme 1.

Divergent building block syntheses from common intermediate 1.
The protecting group initially chosen for the 2-OH position was a levulinoyl (Lev) ester to provide both neighboring group participation and ease of removal using the proven automated method. The protection of the 2-OH position with Lev was carried out in high yield (75%). However, selecting a protecting group to occupy the 3-OH position posed a greater difficulty, considering a selective deprotection would be needed to form the desired α 1→ 3 linkage in a subsequent step. Initial protection at the 3-OH position using a PMB ester proved the glycosylation conditions were too acidic, resulting in loss of the PMB altogether in a nonproductive manner. Further investigation using a fluorenylmethyloxycarbonyl (Fmoc) protecting group also proved problematic as the reaction was low yielding and difficult to purify. Finally, it was determined that a levulinoyl (Lev) ester was sufficient to protect the 3-OH position with a benzoyl (Bz) ester present at 2-OH (Scheme 1). The Bz protecting group provided neighboring group participation to obtain the desired alpha stereoselectivity, while also providing orthogonality to the Lev ester. In addition, the Bz ester reduces the amount of orthoester side product formation compared to use with an acetyl (Ac) group and should be easier to remove than a pivaloyl (Piv) ester. Benzoylation was performed in moderate yields (60%) from intermediate 4 and was followed by an oxidative cleavage that removed the PMB group in good yield (81%). The final Lev deprotection was completed as expected to provide the final donor 6 in moderate yield (64 %).
2.2. Solution-Phase Automation
With all the building blocks in hand, we first tested to see if the previously developed automated approach [21] could be employed without modification to expand the rhamnan fragment library. First, the solution-phase process was tested for its viability to form disaccharide 7. The automation protocol was performed with acceptor 3 and donor 6, a less activated donor than the original differentially protected donor 2. Only small reaction time modifications were made to the previously developed program for use on this glycosylation system. Analytical HPLC traces and mass spectroscopy revealed the presence of the desired disaccharide and upon isolation via preparative HPLC, the rhamnan disaccharide 7 was isolated in 62% overall yield with an average step yield of 79% (Scheme 2 and SI). The success of this run demonstrated that a modified system could be used to create a different rhamnan disaccharide with a less activated donor. Upon successful synthesis of the disaccharide, the next step involved formation of the trisaccharide 8 with an α 1→2, α 1→3 linkage pattern. The same automated program was utilized with acceptor 3, donor 6 (used in the first glycosylation), and donor 2 (used for the second glycosylation). Traditional flash chromatography was performed to isolate trisaccharide 8 in 23% overall yield with an average step yield of 69%.
Scheme 2.

Automated solution-phase syntheses of rhamnan fragments for alpha 1→2 and 1→3 linkage types.
2.3. Solution-Phase Automation Protocol for Carbohydrate Sulfation
Previous studies using solid- or solution-phase automation platforms have focused solely on unmodified rhamnan backbone fragments.[32, 33] However, no rhamnan sulfate fragments syntheses have been reported to date. Considering sulfation of biologically isolated polysaccharides typically occurs at the 2-OH and 3-OH positions, these sites were the main focus of our proposed modifications to rhamnan disaccharides. In order to access these derivatives, however, sulfation conditions amenable to a solution-phase handling platform needed to be developed. In the literature, oligosaccharide sulfation is generally carried out via addition of a sulfur trioxide (SO3) complex in a nitrogen-containing basic solvent (pyridine or DMF).[34, 35] The most commonly reported conditions include SO3•pyridine (Py) complex in pyridine, SO3•trimethylamine (TME) or SO3•triethylamine (TEA) complex in DMF at 50–70 °C.[17, 34, 36–40]
Using procedures reported in literature, both types of conditions were evaluated to assess the stability of the rhamnan disaccharide and physical properties of the reactions. Liquid-phase handling platforms require the reaction components to be in solution, in the case of the fluoroustag-based approached specifically at two different points in the automated process (starting reagents and final reaction mixture). Ideally, the starting materials/reagents would be stable in solution for an extended period of time at ambient temperature. This criteria is necessary to allow for the correct amount of compound to be transferred at the programmed point in the automation sequence. Once the reaction is complete, the final reaction mixture is removed from the reaction vessel by the liquid-handling system. The solvent is then evaporated from the final reaction mixture and can be dissolved in an alternative solvent to allow for facile transfer by the liquid handling system.
Both sulfation conditions (SO3•Py complex in pyridine and SO3•TME complex in DMF) were carried out manually on bench using the previously synthesized rhamnan disaccharide 9 (Scheme 3 and SI). It was observed that both sets of conditions started as suspensions that solubilized over time as the reaction was heated. With the considerable lack of solubility at ambient temperature observed, alternative sulfation conditions amendable to the solution-phase automated platform needed to be explored. Further investigation revealed that the automated solid-phase synthesis of keratan sulfate fragments utilized 0.5 M SO3• Py in pyridine:DMF (1: 1) solution at 50 °C.[41] The 0.5 M SO3•Py solution was prepared as reported, but was also observed to be a suspension at ambient temperature. While heating the SO3•Py suspension solubilized the complex, a suspension reformed upon cooling. Once again, solubility issues hindered use with the solution-phase handling system. Additional exploration resulted in discovery of a less frequently used method involving SO3•Py in DMF at 50 °C for oligosaccharide sulfation.[42] When implemented, the SO3•Py complex was found to be soluble in DMF at ambient temperature. Subsequently, the compatible conditions were tested on the sulfate disaccharide intermediate 9. Fortunately, this sulfation solution did allow the formation of the desired sulfated rhamnan disaccharide, as confirmed by mass spectroscopy analysis.
Scheme 3.

Manual sulfation trial reactions.
A new automation protocol incorporating these compatible conditions was then developed. The automated sulfation protocol included a 4 h reaction at 60 °C, followed by a cooling macro to 25 °C, and finally a purification by the in-line FSPE (see Experimental Table S3 and S4). Unlike previous reaction protocols, no evaporation cycle was included due to the reversible nature of the reaction and FPSE purification method. Considering that dimethylformamide (DMF) is a commonly used loading solvent for FPSE purifications, the sulfation reaction mixture could be directly loaded on the column without the need to remove a non-FSPE compatible solvent. The sulfation protocol was appended to the end of the modified NIS/TMSOTf-promoted glycosylation and Lev deprotection process to efficiently synthesize the desired rhamnan sulfate disaccharides. This new protocol was initially utilized to create 10 from acceptor 3 and donor 2 (Scheme 4A). Sequential automation runs afforded the mono-sulfated disaccharide 10, which was confirmed via analytical HPLC and mass spectroscopy analysis (SI). Purification conditions were then developed to provide a pure compound (10) in 31% overall yield with an average step yield of 68%. The automation process was further extended to obtain disaccharide 11 from acceptor 3 and donor 6. Gratifyingly, the desired rhamnan sulfate disaccharide 11 was effectively isolated in 33% overall yield with an average step yield of 69% (Scheme 4B). No further process optimization was required to achieve useable quantities of the desired products using automated synthesis, a criteria required for automation of oligosaccharide synthesis to become as valuable as automated nucleic acid and peptide synthesies.
Scheme 4.

Automated solution-phase synthesis of monosulfated rhamnan disaccharides 10 and 11.
3. Conclusion
This work demonstrates the feasibility of using an automated solution-phase-based approach for the first synthesis of any rhamnan sulfate fragments. This work expands the target scope of the originally developed NIS/TMSOTf and Lev deprotection automated approach to provide access to several different rhamnan and rhamnan sulfate fragments. An unmodified rhamnan disaccharide as well as a trisaccharide with a α 1→2, α 1→3 linkage were synthesized in 62% and 23% overall yield, respectively, with only minor reaction time modifications from the programs originally used in the prior automation work. The prior protocol was extended through the development of the first automated carbohydrate sulfation protocol for a solution phase-based automation platform to thereby allow for the synthesis of two disaccharide rhamnan sulfate fragments. Importantly, extensive optimization was not required to This work should enable not only the production of chemically well-defined rhamnan and rhamnan sulfate structures for biological assay and tool development, but also the solution-phase-based automation of other sulfated carbohydrates.
4. Experimental Section
4.1. General Experimental
The chemicals used were purchased from Sigma-Aldrich, Oakwood Chemical, TCIAmerica, Fluorous Technologies, Inc, or Alfa Aesar. These chemicals were used as they were packaged unless otherwise stated. Any bench reactions that were moisture or air sensitive were performed in clean, oven-dried glassware under an argon environment. Ambient temperature was considered between 21–23 °C. Reaction monitoring was achieved by analytical thin layer chromatography (TLC) with Sorbent Technologies, Inc. silica hard layer (HL) TLC plates with unmodified silica 60 (250 μm) with 254 μm fluorescent indicator. Purifications using flash chromatography utilized ZEOprep 60 ECO flash silica gel (40–63 Å) purchased from ZEOCHEM or with a Teledyne ISCO Combiflash Rf 200 column chromatography unit and columns. Fluorous solid-phase extractions (FSPE) were performed with FluoroFlash cartridges (2 grams of silica gel bonded with perfluorooctylethylsilyl chains) acquired from Fluorous Technologies, Inc. The automation solution-phase platform used was a Chemspeed ASW2000 (Chemspeed Technologies AG, Augst, Switzerland) synthesis platform with a hood, 16 reactor vials (13 mL capacity for each vial), vacuum pump, and a heating/ cooling unit (200 °C to −20 °C). NMR spectra were recorded on a Varian Inova instrument at 500 or 600 MHz for 1H spectra while 13C spectra were recorded at 125 MHz. The reported chemical shifts (δ) are in parts per million (ppm) relative to deuterated chloroform. ESI-HRMS mass spectra were obtained on a Thermo Scientific LTQ-Orbitrap XL. Analytical HPLC chromatographs were obtained from an Agilent 1200 Analytical HPLC instrument and one separation was performed with an Agilent 1200 Preparative HPLC instrument.
4.2. Synthetic procedures
4.2.1. Propyl 4-O-benzyl-3-O-(4-methoxybenzyl)-1-thio-β-L-rhamnopyranoside (4)
Intermediate 1 (2.1 g, 6.72 mmol) was co-evaporated with toluene (25 mL × 2) and then was diluted in anhydrous toluene (67 mL). Dibutyltin oxide (2.02 g, 8.11 mmol) was poured into the reaction solution and the resulting suspension was heated at 110 °C overnight. After overnight heating, the suspension had become a solution that was cooled to ambient temperature and concentrated down to a crude brown oil. The crude brown oil was dissolved in anhy. N, N-dimethylformamide (67 mL) then had cesium fluoride (2.07 g, 13.63 mmol) and 4methoxybenzyl chloride (1 mL, 7.53 mmol) added. The resulting suspension was heated at 85 °C for 4 h. After 4 h, the reaction solution was cooled to ambient temperature and diluted with ethyl acetate (60 mL) and DI water (60 mL). The extracted aqueous layer was further washed with ethyl acetate (2 × 35 mL) while the combined organic layers were washed with deionized (DI) water (2 × 60 mL) and brine (60 mL). The organic layer was dried with Na2SO4, filtered, and the solvent removed under reduced pressure. Column chromatography was performed and gave compound 4 as a colorless, transparent oil (1.18 g, 2.73 mmol, 40%).1H NMR (400 MHz, CDCl3) δ 7.38 – 7.26 (m, 7H), 6.86 (d, J = 8.7 Hz, 2H), 5.24 (s, 1H), 4.87 (d, J = 11.0 Hz, 1H), 4.62 (d, J = 11.1 Hz, 1H), 4.60 (s, 2H), 4.10 – 4.03 (m, 2H), 3.80 (s, 3H), 3.79–3.77 (m, 1H), 3.45 (t, J = 9.3 Hz, 1H), 2.65 – 2.45 (m, 3H), 1.63 (m, 2H), 1.30 (d, J = 6.2 Hz, 3H), 0.97 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) 159.63, 138.57, 129.96, 129.78, 128.53, 128.07, 127.86, 114.13, 83.69, 80.36, 80.11, 75.48, 71.90, 70.32, 68.01, 55.43, 33.24, 23.11, 17.98, 13.53. HRMS (ESI) m/z: C24H32O5SNa calculated for: 455.1863; HRMS Found: 425.1863 [M+Na]+
4.2.2. Propyl 2-O-benzoyl-4-O-benzyl-3-O-(4-methoxybenzyl)-1-thio-β- L-rhamnopyranoside (S4)
Intermediate 4 (1.18g, 2.73 mmol) was dissolved in anhydrous pyridine (5.5 mL) and cooled to 0 °C via an ice-bath. To the cooled solution, benzoyl chloride (0.48 mL, 4.09 mmol) was added and the ice-bath was removed. The solution was allowed to stir at ambient temperature for 2 hours and then was quenched with methanol (0.3 mL). The quenched solution had the solvent removed under reduced pressure and was coevaporated with toluene. The resulting residue was column purified to provide S4 (1.22 g, 2.27 mmol, 84%) as a transparent, colorless oil. 1H NMR (600 MHz, CDCl3) δ 8.10 – 8.06 (m, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.47 (m, 2H), 7.36–7.27 (m, 5H), 7.21 (d, J = 8.6 Hz, 2H), 6.78 (d, J = 8.6 Hz, 2H), 5.67 (dd, J =3.0, 1.7, 1H), 5.29 – 5.27 (m, 1H), 4.90 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 11.0 Hz, 1H), 4.62 (d, J = 10.9 Hz, 1H), 4.47 (d, J = 11.0 Hz, 1H), 4.13 (m, 1H), 3.96 (dd, J = 9.3, 3.2 Hz, 1H), 3.76 (s, 3H), 3.54 (t, J = 9.4 Hz, 1H), 2.67 – 2.53 (m, 2H), 1.65 (ddt, J = 10.5, 7.2, 3.2 Hz, 1H), 1.36 (d, J = 6.2 Hz, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.89, 159.36, 138.60, 133.30, 130.20, 130.09, 130.04, 129.88, 128.54, 128.46, 128.19, 127.79, 113.88, 82.98, 80.40, 78.30, 75.46, 71.52, 71.32, 68.54, 55.35, 33.90, 23.22, 18.21, 13.50. HRMS (ESI) m/z: C31H36O6SNa calculated for: 559.2125; HRMS Found: 559.2125 [M+Na]+
4.2.3. Propyl 2-O-benzoyl-4-O-benzyl-1-thio-β- L-rhamnopyranoside (5)
Intermediate S4 (0.60 g, 1.12 mmol) was dissolved in a biphasic mixture of anhydrous dichloromethane (37.5 mL) and DI water (2.1 mL). To the biphasic solution, 2, 3-dichloro-5, 6dicyano-1, 4-benzoquinone (0.6 g, 2.64 mmol) was poured in and the reaction was stirred at ambient temperature for 4 hours. After 4 hours, the reaction solution was washed with saturated aqueous NaHCO3 (3 × 35 mL) and the organic layer was dried with Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting crude product was purified via column chromatography to provide 5 (0.38 g, 0.91 mmol, 81 %) as a colorless, transparent oil. 1H NMR (500 MHz, CDCl3) δ 8.07 – 8.04 (m, 2H), 7.63–7.57 (m, 1H), 7.47 (m, 2H), 7.40 – 7.28 (m, 5H), 5.45 (dd, J = 3.4, 1.5 Hz, 1H), 5.30 (d, J = 1.1 Hz, 1H), 4.85 (d, J = 11.2 Hz, 1H), 4.77 (d, J = 11.2 Hz, 1H), 4.20 – 4.13 (m, 2H), 3.51 (t, J = 9.4 Hz, 1H), 2.68 – 2.53 (m, 2H), 2.17 (d, J = 5.0 Hz, 1H), 1.65 (m, 2H), 1.40 (d, J = 6.2 Hz, 3H), 0.99 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 166.33, 138.27, 133.50, 130.00, 129.91, 128.70, 128.60, 128.18, 128.12, 82.76, 82.14, 75.33, 75.28, 71.22, 68.29, 33.92, 23.22, 18.26, 13.49. HRMS (ESI) m/z: C23H28O5SNa calculated for: 439.1550; HRMS Found: 439.1551 [M+Na]+
4.2.4. Propyl 2-O-benzoyl-4-O-benzyl-3-O-levolinoyl-1-thio-β- L-rhamnopyranoside (6)
Compound 5 (0.47 g, 1.13 mmol) was placed under argon and dissolved in anhydrous dichloromethane (11.3 mL). The solution then had the following reagents added in the listed order: levulinic acid (0.175 mL, 1.69 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.446 g, 2.32 mmol), and 4-dimethylaminopyridine (0.082 g, 0.68 mmol). The reaction solution was stirred at ambient temperature overnight. The reaction solution was washed with DI water (3 × 25 mL), 1 N HCl (3 × 25 mL), satd. aq. NaHCO3 solution (3 × 25 mL), and brine (25 mL). The organic layer was extracted, dried with Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting crude material was purified by column chromatography (slow gradient of 20% diethyl ether in pentane to 50% diethyl ether in pentane). Compound 6 was isolated as a clear, colorless oil (0.37 g, 0.72 mmol, 64 %). 1H NMR (600 MHz, CDCl3) δ 8.07 – 8.04 (m, 2H), 7.62 (m, 1H), 7.49 (m, 2H), 7.36 – 7.27 (m, 5H), 5.58 (dd, J = 3.2, 1.6 Hz, 1H), 5.37 (dd, J = 9.7, 3.3 Hz, 1H), 5.26 (d, J = 1.2 Hz, 1H), 4.75 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 11.2 Hz, 1H), 4.28 – 4.22 (m, 1H), 3.65 (t, J = 9.5 Hz, 1H), 2.76–2.69 (m, 1H), 2.68 – 2.53 (m, 3H), 2.53 – 2.39 (m, 2H), 2.09 (s, 3H), 1.65 (m, 2H), 1.38 (d, J = 6.2 Hz, 3H), 0.99 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 206.32, 171.83, 165.66, 138.11, 133.54, 129.97, 129.88, 128.68, 128.56, 128.06, 127.95, 82.54, 79.08, 75.12, 72.82, 72.77, 68.50, 37.99, 33.69, 29.87, 28.10, 23.13, 18.19, 13.49. HRMS (ESI) m/z: C28H34O7SNa calculated for: 537.1917; HRMS Found: 537.1919 [M+Na]+.
4.3. Automation Platform Procedures and Compounds
4.3.1. General Procedures and Automation Set-Up
These procedures and the automation set-up information can be found in the supporting information on page S5–8.
4.3.2. 2-(4-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)oxy)phenoxy)ethyl 2-O-benzoyl-4-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (7)
This compound was formed using the automation platform protocol outlined in Table S1. Purification was achieved utilizing a Phenomenex Luna 5 μm C18(2), 100 Å, LC Column 250 × 21.2 mm on the Agilent 1200 Preparatory HPLC. Elute gradient: 0–30 min 95:5 acetonitrile (ACN):H2O (with 0.1% TFA), 30–50 min 95–100:5–0 ACN:H2O, 50–62 min 100% ACN. The collected material had the solvent removed under reduced pressure and compound 7 was isolated as a clear, colorless oil (0.017 g, 0.013 mmol, 62% overall yield). 1H NMR (600 MHz, CDCl3) 8.08 – 8.03 (m, 2H), 7.61 (m, 1H), 7.48 (m, 2H), 7.39 – 7.26 (m, 12H), 7.19 (d, J = 7.3 Hz, 2H), 7.13 (m, 1H), 6.86 – 6.78 (m, 4H), 5.52 (dd, J = 3.2, 1.7 Hz, 1H), 5.12 (d, J = 1.3 Hz, 1H), 4.88 (d, J = 10.9 Hz, 1H), 4.86 – 4.83 (m, 2H), 4.73 (d, J = 11.1 Hz, 1H), 4.65 (s, 2H), 4.62 (d, J = 10.8 Hz, 1H), 4.31 (dd, J = 9.4, 3.4 Hz, 1H), 4.09 – 4.02 (m, 3H), 3.96 (t, J = 5.9 Hz, 2H), 3.94 – 3.84 (m, 3H), 3.79 – 3.70 (m, 2H), 3.47 (dt, J = 13.8, 9.4 Hz, 2H), 2.29 (m, 2H), 2.06 (m, 2H), 1.37 (d, J = 6.2 Hz, 3H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.34, 153.34, 153.11, 138.65, 138.46, 138.35, 133.44, 130.06, 129.96, 128.67, 128.57, 128.49, 128.47, 128.22, 128.20, 128.08, 127.78, 127.68, 127.61, 115.92, 115.60, 99.22, 99.19, 81.81, 80.32, 79.88, 75.55, 75.37, 74.98, 73.39, 72.35, 70.69, 68.23, 67.93, 67.12, 66.01, 28.13, 20.82, 18.37, 18.17. HRMS (ESI) m/z: C59H57O12F17Na calculated for: 1303.3471; HRMS Found: 1303.3452 [M+Na]+
4.3.3. 2-(4-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)oxy)phenoxy)ethyl 3,4-di-O-benzyl-α-L-rhamnopyranosyl-(1→3)- 2-O-benzoyl-3-O-benzyl-α-Lrhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (8)
This compound was formed using the automation platform protocol outlined in Table S2. Purification was achieved utilizing flash chromatography and 8 was isolated as a clear, colorless oil (0.0119 g, 6.97 μmol, 23 % overall yield). 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 7.3 Hz, 2H), 7.59 (m, 1H), 7.47 (m, 2H), 7.37 – 7.23 (m, 16H), 7.23–7.18 (m, 8H), 7.13 (m, 1H), 6.84 – 6.762(m, 4H), 5.56 – 5.54 (m, 1H), 5.17 – 5.14 (m, 1H), 5.13 (s, 1H), 4.88 – 4.44 (m, 12H), 4.30 (dd, J = 9.3, 3.1 Hz, 1H), 4.05 – 4.00 (m, 3H), 3.94 (t, J = 5.9 Hz, 2H), 3.92 – 3.80 (m, 5H), 3.77 – 3.65 (m, 3H), 3.55 (t, J = 9.4 Hz, 1H), 3.48 (t, J = 9.4 Hz, 1H), 3.39 (t, J = 9.3 Hz, 1H), 2.28 (m, 2H), 2.04 (m, 2H), 1.28 (m, J = 9.4, 6.2 Hz, 6H), 1.14 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.48, 153.34, 153.09, 138.72, 138.66, 138.59, 138.09, 133.24, 129.97, 128.67, 128.58, 128.47, 128.43, 128.38, 128.32, 128.06, 127.99, 127.92, 127.84, 127.74, 127.71, 127.64, 127.47, 115.92, 115.58, 99.20, 98.95, 80.77, 80.29, 79.91, 79.76, 75.55, 75.17, 74.88, 72.81, 72.27, 72.19, 69.28, 68.50, 68.38, 68.26, 67.94, 67.12, 65.98, 29.82, 20.81, 18.29, 18.14, 17.82. HRMS (ESI) m/z: C79H79O16F17Na calculated for: 1629.4989; HRMS Found: 1629.4999 [M+Na]+
4.3.4. 2-(4-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)oxy)phenoxy)ethyl 3,4-di-O-benzyl-2-O-sulfonate-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (10)
This compound was formed using the automation platform protocol outlined in Table S3. The compound was purified via column chromatography (elute 92:5:3 EtOAc: MeOH: DI H2O) and the like fractions were combined and the solvent removed under reduced pressure to give 10 as a transparent, colorless oil (0.009 g, 6.6 μmol, 31% overall yield). 1H NMR (500 MHz, CDCl3) δ 7.38–7.26 (m, 12H), 7.25 – 7.11 (m, 8H), 6.84 – 6.77 (m, 4H), 5.40 (s, 1H), 4.98 (s, 1H), 4.83 (m, 2H), 4.77 (d, J = 11.7 Hz, 2H), 4.68 – 4.52 (m, 3H), 4.45 (m, 2H), 4.02–3.89 (m, 6H), 3.88–3.82 (m, 2H), 3.79 (d, J = 9.2 Hz, 1H), 3.71–3.64 (m, 2H), 3.61 (t, J = 9.5 Hz, 1H), 3.23 (t, J = 9.2 Hz, 1H), 2.38–2.23 (m, 2H), 2.10–2.02 (m, 2H), 1.30 (d, J = 6.4 Hz, 4H), 1.21 (d, J = 6.0 Hz, 4H).13C NMR (126 MHz, CDCl3) δ 153.26, 153.06, 138.61, 138.48, 138.42, 137.24, 128.88, 128.64, 128.51, 128.49, 128.45, 128.25, 128.17, 128.15, 128.11, 127.76, 127.75, 127.72, 115.80, 115.55, 99.63, 98.82, 80.23, 79.86, 78.57, 76.66, 76.39, 75.41, 75.26, 74.53, 71.85, 71.69, 68.59, 67.88, 67.77, 67.06, 66.05, 28.29, 28.11, 27.93, 20.79, 18.10, 17.78. HRMS (ESI) m/z: C59H58O14F17 S calculated for: 1345.3281; HRMS Found: 1345.3244 [M−]
4.3.5. 2-(4-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)oxy)phenoxy)ethyl 2-O-benzoyl-4-O-benzyl-3-O-sulfonate-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (11)
This compound was formed using the automation platform protocol outlined in Table S4. The compound was purified via column chromatography (silica, elute 92:5:3 EtOAc: MeOH: DI H2O) and the like fractions were combined and the solvent removed under reduced pressure to give 11 as a transparent, colorless oil (0.01 g, 7.1 μmol, 33% overall yield). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 7.5 Hz, 2H), 7.41 (t, J = 6.9 Hz, 1H), 7.30–7.27 (m, 3H), 7.25–7.21 (m, 3H), 7.20 – 7.12 (m, 9H), 7.11–7.04 (m, 3H), 6.80 – 6.72 (m, 4H), 5.96 (s, 1H), 5.26 (s, 1H), 5.02 (d, J = 9.3 Hz, 1H), 4.95 (d, J = 10.8 Hz, 1H), 4.84 (s, 1H), 4.72 (d, J = 10.7 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.57 – 4.52 (m, 2H), 4.46 (d, J = 10.8 Hz, 1H), 4.06 (s, 1H), 3.92 (t, J = 5.7 Hz, 4H), 3.84–3.77 (m, 3H), 3.73 – 3.60 (m, 4H), 3.51 – 3.45 (m, 1H), 2.32–2.22 (m, 4H), 2.06–2.00 (m, 2H), 1.32 (d, J = 6.0 Hz, 3H), 1.23 (d, J = 5.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.42, 153.34, 153.00, 138.83, 138.22, 138.01, 133.35, 130.29, 129.67, 129.12, 128.38, 128.29, 128.04, 128.01, 127.76, 127.64, 127.43, 115.80, 115.52, 99.05, 80.28, 79.91, 75.23, 74.96, 72.42, 71.91, 68.57, 67.75, 67.08, 66.03, 28.11, 20.80, 18.28, 17.72. HRMS (ESI) m/z: C59H56O15F17S calculated for: 1359.3074; HRMS Found: 1359.3037 [M−]
Supplementary Material
Highlights.
First synthesis of any sulfated rhamnan natural product
Development of an automated solution-phase process for carbohydrate sulfation
Automated solution-phase syntheses of monosulfated rhamnan dimers
Automated solution-phase syntheses of alternating 1,2 and 1,3-linked rhamnans
5. Acknowledgements
This work was supported by Indiana University and the National Institutes of Health (5U01GM116248-02). We would like to thank Dr. Régis Saliba for his input and assistance with the automation platform. We also want to thank Prof. Amar Flood and his research group for use of their Agilent 1200 Preparatory HPLC system. Finally, we would like to thank Dr. Alison Vickman for her suggestions and edits on the initial drafts of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- [1].Wong S, Shim MS, Kwon YJ, Synthetically designed peptide-based biomaterials with stimuli-response and membrane-active properties for biomedical applications, J. Mater. Chem. B, 2 (2014) 595–615. 10.1039/C3TB21344G. [DOI] [PubMed] [Google Scholar]
- [2].Lee AC-L, Harris JL, Khanna KK, Hong J-H, A comprehensive review on current advances in peptide drug development and design, Int. J. Mol. Sci, 20 (2019) 2383 10.3390/ijms20102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Laine RA, Information capacity of the carbohydrate code, Pure Appl. Chem, 69 (1997) 1867–1873. 10.1351/pac199769091867. [DOI] [Google Scholar]
- [4].Merrifield RB, Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc, 85 (1963) 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- [5].Seeberger PH, Haase W-C, Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries, Chem. Rev 100 (2000) 4349–4394. 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]
- [6].Plante OJ, Palmacci ER, Seeberger PH, Automated solid-phase synthesis of oligosaccharides, Science, 291 (2001) 1523–1527. 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- [7].Panza M, Pistorio SG, Stine KJ, Demchenko AV, Automated chemical oligosaccharide synthesis: novel approach to traditional challenges, Chem. Rev 118(2018) 8105–8150. 10.1021/acs.chemrev.8b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nokami T, Isoda Y, Sasaki N, Takaiso A, Hayase S, Itoh T, Hayashi R, Shimizu A, Yoshida J, Automated electrochemical assembly of the protected potential TMG-chitotriomycin precursor based on rational optimization of the carbohydrate building block, Org. Lett, 17 (2015) 1525–1528. 10.1021/acs.orglett.5b00406. [DOI] [PubMed] [Google Scholar]
- [9].Manmode S, Sato T, Sasaki N, Notsu I, Hayase S, Nokami T, Itoh T, Rational optimization of the mannoside building block for automated electrochemical assembly of the core trisaccharide of GPI anchor oligosaccharides, Carbohydr. Res, 450 (2017) 4–48. 10.1016/j.carres.2017.08.009. [DOI] [PubMed] [Google Scholar]
- [10].Sasaki N, Nokami T, Itoh T, Synthesis of a TMG-chitotriomycin precursor based on electrolyte-free electrochemical glycosylation using an ionic liquid tag, Chem. Lett, 46 (2017) 683–685. 10.1246/cl.170126. [DOI] [Google Scholar]
- [11].Manmode S, Kato M, Ichiyanagi T, Nokami T, Itoh T, Automated electrochemical assembly of the β-(1,3)-β-(1,6)-glucan hexasaccharide using thioglucoside building blocks, Asian J. Org. Chem, 7 (2018) 1802–1805. 10.1002/ajoc.201800345. [DOI] [Google Scholar]
- [12].Li T, Liu L, Wei N, Yang JY, Chapla DG, Moremen KW, Boons GJ, An automated platform for the enzyme-mediated assembly of complex oligosaccharides, Nat. Chem, 11 (2019) 229–236. 10.1038/s41557-019-0219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang SL, Pohl NLB, Automated fluorous-assisted solution-phase synthesis of β−1,2-, 1,3-, and 1,6-mannan oligomers, Carbohydr. Res, 430 (2016) 8–15. 10.1016/j.carres.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tang SL, Linz LB, Bonning BC, Pohl NL, Automated solution-phase synthesis of insect glycans to probe the binding affinity of pea enation mosaic virus, J. Org. Chem, 80 (2015) 10482–10489. 10.1021/acs.joc.5b01428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saliba RC, Wooke ZJ, Nieves GA, Chu AA, Bennett CS, Pohl NLB, Challenges in the conversion of manual processes to machine-assisted syntheses: Activation of thioglycoside donors with aryl(trifluoroethyl)iodonium triflimide, Org. Lett, 20 (2018) 800–803. 10.1021/acs.orglett.7b03940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nagy G, Peng T, Kabotso DE, Novotny MV, Pohl NL, Protocol for the purification of protected carbohydrates: toward coupling automated synthesis to alternative-pump recycling high-performance liquid chromatography, Chem. Commun, 52 (2016) 13253–13256. 10.1039/C6CC07584C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bhaduri S, Pohl NLB, Fluorous-tag assisted syntheses of sulfated keratan sulfate oligosaccharide fragments, Org. Lett, 18 (2016) 1414–1417. 10.1021/acs.orglett.6b00344. [DOI] [PubMed] [Google Scholar]
- [18].Liu L, Pohl NLB, Synthesis of a series of maltotriose phosphates with an evaluation of the utility of a fluorous phosphate protecting group, Carbohydr. Res, 369 (2013) 14–24. 10.1016/j.carres.2012.12.015. [DOI] [PubMed] [Google Scholar]
- [19].Jaipuri FA, Pohl NL, Toward solution-phase automated iterative synthesis: fluorous-tag assisted solution-phase synthesis of linear and branched mannose oligomers, Org. Biomol. Chem, 6 (2008) 2686–2691. 10.1039/B803451F. [DOI] [PubMed] [Google Scholar]
- [20].Park G, Ko KS, Zakharova A, Pohl NL, Mono- vs. di-fluorous-tagged glucosamines for iterative oligosaccharide synthesis, J. Fluor. Chem, 129 (2008) 978–982. 10.1016/j.jfluchem.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kohout VR, Pirinelli AL, Pohl NLB, Acid-mediated N-iodosuccinimide-based thioglycoside activation for the automated solution-phase synthesis of α−1,2-linkedrhamnopyranosides, Pure. Appl. Chem, 91 (2019) 1243–1255. 10.1515/pac-20190307. [DOI] [Google Scholar]
- [22].Molinaro A, Newman M-A, Lanzetta R, Parrilli M, The structures of liposaccharides from plant-associated Gram-negative bacteria, Eur. J. Org. Chem, 2009 (2009) 5887–5896. 10.1002/ejoc.200900682. [DOI] [Google Scholar]
- [23].Mistou MY, Sutcliffe IC, van Sorge NM, Bacterial glycobiology: rhamnose-containing cell wall polysaccharides in Gram-positive bacteria, FEMS Microbiol. Rev, 40 (2016) 464–479. 10.1093/femsre/fuw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].St. Michael F, Yang Q, Cairns C, Vinogradov E, Fleming P, Hayes AC, Aubry A, Cox AD, Investigating the candidacy of the serotype specific rhamnan polysaccharide based glycoconjugates to prevent disease caused by the dental pathogen Streptococcus mutans, Glycoconj. J, 35 (2018) 53–64. 10.1007/s10719-017-9798-z. [DOI] [PubMed] [Google Scholar]
- [25].Wang L, Wang X, Wu H, Liu R, Overview on biological activities and moleculear characteristics of sulfated polysaccharides from marine green algae in recent years, Mar. Drugs, 12 (2014) 4984–5020. 10.3390/md12094984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yamashiro Y, Nakamura M, Yogi T, Teruya T, Konishi T, Uechi S, Tako M, Anticoagulant activity of rhamnan sulfate isolated from commercially cultured Monostroma nitidum, Int. J. Biomed. Mater. Res, 5 (2017) 37–43. 10.11648/j.ijbmr.20170503.12. [DOI] [Google Scholar]
- [27].Wang S, Wang W, Hao C, Yunjia Y, Qin L, He M, Mao W, Antiviral activity against enterovirus 71 of sulfated rhamnan isolated from the green alga Monostroma latissimum, Carbohydr. Polym, 200 (2018) 43–53. 10.1016/j.carbpol.2018.07.067. [DOI] [PubMed] [Google Scholar]
- [28].Ropellato J, Carvalho MM, Ferrira LG, Noseda MD, Zuconelli CR, Goncalves AG, Ducatti DR, Kenski JC, Nasato PL, Winnischofer SM, Duarte ME, Sulfated heterorhamnans from the green seaweed Gayralia oxysperma: partial depolymerization, chemical structure and antitumor activity, Carbohydr. Polym, 117 (2015) 476–485. 10.1016/j.carbpol.2014.09.089. [DOI] [PubMed] [Google Scholar]
- [29].Lee J-;B, Koizumi S, Hayashi K, Hayashi T, Structure of rhamnan sulfate from the green alga Monostroma nitidum and its anti-herpetic effect, Carbohydr. Polym, 81 (2010) 572–577. 10.1016/j.carbpol.2010.03.014. [DOI] [Google Scholar]
- [30].Herczeg M, Mezo E, Molnar N, Ng SK, Lee YC, Dah-Tsyr Chang M, Borbas A, Inhibitory effect of multivalent rhamnobiosides on recombinant horseshoe carb plasma lectin interactions with Pseudomonas aeruginosa PAO1, Chem. Asian J; 11 (2016) 3398–3413. 10.1002/asia.201601162. [DOI] [PubMed] [Google Scholar]
- [31].Volbeda AG, Reintjens NRM, Overkleeft HS, van der Marel GA, Codée JDC, The cyanopivaloyl ester: A protecting group in the assembly of oligorhamnans, Eur. J. Org. Chem 2016 (2016) 5282–5293. 10.1002/ejoc.201600956. [DOI] [Google Scholar]
- [32].Volbeda AG, van Mechelen J, Meeuwenoord N, Overkleeft HS, van der Marel GA, Codée JDC, Cyanopivaloyl ester in the automated solid-phase synthesis of oligorhamnans, J. Org. Chem, 82 (2017) 12992–13002. 10.1021/acs.joc.7b02511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Palmaai ER, Plante OJ, Hewitt MC, Seeberger PH, Automated synthesis of oligosaccharides, 86 (2003) 3975–3990. 10.1002/hlca.200390331. [DOI] [Google Scholar]
- [34].Dulaney SB, Huang X, Strategies in synthesis of heparin/heparan sulfate oligosaccharides: 2000-present, Adv. Carbohydr. Chem. Biochem, 67 (2012) 95–136. 10.1016/B978-0-12-396527-1.00003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Al-Horani RA, Desai UR, Chemical sulfation of small molecules-advances and challenges, Tetrahedron, 66 (2010) 2907–2918. 10.1016/j.tet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sheng GJ, Oh YI, Chang SK, Hsieh-Wilson LC, Tunable heparan sulfate mimetics for modulating chemokine activity, J. Am. Chem. Soc, 135 (2013) 10898–10901. 10.1021/ja4027727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu R, Morales-Collazo O, Wei A, Sulfoform generation from an orthogonally protected disaccharide, Carbohydr. Res, 355 (2012) 19–27. 10.1016/j.carres.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Matta K, Kumar V, Locke R, Syntheses of novel sulfated glycans for cell-adhesion interaction studies, Synlett, 16 (2009) 2633–2636. 10.1055/s-0029-1217972. [DOI] [Google Scholar]
- [39].Fan R-H, Achkar J, Hernandex-Torres JM, Wei A, Orthogonal sulfation strategy for synthetic heparan sulfate ligands, Org. Lett, 7 (2005) 5095–5098. 10.1021/ol052130o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang B, Yoshida K, Yin Z, Dai H, Kavunja H, El-Dakdouki MH, Sungsuwan S, Dulaney SB, Huang X, Chemical synthesis of a heparan sulfate glycopeptide: Syndecan-1, Angew. Chem. Int. Ed, 51 (2012) 10185–10185–10189. 10.1002/anie.201205601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hahm HS, Broecker F, Kawasaki F, Mietzsch M, Heilbronn R, Fukuda M, Seeberger PH, Automated glycan assembly of oligo-N-acetyllactosamine and keratan sulfate probes to study virus-glycan interactions, Chem, 2 (2017) 114–124. 10.1016/j.chempr.2016.12.004. [DOI] [Google Scholar]
- [42].Guiseley KB, Ruoff PM, Monosaccharide sulfates. I. Glucose 6-sulfate. Preparations, characterizations of the crystalline postassium salt, and kinetic studies, J. Org. Chem, 26 (1961) 1248–1254. 10.1021/jo01063a064. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.