

Abstract
Fragmentation of natural environments as a result of human interference has been associated with a decrease in species richness and increase in abundance of a few species that have adapted to these environments. The Brazilian Atlantic Forest, which has been undergoing an intense process of fragmentation and deforestation caused by human-made changes to the environment, is an important hotspot for malaria transmission. The main vector of simian and human malaria in this biome is the mosquito Anopheles cruzii. Anthropogenic processes reduce the availability of natural resources at the tree canopies, An. cruzii primary habitat. As a consequence, An. cruzii moves to the border of the Atlantic Forest nearing urban areas seeking resources, increasing their contact with humans in the process. We hypothesized that different levels of anthropogenic changes to the environment can be an important factor in driving the genetic structure and diversity in An. cruzii populations. Five different hypotheses using a cross-sectional and a longitudinal design were tested to assess genetic structure in sympatric An. cruzii populations and microevolutionary processes driving these populations. Single nucleotide polymorphisms were used to assess microgeographic genetic structure in An. cruzii populations in a low-endemicity area in the city of São Paulo, Brazil. Our results show an overall weak genetic structure among the populations, indicating a high gene flow system. However, our results also pointed to the presence of significant genetic structure between sympatric An. cruzii populations collected at ground and tree-canopy habitats in the urban environment and higher genetic variation in the ground-level population. This indicates that anthropogenic modifications leading to habitat fragmentation and a higher genetic diversity and structure in ground-level populations could be driving the behavior of An. cruzii, ultimately increasing its contact with humans. Understanding how anthropogenic changes in natural areas affect An. cruzii is essential for the development of more effective mosquito control strategies and, on a broader scale, for malaria-elimination efforts in the Brazilian Atlantic Forest.
Subject terms: Computational biology and bioinformatics, Evolution, Genetics, Molecular biology
Introduction
Anthropogenic interference in the environment is a major driving force of evolution1,2. The ecology and behavior of many insect species are greatly affected by urbanization and the global increase in temperature3. This is supported by a number of studies that have shown evidence of a decrease in overall species richness and increase in the abundance of a few species that can thrive in urban environments1,4–8. While urbanization processes have a major impact on vector mosquito communities, some species such as Aedes aegypti and Culex quinquefasciatus have adapted to urban environments, which enabled them to thrive and benefit from these anthropogenic changes. Over the last decades, the distribution and abundance of these species have increased substantially9–11, leading to the emergence and re-emergence of many vector-borne diseases12–15. While some malaria vector species tend to seek hosts and breeding habitats in forested areas (e.g., Anopheles triannulatus, An. oswaldoi, and An. cruzii), others, such as Anopheles darlingi, are rarely found in forested areas but are frequently found in villages and can benefit from deforestation16,17.
The Brazilian Atlantic Forest is one of the most endangered ecosystems in the world18. This important biome is characterized by dense, humid forest with a high degree of biodiversity and endemism19,20. Historically, the Brazilian Atlantic Forest has undergone deforestation and fragmentation as a result of urbanization, industrialization, and agricultural expansion21. However, this process has intensified over the last decades, leading to increasing devastation and biodiversity loss in this biome21,22.
The Atlantic Forest is an important malaria transmission hotspot23,24 and is home to Anopheles (Kerteszia) cruzii Dyar and Knab, a neotropical mosquito found abundantly in this biome, where it is the main vector of simian and human malaria. This mosquito has a close relationship with plants from the Bromeliaceae family (bromeliads)25 as it uses the water accumulated in the bromeliad water tanks as its primary/exclusive breeding habitat26. For this reason, malaria in the Atlantic Forest has been known historically as “bromeliad malaria”25,27. Fragmentation of the Atlantic Forest ultimately reduces the number of natural malaria hosts (monkeys) and dead-end hosts in this region, increasing the contact between mosquito vectors and humans23,28. Some studies have found an association between anthropogenic changes in the environment and the occurrence of human malaria, asymptomatic plasmodial infections and zoonotic malaria transmission in the Atlantic Forest28–31.
Because of this species’ close relationship with the Atlantic Forest environment and because of its acrodendrophilic habits (i.e., its preference for inhabiting tree canopies), the ecology and phenotype of An. cruzii are believed to be affected by the fragmentation of the Atlantic Forest biome30,32,33. Interestingly, in environments where An. cruzii has been found in abundance only in tree canopies but not at ground level, cases of human malaria are rarely reported despite a high prevalence of malaria among nonhuman primates in the same areas34. However, in areas where the mosquito is found not only in the tree canopy but also at ground level, a higher prevalence of both human and simian malaria has been observed34. Anopheles cruzii blood feeds primarily on howler monkeys in tree canopies and on monkeys and humans at ground level. Hence, the species’ vertical dispersal increases the exposure of humans to Plasmodium31,34–36. The species was also found to be more active at ground level in areas with a greater edge effect and greater loss of forested areas37, a factor which may contribute to the zoonotic transmission of Plasmodium.
The presence of An. cruzii breeding sites in bromeliads in the urban environment has been reported previously38,39; however, this mosquito is considered a non-synanthropic species because although it can invade houses in search of blood meals, its natural breeding habitat is in forested areas40,41. Previous studies reported a high frequency of An. cruzii in peridomestic sites during an asymptomatic malaria outbreak in the Atlantic Forest and found that humans were a primary blood-meal source, indicating that this mosquito exhibits anthropophilic behavior42. The presence of An. cruzii in urban, peri-urban and sylvatic environments demonstrates the dispersive behavior of the species, as it can be found in different ecotypes, and its adaptation to anthropogenically modified areas close to forest fragments33,37,39.
Anthropogenic changes in Atlantic Forest ecosystems may have led to 185 cases of autochthonous human malaria between 2007 and 2017 in the city of São Paulo, the largest and most populous city in Brazil43,44. This hypothesis is supported by several autochthonous P. vivax malaria cases detected in the southern region of the city in the last decades23,29 and by previous studies in which An. cruzii was collected in abundance in tree canopies and at ground level in the region37,45. Furthermore, in a previous study carried out in the same region, An. cruzii was found naturally infected with P. vivax and P. malariae29.
Despite the epidemiological importance of this mosquito species in Plasmodium transmission and its high abundance in densely populated areas in the city of São Paulo, there is a dearth of studies assessing how anthropogenic changes in the environment affect the ecology and behavior of An. cruzii and how such changes may be driving patterns of malaria transmission. Moreover, to our knowledge no genetic studies have yet attempted to understand how An. cruzii is adapting locally to the increased anthropogenic modification and destruction of its natural habitats24. Single nucleotide polymorphisms (SNPs) are informative molecular markers used to analyze genetic differences between populations on micro- and macro-geographic and evolutionary scales46,47 and can be used to examine neutral genome regions and regions under selection, increasing the sensitivity and specificity of this type of analysis48. Various population studies in the literature have analyzed SNPs in Anopheles species from around the world49–51, and the structure of populations of An. darlingi, the main malaria vector in the Amazon region, has been analyzed on microgeographic and macrogeographic scales with the aid of SNPs in several Brazilian studies52–55. These studies show that this genetic tool is reliable and sensitive enough to reveal genetic differences using a fraction of the samples required for other molecular markers.
Understanding how anthropogenic changes in natural areas affect An. cruzii is essential for the development of more effective mosquito control strategies and, on a broader scale, for malaria-elimination efforts in Brazil. The objective of this study was therefore to genetically characterize populations of An. cruzii collected over 17 months in three different environments that have undergone different degrees of anthropogenic impacts and were classified as Natural, Suburban/Rural, and Urban (Fig. 1). We tested five hypotheses using a cross-sectional and longitudinal design to assess genetic structure in sympatric An. cruzii specimens from tree-canopy and ground-level habitats. Additionally, we assessed cross-sectional genetic structure and microevolution of An. cruzii specimens from environments with different degrees of anthropogenic modifications. Hypothesis-1—There is genetic structure between tree-canopy and ground-level An. cruzii populations; Hypothesis-2—There is genetic structure between tree-canopy and ground-level An. cruzii populations in each environment (Natural, Suburban/Rural and Urban); Hypothesis-3—There is cross-sectional genetic structure between the different environments (Natural, Suburban/Rural and Urban); Hypothesis-4—There is microevolutionary structure between populations over time; and Hypothesis-5—There is microevolutionary structure between populations in each environment (Natural, Suburban/Rural, and Urban).
Figure 1.

Map of the Anopheles cruzii sampling locations in the city of São Paulo, Brazil. (a) Map of Brazil showing the locality of the city of São Paulo in the state of São Paulo in the southeastern region of the country. (b) Map of the city of São Paulo showing the locality of the subdistrict of Parelheiros where the An. cruzii collections occurred. (c) Satellite images of the An. cruzii sampling locations. From left to right: Urban—the Engenheiro Marsilac neighborhood; Suburban/Rural—a transition area between Atlantic Forest remnants and a cattle range; and Natural—Atlantic Forest remnants on private property. The map of vegetation remnants of the Atlantic Forest Biome in the municipality of São Paulo is
available at https://geosampa.prefeitura.sp.gov.br/PaginasPublicas/_SBC.aspx.
Results
A total of 21,678 SNPs were genotyped in 380 specimens of An. cruzii (Table 1) by SNPsaurus (SNPsaurus, Oregon, USA). Filtering of this dataset (for quality, read depth, LD, MAF and indels) resulted in a set of markers with 1,313 SNPs. Outlier detection based on the fsthet method identified 78 (approx. 6%) loci that are possibly under positive selection56 (Fig. 2). These loci were removed from the subsequent analysis. The remaining 1235 SNPs were assumed to be neutral although their neutrality could not be directly proven.
Table 1.
Anopheles cruzii sampling information.
| Collection site | Trap | Number of specimens* | Geographic coordinates | Vegetation cover (%) | ||
|---|---|---|---|---|---|---|
| 2016 | 2017 | Latitude | Longitude | |||
| Natural | CDC canopy | 30 | 25 | 23° 56.378′S | 46° 41.659′W | 92 |
| CDC ground | 30 | – | ||||
| Shannon | 30 | 26 | ||||
| Suburban/rural | CDC canopy | 22 | 25 | 23° 54.556′S | 46° 42.167′W | 71 |
| CDC ground | 30 | – | ||||
| Shannon | 30 | 30 | ||||
| Urban | CDC canopy | 18 | 9 | 23° 54.395′S | 46° 42.486′W | 63 |
| CDC ground | 15 | – | ||||
| Shannon | 30 | 30 | ||||
*Specimens randomly selected from collections conducted once a month from February 2016 to July 2017.
Figure 2.
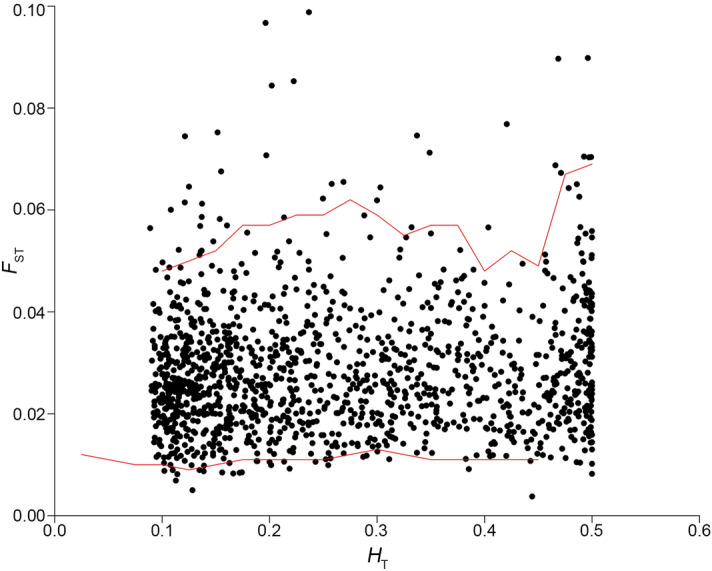
Detection of outlier SNPs. Graph of the distribution of heterozygosity values in relation to FST values of all filtered SNPs. Each locus is represented by a black dot in the graph. The red lines show 95% smoothed quantiles calculated by fsthet.
Comparisons of the test for Hardy–Weinberg equilibrium (HWE) for all the populations showed that expected heterozygosity was higher than observed heterozygosity (Ho) (Table 2). However, according to the P values, the deviations from HWE were not statistically significant.
Table 2.
Genetic descriptors for 1235 SNPs in Anopheles cruzii populations for all tested hypotheses.
| Hypothesis | Area | Population | Ho | He | HWE (non-corrected P value) | HWE (corrected P value) |
|---|---|---|---|---|---|---|
| Hypothesis 1 | All areas | Canopy | 0.192 | 0.2647 | 0.2592 | 0.3099 |
| Ground | 0.1907 | 0.2638 | 0.2442 | 0.2888 | ||
| Hypothesis 2 | Natural | Canopy | 0.1963 | 0.269 | 0.4423 | 0.5642 |
| Ground | 0.1862 | 0.2643 | 0.4349 | 0.5367 | ||
| Suburban/rural | Canopy | 0.187 | 0.2717 | 0.4487 | 0.5562 | |
| Ground | 0.1994 | 0.2638 | 0.4855 | 0.6297 | ||
| Urban | Canopy | 0.2119 | 0.2709 | 0.618 | 0.7587 | |
| Ground | 0.2057 | 0.2872 | 0.5587 | 0.6597 | ||
| Hypothesis 3 | Natural | 0.1892 | 0.2648 | 0.133 | 0.149 | |
| Suburban/rural | 0.1889 | 0.2634 | 0.1342 | 0.1499 | ||
| Urban | 0.1897 | 0.2626 | 0.1927 | 0.2233 | ||
| Hypothesis 4 | All areas | 2016 | 0.1842 | 0.2627 | 0.122 | 0.1357 |
| 2017 | 0.1921 | 0.2645 | 0.0833 | 0.0898 | ||
| Hypothesis 5 | Natural | 2016 | 0.1853 | 0.2639 | 0.1895 | 0.2172 |
| 2017 | 0.1966 | 0.2658 | 0.3523 | 0.4389 | ||
| Suburban/rural | 2016 | 0.1853 | 0.2622 | 0.2039 | 0.2354 | |
| 2017 | 0.1946 | 0.265 | 0.3206 | 0.3938 | ||
| Urban | 2016 | 0.1853 | 0.2639 | 0.1894 | 0.2171 | |
| 2017 | 0.1966 | 0.2658 | 0.3523 | 0.4389 |
Observed heterozygosity (Ho), Expected heterozygosity (He), Hardy–Weinberg equilibrium (HWE).
Hypothesis 1 and Hypothesis 2
Genetic structure
Global statistics performed to measure population structure (D57, FST and G’’ST58) and the inbreeding coefficient (FIS) between populations, yielded low values that were not statistically significant for the tests of hypothesis 1, indicating that there is no genetic structure between pooled canopy and ground-level populations from the Natural, Suburban/Rural and Urban environments (Table 3). All population structure estimates for the tests of hypothesis 2 for populations from Natural and Suburban/Rural environments also yielded low values that were not statistically significant (Table 3). In contrast, estimates for the Urban population revealed low and statistically significant values for D, FST and FIS, except for the estimator G”ST, which showed moderate genetic structure, albeit with a non-significant P value (Table 3). Pairwise estimates of FST, G”ST and D among all populations in hypothesis 2 revealed significant estimates for the same pairs of populations, Natural ground x Suburban/Rural ground, Natural ground x Urban ground, and Urban canopy x Urban ground (S3 Table). Suggesting that the ground-level population in all areas is more structured than the tree-canopy populations. However, after the correction for multiple tests, all P values for all tests became non-significant. Analysis of molecular variance (AMOVA) carried out to detect population differentiation, showed that 99.83% and 99.24% of the variance for the tests of hypothesis 1 and 2, respectively, were estimated to be within populations, with non-significant P values (S4 Table). These results indicate that there is no population structure between the populations from the Natural, and Suburban/Rural environment. For the Urban environment, however, the results revealed that there may be genetic structure at a low hierarchical level.
Table 3.
Population structure statistics. Global estimates of D, FST, FIS and G”ST for all SNPs for the tests of Hypothesis 1 and Hypothesis 2.
| Hypothesis | Area | Population structure | |||
|---|---|---|---|---|---|
| Statistics | Estimate | Non-corrected P value | Corrected P value | ||
| Hypothesis 1 | All areas | D | 0.000229923 | 0.6183816 | 0.848255 |
| FST | − 0.000702856 | 0.8941059 | 0.895105 | ||
| FIS | − 0.000989916 | 0.8951049 | 0.895105 | ||
| G''ST | 0.02421096 | 0.8941059 | 0.895105 | ||
| Hypothesis 2 | Natural | D | 0.000526055 | 0.7582418 | 0.863137 |
| FST | 3.46145E − 05 | 0.4335664 | 0.846953 | ||
| FIS | 4.7792E − 05 | 0.4335664 | 0.846953 | ||
| G''ST | 0.06047334 | 0.4285714 | 0.846953 | ||
| Suburban/rural | D | 0.000759986 | 0.3396603 | 0.846953 | |
| FST | 0.000353953 | 0.3876124 | 0.846953 | ||
| FIS | 0.000509479 | 0.3876124 | 0.846953 | ||
| G''ST | 0.07533064 | 0.5314685 | 0.848255 | ||
| Urban | D | 0.001501095 | 0.008991009 | 0.210646 | |
| FST | 0.004968052 | 0.040959041 | 0.210646 | ||
| FIS | 0.007001884 | 0.040959041 | 0.210646 | ||
| G''ST | 0.1256977 | 0.065934066 | 0.296703 | ||
Statistically significant P values (> 0.05) are shown in bold. Hypothesis 1: Comparison of tree-canopy (70) and ground-level (75) Anopheles cruzii populations from all areas. Hypothesis 2: comparison of Anopheles cruzii populations from tree canopy and ground level separated by area classified according to the degree of anthropogenic modification (Natural: 30/30, suburban/rural: 22/30 and urban: 18/15).
Multivariate statistics
Principal component analysis (PCA) performed to assess genetic diversity among sampled populations revealed a high degree of overlapping in the test to verify hypothesis 1. The data variance explained by PC1 and PC2 of PCA was 2.4% and 2%, respectively (Fig. 3a). Overall, the population from the canopy habitat was more diverse and had more outliers. In the test of hypothesis 2, the data variances explained by PC1 and PC2 for the Natural population were 3.8% and 3.4%, respectively (Fig. 3b). There is a high degree of overlapping in the plot, and the mosquitoes collected at the canopy are genetically more diverse than the ones collected at ground-level. The data variances explained by PC1 and PC2 for the Suburban/Rural population were 3.8% and 3.5%, respectively (Fig. 3b). The plot shows overlapping, indicating similarity, and that both populations were highly dispersed. For the Urban population, the data variances explained by PC1 and PC2 were 5.1% and 4.9%, respectively (Fig. 3b). In the urban environment plot the ground-level population is more diverse than the canopy population, contrasting with the trend seen in the Natural and Suburban/Rural environments.
Figure 3.

Genetic variation of tree-canopy and ground-level Anopheles cruzii populations using principal component analysis. (a) Hypothesis 1: comparison of tree-canopy (70) and ground-level (75) Anopheles cruzii populations from all areas. (b) Hypothesis 2: comparison of Anopheles cruzii populations from tree canopy and ground level separated by area classified according to the degree of anthropogenic modification (natural: 30/30, suburban/rural: 22/30 and urban: 18/15). Data were transformed using PCA and plotted as a function of the first two principal components. In parenthesis: number of specimens used in the analyses.
Bayesian cluster analysis
The results of the Bayesian cluster analysis performed to infer population structure and subsequent application of the Evanno method, which identifies genetically homogeneous groups of individuals, revealed low structure in the An. cruzii populations studied (Fig. 4). For hypothesis 1, the most likely number of genetic groups was K = 2 according to the ΔK estimator. The STRUCTURE plot revealed two genetic groups across the two populations without a significant genetic structure between them (Fig. 4a). For hypothesis 2, the most likely number of groups for the Natural population was K = 2 according to the ΔK estimator. The STRUCTURE plot showed genetic groups across the two populations without a clear genetic structure between them.
Figure 4.

Genetic structure of tree-canopy and ground-level Anopheles cruzii populations in São Paulo. (a) Hypothesis 1: comparison of tree-canopy (70) and ground-level (75) Anopheles cruzii populations from all areas. (b) Hypothesis 2: comparison of tree-canopy and ground-level Anopheles cruzii populations separated by area classified according to the degree of anthropogenic modification (natural: 30/30, suburban/rural: 22/30 and urban: 18/15). Each of the individuals from the two populations collected in different areas is represented by a vertical line divided into different colored segments. The length of each segment represents the probability of the individual belonging to the genetic cluster represented by that color. In parenthesis: number of specimens used in the analyses.
The most likely number of groups for the Suburban/rural population was K = 3 according to the ΔK estimator. The STRUCTURE plot revealed three genetic groups well distributed between the two populations, and the canopy population showed slightly more variation in group membership than the ground-level population. The most likely number of groups for the Urban population was K = 3 according to the ΔK estimator. The STRUCTURE plot revealed three genetic groups which varied in membership between sympatric canopy and ground-level populations. While the canopy population is less structured with a predominance of the color green, the ground-level population appears to be more structured with an equal prevalence of the three genetic groups (Fig. 4b).
Hypothesis 3, Hypothesis 4 and Hypothesis 5
The tests for hypotheses 3, 4, and 5 did not reveal significant genetic structuring between populations, indicating that the three hypotheses should be rejected. Although genetic estimators (D, FST, and G”ST) and inbreeding coefficient (FIS) showed low estimates for the tests of hypothesis 3 with significant P value, after correction for multiple tests all P values became non-significant. Furthermore, the lack of structuring was corroborated by PCA and Bayesian analysis, which showed no signs of cross-sectional structuring between the populations from the Natural, Suburban/Rural and Urban environments (S1 Table; S1 Fig). Mantel test showed no correlation between genetic distance (FST/(1 − FST)) and geographic distance between Natural, Suburban/Rural and Urban environments, indicating no evidence of isolation by distance (IBD) between these populations (r = − 0.1449955; P = 0.6689). The analyses indicated that the An. cruzii populations collected in 2016 and 2017 did not show temporal variation when the environments with different degrees of anthropogenic modifications were analyzed pooled and separately (S2 Table; S2 Fig). AMOVA showed that more than 99% of the variance was estimated to be within populations in the tests for all hypotheses with non-significant P values (S4 Table).
Discussion
Deforestation and fragmentation of forests can have a negative impact on the abundance and survival of An. cruzii, a primary vector of human malaria in the Atlantic Forest, as this species is closely associated with forested areas59. However, because of its extremely anthropophilic nature, anthropogenic modifications can increase contact between this species and humans. We tested five hypotheses using a cross-sectional and longitudinal design to assess genetic structuring in sympatric An. cruzii populations and cross-sectional genetic structuring and microevolution in populations of this species from environments with different degrees of anthropogenic modifications.
Our findings suggest that An. cruzii has weaker fine-scale genetic structure than other Anopheles vectors in Brazil53,55. Nevertheless, the tests of hypothesis 2, in which An. cruzii populations collected in tree canopies and at ground level habitats in each environment were compared, yielded interesting results. PCA showed a different pattern of diversity in the Natural and Suburban/Rural environments, where the canopy habitat populations were more diverse, while in the Urban environment the ground-level habitat population appeared to be more diverse. To our knowledge, this is the first study in which An. cruzii populations collected in tree canopies and at ground level have been compared genetically. Our results show a clear structuring pattern between tree-canopy and ground-level populations from the Urban environment, and this was corroborated by all the analyses carried out. PCA and Bayesian cluster analyses showed that the urban ground-level population is more diverse and structured than the urban tree-canopy population.
While SNPs are reliable molecular markers for analyzing structure in mosquito populations on a microgeographic scale53,60, the SNP analyses showed higher expected heterozygosity than observed heterozygosity in all the hypotheses tested, indicating low levels of heterozygosis among the populations. Moreover, there was no evidence of cross-sectional structure between populations from the Natural, Suburban/Rural or Urban environments according to the PCA, Bayesian analysis and AMOVA, indicating that the differences between populations are very subtle, and only when closely related populations were compared were signs of local-level structuring observed. Although the genetic structure statistics (D, FST and G”ST) and the inbreeding coefficient (FIS) yielded statistically significant values for the test of hypothesis 3, all the estimated values were quite low indicating low genetic structure. Furthermore, the significant FIS values could indicate that the heterozygosity in An. cruzii populations could be reduced by inbreeding of individuals. However, after correction for multiple tests, all P values became non-significant. It is important to consider that this study is investigating subtle variations in sympatric An. cruzii populations in both microgeographic and fine-temporal scales, which in turn can impact the significance of P values for multiple tests as it does not consider the power of the tests61,62.
The specimens analyzed here were collected in three different environments and were considered three different populations. However, as there are no geographic barriers, such as mountains, bodies of water, or a large canyon between the environments, our results indicated that the specimens represent a large, well-distributed population with a high overall genetic homogeneity. Previous studies carried out with mosquitoes from Brazil on a micro-geographic scale, suggest that native species exhibited weaker genetic structure while invasive species were shown to be more genetically structured and diverse10,11,63,64.
In the present study, An. cruzii populations showed no signs of genetic structure and variation when analyzed over time although microevolutionary changes, reflected as a variation in wing-shape over time in this species in the same areas, appear to vary greatly in urban environments compared with sylvatic and peri-urban environments33. Molecular and phenotypic markers are often contradictory, and in a previous study by our group geometric morphometrics showed clear structuring over time in An. cruzii but genetic analysis did not indicate significant structuring in the same populations33. Studies comparing the results of analyses using wing geometry and microsatellite markers in mosquitoes have shown that wing patterns change faster than genetic patterns65. While shape in mosquitoes is determined by multiple genes and their expression, SNPs can be filtered to ensure that they are independent, neutral and bi-allelic markers. Hence, differences in outcomes are to be expected for these two approaches66,67.
The loss of genetic diversity in native populations may be associated with anthropogenic modifications in the environment (e.g., deforestation, agriculture and urbanization)1,53,68. In addition, there is growing evidence that animal and plant populations experience divergent selection in urban and nonurban environments, which can explain the increased levels of genetic diversity in the population from ground-level habitat in the urban environment in the present study1,5. While there is no evidence of the presence of An. cruzii in highly urbanized metropolitan areas69, this species is constantly found in human dwellings close to forest fragments and in ornamental bromeliads in the anthropic environment32,33,39,70. Although An. cruzii is a bromeliad-specialist and its abundance is clearly associated with humidity, forest cover and bromeliad availability, studies have shown that anthropogenic modifications in natural areas can drive vertical dispersal in this mosquito and a variation in its phenotype and genotype, which in turn may drive the dynamics of Plasmodium transmission involving this important vector33,37.
A previous study showed that although anthropogenic changes in the environment can lead to a reduction in the abundance of An. cruzii, this species is found more frequently at ground level habitat in environments with increased deforestation due to human use37, indicating that the acrodendrophily of An. cruzii can be affected by human modifications in natural areas and areas close to dwellings37,71. These ecological findings corroborate the findings of our genetic study, suggesting that the presence of An. cruzii at ground level could be associated with proximity to anthropogenically modified environments. This varying acrodendrophilic behavior of An. cruzii is known to have important epidemiological implications for the dynamics of malaria transmission28,34.
Our findings suggest that the changes promoted by human activities play an essential role in the vertical dispersal of An. cruzii. Anthropogenic fragmentation of the Atlantic Forest biome leads to a reduction in natural hosts for this mosquito to blood feed on, which in tree canopies would be mainly birds and nonhuman primates24,71,72. This reduction may be driving An. cruzii to look for blood sources at ground level in places with human dwellings close to forest patches, which corroborates the anthropophilic behavior of An. cruzii and provides further evidence for the transmission of human and simian Plasmodium to humans living in the vicinity of forested areas28,37,71.
Furthermore, previous studies have shown that in some areas An. cruzii is more present inhabiting tree canopies, while in other areas it can be found in similar numbers in tree canopies and at ground level37,73. In a study conducted at the Horto Florestal da Cantareira, São Paulo, 99% of An. cruzii specimens collected were found in the tree canopies and despite the high malaria prevalence among the simian population there were no human cases of malaria73, whereas in the state of Santa Catarina, in the south of Brazil, a region where simian and human malaria have been reported, An. cruzii was found abundantly in both tree canopies and at ground level73. The same phenomenon is likely occurring in Parelheiros, a region with low malaria endemicity in the city of São Paulo where An. cruzii was collected in high numbers in both tree canopies and at ground level habitats29,37,45.
The different patterns of An. cruzii acrodendrophily led previous authors to believe that this species could represent two different species34. After collecting An. cruzii specimens from tree canopies and ground level, Deane et al. (1984) marked them with dye and released them to test their pattern of vertical dispersal. When they had collected the marked specimens again, the authors noticed that mosquitoes released at ground level were collected in tree canopies and vice-versa. They suggested that the mosquitoes in the study area belonged to the same species but did not dismiss the possibility of An. cruzii being two species34. Although there is probably still movement between An. cruzii from tree canopies to ground level and vice-versa, our results suggest that in areas close to human dwellings, An. cruzii populations are more diverse and genetic structured at ground level.
Contrarily to this study, previous studies conducted in different regions of Brazil found significant genetic variation between populations of An. cruzii74–76. Evidence of structuring in An. cruzii populations from high altitudes and low altitudes were observed using both nuclear and mitochondrial markers74,75. Furthermore, it has been suggested that An. cruzii may be a complex with at least two sibling species occurring in the southern and southeastern regions of Brazil76,77. However, these studies were conducted using molecular markers other than SNPs and on a larger scale than the present study. The present study is not without limitations. The sampling was conducted in only one site of each type of habitat (Natural, Suburban/Rural and Urban) with no replications, therefore, the genetic differences found in this study, even though unlikely, could be due to a factor unique to that one site.
Effective vector-control strategies are very important to eradicate malaria. Because of their complex biology and ecology and similar genetic characteristics, An. cruzii populations are particularly difficult to control. The species moves from natural areas to domestic and peridomestic areas in search of blood sources, posing a challenge for control strategies based on integrated vector management28,78. In the past, deforestation and bromeliad destruction were common approaches adopted to reduce the population of this vector mosquito during malaria outbreaks, but these strategies are no longer acceptable79. Deforestation leads to loss of biodiversity, resulting in a reduction in the number of dead-end Plasmodium hosts in the Atlantic Forest and an increase in the likelihood of Plasmodium transmission to humans23,28.
Considering that An. cruzii is not a synanthropic species, a significant genetic structure between populations from Natural, Suburban/Rural, and Urban areas as well as tree-canopy and ground-level at the microgeographic scale indicates the disruption of its natural environments. Such presence of genetic variation in sympatric An. cruzii populations can be used as a proxy for ecosystem health, and to infer both the impact of deforestation and defaunation in its distribution as well as the extent of human exposure to this mosquito vector species. Environmental planning, forest conservation, and control of illegal human settlements and ecotourism in protected areas could play an important role in reducing the presence of this vector at ground level, resulting in turn in a likely reduction in plasmodial infections in the Atlantic Forest23,28.
Conclusions
Our findings point to the existence of structuring between populations of An. cruzii from tree canopies and ground level habitats in the urban environment, with the ground-level population showing higher genetic variation. Although this study has some limitations, our results point to the possibility that anthropogenic modifications leading to habitat fragmentation may be driving the biology of An. cruzii and maintaining genetic diversity and structure in populations more present at ground level. Our study design could be used to assess the genetics of An. cruzii populations in malaria-transmission areas where this species is abundant at ground level.
Material and methods
Study area
The chosen study area was the Capivari-Monos conservation area in the subdistrict of Parelheiros, São Paulo, Brazil. Three environments with different levels of anthropogenic environmental modifications were chosen: Natural—Atlantic Forest remnants on private property; Suburban/Rural—a transition area between Atlantic Forest remnants and a cattle range; and Urban—the Engenheiro Marsilac neighborhood. The map of sampling locations was produced using ArcGIS 10.2 (Esri, Redlands, CA).
Vegetation cover at each collection site was identified using the "Map of vegetation remnants of the Atlantic Forest Biome in the municipality of São Paulo" available at https://geosampa.prefeitura.sp.gov.br/PaginasPublicas/_SBC.aspx. A one-kilometer buffer, defined around each georeferenced collection site, was used to characterize vegetation cover at each collection site. The area within each buffer was then used to measure the proportion of vegetation cover around each collection site. Measurements were made in QGIS 2.18 (https://www.qgis.org)37.
Mosquito collections and identification of specimens
Mosquito collections were conducted once a month from February 2016 to July 2017. Adult specimens were collected using CO2-baited CDC light traps (CDC-LT)80 placed in tree canopies (at heights > 12 m) and at ground level (at a height of 1 m) for 14 h including dusk and dawn, when An. cruzii blood-feeding activity peaks. A Shannon light trap was also set up at each collection site for 2 h after dusk. Mosquitoes were collected over 17 months with the traps set in different locations within each collection site. When possible, 30 specimens collected throughout 17 months were randomly selected from each trap in each collection site to avoid bias introduced by analyzing siblings81,82. Mosquitoes were identified with the aid of taxonomic keys26 and stored in 1.5 mL tubes with isopropyl alcohol at room temperature until DNA was extracted.
SNP genotyping
Genomic DNA was extracted from mosquitoes’ whole body using the DNeasy Blood and Tissue kit (Qiagen) following the manufacturer’s protocol. A Qubit Fluorometer (ThermoFisher, Dietikon, Switzerland) was used to determine DNA concentration, and electrophoresis on 1% agarose gel was performed to assess the quality and purity of the samples. Raw genomic DNA was then sent to SNPsaurus (SNPsaurus, Oregon, USA) to be converted into nextRAD genotyping-by-sequencing libraries83.
Using Nextera technology (Illumina, Inc), genomic DNA was simultaneously fragmented and tagged with short adapter sequences to the cleaved ends. The Nextera reaction was scaled to fragment 10 ng of genomic DNA, of which up to 5 ng was used for input to help dilute inhibitors. Fragmented DNA was then amplified using Phusion Hot Start Flex DNA Polymerase (New England Biolabs, Inc., Ipswich, MA) with one of the primers matching the adapter and extending 8 nucleotides into the genomic DNA with the selective sequence TGCAGGAG. Thus, only fragments starting with a sequence that can be hybridized by the selective sequence of the primer will be efficiently amplified. PCR conditions were as follows: 3 min at 72 °C, 3 min at 98 °C, followed by 25 cycles of 45 s at 98 °C and 1 min at 75 °C. The nextRAD libraries were sequenced on a HiSeq 4000 with four lanes of single-end 150 bp reads (University of Oregon).
The genotyping analysis were processed using SNPsaurus nextRAD custom scripts (SNPsaurus, Oregon, USA)83,84 that trimmed the reads with BBDuk (BBMap tools, https://sourceforge.net/projects/bbmap/). Next, a de novo reference genome was created by collecting 10 million reads equally divided between the samples and excluding reads using the thresholds of 7 and 700, which have been empirically determined to produce de novo reference loci that are useful for alignment83. The remaining loci were then aligned to each other to identify allelic loci and collapse allelic haplotypes into a single representative. All reads were mapped to the reference with an alignment identity threshold of 0.95 using BBMap (BBMap tools). Genotype calling was performed using SAMtools85 and BCFtools86. The variant call format (VCF) file was filtered to remove alleles with a population frequency of less than 3% and loci that were heterozygous in all samples or had more than 2 alleles in a sample, suggesting collapsed paralogs. The absence of artifacts was checked by counting SNPs at every nucleotide position read and verifying that the number of SNPs did not increase with reduced base quality toward the end of the read87.
Statistical and genetic analysis
VCFtools (https://github.com/vcftools)88 was used to perform the final SNP filtering of the genotype data. The criteria established for locus filtering were based on published data83,87: loci were filtered to assess minimum base quality (30), read depth (20), linkage disequilibrium to select a single independent SNP per locus, minimum allele frequency of 5% across all populations, exclusion of loci with more than 5% of missing data, and removal of indels.
The filtered dataset was entered in PGDSpider to convert the data into STRUCTURE 2.3.489 and Arlequin 3.590 formats. Filtered loci were analyzed to produce smoothed quantiles that enabled the relationship between FST and heterozygosity in each locus to be determined. This analysis uses the raw empirical data56 to identify outlier loci which can be under selection. The analysis was carried out with the R package fsthet56,91.
After filtering and identification of outlier loci, the resulting independent and neutral SNPs were used to characterize the populations genetically. Anopheles cruzii specimens collected in three different environments with different levels of anthropogenic changes (Natural, Suburban/Rural and Urban) were considered three different populations and its respective SNPs were then separated according to each of the five hypotheses tested. In the tests of each hypothesis, each sample being compared was considered a different ‘population’. Data corresponding to each hypothesis were analyzed with the statistical and genetic analysis software. All analyses comparing tree-canopy and ground-level samples were carried out with specimens collected only in the year of 2016.
Observed heterozygosity (Ho), expected heterozygosity (He) and Hardy–Weinberg equilibrium (HWE) tests for all SNPs per population per hypotheses were performed in Arlequin v3.590 to investigate heterozygosis among the populations. To assess the genetic variance between populations, we used the genetic structure estimators D57, FST and G’’ST58, and the inbreeding coefficient FIS were estimated globally in R using the StrataG package92. Pairwise estimates of D, FST and G’’ST among all populations in hypothesis 2 were conducted in R using the StrataG package92 to further investigate genetic structure between tree-canopy and ground-level populations. P values for all multiple tests were adjusted by applying the false discovery rate correction93 using the function p.adjust in R91. To assess genetic diversity among sampled population, we used the multivariate principal component analysis (PCA) carried out with adegenet94 and ade495 in R. Analysis of molecular variance (AMOVA) was performed in Arlequin 3.590 to detect population differentiation in all hypotheses. To test for isolation by distance for the populations in hypothesis 3, a Mantel test between genetic distance (FST/(1 − FST)) and geographic distance in kilometers was calculated with ade495 package in R91 using 9,999 permutations.
Bayesian cluster analysis in STRUCTURE was performed with the StrataG package in R to infer population structure between populations in all tested hypotheses. The analysis was carried out assuming the Admixture model and correlated allelic frequencies among populations52–54. Run length was set to 100,000 MCMC replicates after a burn-in period of 100,000. The number of clusters (K) varied from 1 to 10, with 10 replicates for each value of K83. The most likely number of clusters was determined by the ΔK method96, which identifies genetically homogeneous groups of individuals, used in Structure Harvester97.
Supplementary information
Acknowledgements
We would like to thank the São Paulo Research Foundation (FAPESP) (BIOTA Program, Grants 2014/50444-5, 2014/10919-4 and 2016/166471) and the National Council for Scientific and Technological Development, Brazil, (CNPq 301466/2015-7) for providing financial support. LCM is the recipient of a doctoral fellowship from FAPESP (Grant No. 2015/23386-7). The authors would like to thank Walter Ceretti Junior and Antônio Ralph Medeiros de Sousa for helping with the field collections and Aristides Fernandes for identifying the specimens. Special thanks are due to Tatiane Marques Porangaba de Oliveira, Luis Filipe Mucci, Ana Maria Ribeiro de Castro Duarte, Fabiana Santos Silva and all the staff at the Department of Epidemiology in the School of Public Health, University of São Paulo, and the Superintendency for the Control of Endemic Diseases (SUCEN).
Author contributions
LCM, ABBW, and MTM designed the study. LCM and ALSS performed the methodology. LCM performed the formal data analysis and wrote the original manuscript draft. ABBW and MTM were responsible for funding and project administration. All authors reviewed and approved the final version of the manuscript.
Data availability
Data supporting the conclusions are included within the article. The dataset analyzed during the current study are available in the Mendeley Data repository, https://doi.org/10.17632/jd72b5v8vv.1.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-74152-3.
References
- 1.Johnson, M. T. J. & Munshi-South, J. Evolution of life in urban environments. Science. 358, eaam8327 (2017). [DOI] [PubMed]
- 2.Pelletier F, Coltman DW. Will human influences on evolutionary dynamics in the wild pervade the anthropocene? BMC Biol. 2018;16:1–10. doi: 10.1186/s12915-017-0476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilke ABB, Beier JC, Benelli G. Complexity of the relationship between global warming and urbanization—an obscure future for predicting increases in vector-borne infectious diseases. Curr. Opin. Insect Sci. 2019;35:1–9. doi: 10.1016/j.cois.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 4.McKinney ML. Urbanization as a major cause of biotic homogenization. Biol. Conserv. 2006;127:247–260. doi: 10.1016/j.biocon.2005.09.005. [DOI] [Google Scholar]
- 5.Knop E. Biotic homogenization of three insect groups due to urbanization. Glob. Change Biol. 2016;22:228–236. doi: 10.1111/gcb.13091. [DOI] [PubMed] [Google Scholar]
- 6.Medeiros-Sousa AR, Fernandes A, Ceretti-Junior W, Wilke ABB, Marrelli MT. Mosquitoes in urban green spaces: using an island biogeographic approach to identify drivers of species richness and composition. Sci. Rep. 2017;7:17826. doi: 10.1038/s41598-017-18208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chadee DD, Martinez R. Aedes aegypti (L.) in Latin American and Caribbean region: With growing evidence for vector adaptation to climate change? Acta Trop. 2016;156:137–143. doi: 10.1016/j.actatropica.2015.12.022. [DOI] [PubMed] [Google Scholar]
- 8.Wilke ABB, et al. Community composition and year-round abundance of vector species of mosquitoes make Miami-Dade County, Florida a receptive gateway for arbovirus entry to the United States. Sci. Rep. 2019;9:8732. doi: 10.1038/s41598-019-45337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rochlin I, Faraji A, Ninivaggi DV, Barker CM, Kilpatrick AM. Anthropogenic impacts on mosquito populations in North America over the past century. Nat. Commun. 2016;7:1–14. doi: 10.1038/ncomms13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Multini LC, Wilke ABB, Suesdek L, Marrelli MT. Population genetic structure of Aedes fluviatilis (Diptera: Culicidae) PLoS ONE. 2016;11:e0162328. doi: 10.1371/journal.pone.0162328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilke ABB, Wilk-da-Silva R, Marrelli MT. Microgeographic population structuring of Aedes aegypti (Diptera: Culicidae) PLoS ONE. 2017;12:e0185150. doi: 10.1371/journal.pone.0185150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaDeau SL, Allan BF, Leisnham PT, Levy MZ. The ecological foundations of transmission potential and vector-borne disease in urban landscapes. Funct. Ecol. 2015;29:889–901. doi: 10.1111/1365-2435.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver SC. Urbanization and geographic expansion of zoonotic arboviral diseases: Mechanisms and potential strategies for prevention. Trends Microbiol. 2013;21:360–363. doi: 10.1016/j.tim.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Araujo RV, et al. São Paulo urban heat islands have a higher incidence of dengue than other urban areas. Braz. J. Infect. Dis. 2015;19:146–155. doi: 10.1016/j.bjid.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gould E, Pettersson J, Higgs S, Charrel R, de Lamballerie X. Emerging arboviruses: Why today? One Health. 2017;4:1–13. doi: 10.1016/j.onehlt.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker Lima JM, Vittor A, Rifai S, Valle D. Does deforestation promote or inhibit malaria transmission in the Amazon? A systematic literature review and critical appraisal of current evidence. Philos. Trans. R. Soc. B Biol. Sci. 2017;372:201. doi: 10.1098/rstb.2016.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira MU, Castro MC. Challenges for malaria elimination in Brazil. Malar. J. 2016;15:284. doi: 10.1186/s12936-016-1335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:853–858. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- 19.Rezende CL, et al. From hotspot to hopespot: an opportunity for the Brazilian Atlantic Forest. Perspect. Ecol. Conserv. 2018;16:208–214. [Google Scholar]
- 20.Morellato LPC, Haddad CFB. Introduction: the Brazilian Atlantic Forest 1. Biotropica. 2000;32:786–792. doi: 10.1111/j.1744-7429.2000.tb00618.x. [DOI] [Google Scholar]
- 21.Ribeiro MC, Metzger JP, Martensen AC, Ponzoni FJ, Hirota MM. The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implic. Conserv. Biol. Conserv. 2009;142:1141–1153. doi: 10.1016/j.biocon.2009.02.021. [DOI] [Google Scholar]
- 22.Joly CA, Metzger JP, Tabarelli M. Experiences from the Brazilian Atlantic Forest: ecological findings and conservation initiatives. New Phytol. 2014;204:459–473. doi: 10.1111/nph.12989. [DOI] [PubMed] [Google Scholar]
- 23.Laporta GZ, De Prado PIKL, Kraenkel RA, Coutinho RM, Sallum MAM. Biodiversity can help prevent malaria outbreaks in tropical forests. PLoS Negl. Trop. Dis. 2013;7:e2139. doi: 10.1371/journal.pntd.0002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marrelli MT, Malafronte RS, Sallum MA, Natal D. Kerteszia subgenus of Anopheles associated with the Brazilian Atlantic rainforest: current knowledge and future challenges. Malar. J. 2007;6:127. doi: 10.1186/1475-2875-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gadelha P. From, “forest malaria” to “bromeliad malaria”: a case-study of scientific controversy and malaria control. Parassitologia. 1994;36:175–195. [PubMed] [Google Scholar]
- 26.Forattini, O. P. Culicidologia Médica. (Editora da Universidade de São Paulo, 2002).
- 27.de Pina-Costa A, et al. Malaria in Brazil: what happens outside the Amazonian endemic region. Mem. Inst. Oswaldo Cruz. 2014;109:618–633. doi: 10.1590/0074-0276140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Multini LC, Marrelli MT, Beier JC, Wilke ABB. Increasing complexity threatens the elimination of extra-Amazonian malaria in Brazil. Trends Parasitol. 2019;35:383–387. doi: 10.1016/j.pt.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Duarte AMR, et al. Natural infection in anopheline species and its implications for autochthonous malaria in the Atlantic forest in Brazil. Parasit. Vectors. 2013;6:58. doi: 10.1186/1756-3305-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sallum MA, et al. Finding connections in the unexpected detection of Plasmodium vivax and Plasmodium falciparum DNA in asymptomatic blood donors: a fact in the Atlantic Forest. Malar. J. 2014;13:337. doi: 10.1186/1475-2875-13-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brasil P, et al. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob. Health. 2017;5:e1038–e1046. doi: 10.1016/S2214-109X(17)30333-9. [DOI] [PubMed] [Google Scholar]
- 32.Laporta GZ, et al. Plasmodium falciparum in the southeastern Atlantic forest: a challenge to the bromeliad-malaria paradigm? Malar. J. 2015;14:1–12. doi: 10.1186/s12936-015-0680-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Multini LC, Wilke ABB, Marrelli MT. Urbanization as a driver for temporal wing-shape variation in Anopheles cruzii (Diptera: Culicidae) Acta Trop. 2019;190:30–36. doi: 10.1016/j.actatropica.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Deane LM, Ferreira Neto JA, Lima MM. The vertical dispersión of Anopheles (Kerteszia) cruzi in a forest in southern Brazil suggests that human cases of malaria of simian origin might be expected. Mem. Inst. Oswaldo Cruz. 1984;79:461–463. doi: 10.1590/S0074-02761984000400011. [DOI] [PubMed] [Google Scholar]
- 35.Ueno HM, Forattini OP, Kakitani I. Vertical and seasonal distribution of Anopheles (Kerteszia) in Ilha Comprida, Southeastern Brazil. Rev. Saude Publica. 2007;41:269–275. doi: 10.1590/S0034-89102007000200014. [DOI] [PubMed] [Google Scholar]
- 36.Yamasaki T, et al. Detection of etiological agents of malaria in howler monkeys from Atlantic Forests, rescued in regions of São Paulo city. Brazil. J. Med. Primatol. 2011;40:392–400. doi: 10.1111/j.1600-0684.2011.00498.x. [DOI] [PubMed] [Google Scholar]
- 37.Medeiros-Sousa AR, et al. Effects of anthropogenic landscape changes on the abundance and acrodendrophily of Anopheles (Kerteszia) cruzii, the main vector of malaria parasites in the Atlantic Forest in Brazil. Malar. J. 2019;18:110. doi: 10.1186/s12936-019-2744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffing SM, Tauil PL, Udhayakumar V, Silva-Flannery L. A historical perspective on malaria control in Brazil. Mem. Inst. Oswaldo Cruz. 2015;110:701–718. doi: 10.1590/0074-02760150041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marques GRAM, Forattini OP. Encontro de imaturos de Anopheles cruzii em bromélias de área urbana, litoral de São Paulo. Rev. Saude Publica. 2009;43:369–372. doi: 10.1590/S0034-89102009005000006. [DOI] [PubMed] [Google Scholar]
- 40.Forattini OP, Kakitani I, Massad E, Marucci D. Studies on mosquitoes (Diptera: Culicidae) and anthropic environment: 4—Survey of resting adults and synanthropic behaviour in South-Eastern. Brazil. Rev. Saude Publica. 1993;27:398–411. doi: 10.1590/S0034-89101993000600002. [DOI] [PubMed] [Google Scholar]
- 41.Guimarães AÉ, Gentile C, Lopes CM, De Mello RP. Ecology of mosquitoes (Diptera: Culicidae) in areas of Serra do Mar State Park, State of São Paulo, Brazil. II -Habitat Distribution. Mem. Inst. Oswaldo Cruz. 2000;95:17–28. doi: 10.1590/S0074-02762000000100002. [DOI] [PubMed] [Google Scholar]
- 42.Kirchgatter K, et al. Anopheles (Kerteszia) cruzii (Diptera: Culicidae) in peridomiciliary area during asymptomatic malaria transmission in the atlantic forest: molecular identification of blood-meal sources indicates humans as primary intermediate hosts. Rev. Inst. Med. Trop. Sao Paulo. 2014;56:403–409. doi: 10.1590/S0036-46652014000500006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ministério da Saúde. Sistema de Informação de Agravos de Notificação Sinan Net. (2018). Available at: https://portalsinan.saude.gov.br/malaria. (Accessed: 1st Oct 2019).
- 44.Meneguzzi VC, et al. Use of geoprocessing to define malaria risk areas and evaluation of the vectorial importance of anopheline mosquitoes (Diptera: Culicidae) in Espírito Santo. Braz. Mem. Inst. Oswaldo Cruz. 2009;104:570–575. doi: 10.1590/S0074-02762009000400006. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro AF, et al. Mosquitoes in degraded and preserved areas of the Atlantic Forest and potential for vector-borne disease risk in the municipality of São Paulo, Brazil. J. Vector Ecol. 2012;37:316–324. doi: 10.1111/j.1948-7134.2012.00233.x. [DOI] [PubMed] [Google Scholar]
- 46.Morin, P. A., Luikart, G., Wayne, R. K. & the SNP workshop group. SNPs in ecology, evolution and conservation. Trends Ecol. Evol.19, 208–216 (2004).
- 47.Helyar SJ, et al. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol. Ecol. Resour. 2011;11:123–136. doi: 10.1111/j.1755-0998.2010.02943.x. [DOI] [PubMed] [Google Scholar]
- 48.Vignal A, Milan D, SanCristobal M, Eggen A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 2002;34:275–305. doi: 10.1186/1297-9686-34-3-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee Y, Marsden CD, Nieman C, Lanzaro GC. A new multiplex SNP genotyping assay for detecting hybridization and introgression between the M and S molecular forms of Anopheles gambiae. Mol. Ecol. Resour. 2014;14:297–305. doi: 10.1111/1755-0998.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y, Seifert SN, Fornadel CM, Norris DE, Lanzaro GC. Single-nucleotide polymorphisms for high-throughput genotyping of Anopheles arabiensis in East and Southern Africa. J. Med. Entomol. 2012;49:307–315. doi: 10.1603/ME11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles A, et al. Genetic diversity of the African malaria vector Anopheles gambiae. Nature. 2017;552:96–100. doi: 10.1038/nature24995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emerson KJ, Conn JE, Bergo ES, Randel MA, Sallum MAM. Brazilian Anopheles darlingi Root (Diptera: Culicidae) clusters by major biogeographical region. PLoS ONE. 2015;10:e0130773. doi: 10.1371/journal.pone.0130773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campos M, et al. Microgeographical structure in the major Neotropical malaria vector Anopheles darlingi using microsatellites and SNP markers. Parasit. Vectors. 2017;10:76. doi: 10.1186/s13071-017-2014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prussing C, et al. Decreasing proportion of Anopheles darlingi biting outdoors between long-lasting insecticidal net distributions in peri-Iquitos. Amazonian Peru. Malar. J. 2018;17:1–14. doi: 10.1186/s12936-018-2234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campos M, et al. Genetic diversity of Nyssorhynchus (Anopheles) darlingi related to biting behavior in western Amazon. Parasit. Vectors. 2019;12:1–9. doi: 10.1186/s13071-019-3498-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flanagan SP, Jones AG. Constraints on the FST–heterozygosity outlier approach. J. Hered. 2017;108:561–573. doi: 10.1093/jhered/esx048. [DOI] [PubMed] [Google Scholar]
- 57.Jost L. G ST and its relatives do not measure differentiation. Mol. Ecol. 2008;17:4015–4026. doi: 10.1111/j.1365-294X.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- 58.Meirmans PG, Hedrick PW. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011;11:5–18. doi: 10.1111/j.1755-0998.2010.02927.x. [DOI] [PubMed] [Google Scholar]
- 59.Dorvillé LFM. Mosquitoes as bioindicators of forest degradation in southeastern Brazil, a statistical evaluation of published data in the literature. Stud. Neotrop. Fauna Environ. 1996;31:68–78. doi: 10.1076/snfe.31.2.68.13331. [DOI] [Google Scholar]
- 60.Schmidt TL, et al. Genome-wide SNPs reveal the drivers of gene flow in an urban population of the Asian Tiger Mosquito, Aedes albopictus. PLoS Negl. Trop. Dis. 2017;11:1–20s. doi: 10.1371/journal.pntd.0006009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sobota RS, et al. Addressing population-specific multiple testing burdens in genetic association studies. Ann. Hum. Genet. 2015;79:136–147. doi: 10.1111/ahg.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fadista J, Manning AK, Florez JC, Groop L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 2016;24:1202–1205. doi: 10.1038/ejhg.2015.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Multini LC, de Souza AL, Marrelli MT, Wilke AB. Population structuring of the invasive mosquito Aedes albopictus (Diptera: Culicidae) on a microgeographic scale. PLoS ONE. 2019;14:e0220773. doi: 10.1371/journal.pone.0220773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilke ABB, de Carvalho GC, Marrelli MT. Retention of ancestral polymorphism in Culex nigripalpus (Diptera: Culicidae) from São Paulo, Brazil. Infect. Genet. Evol. 2018;65:333–339. doi: 10.1016/j.meegid.2018.08.017. [DOI] [PubMed] [Google Scholar]
- 65.Louise C, Vidal PO, Suesdek L. Microevolution of Aedes aegypti. PLoS ONE. 2015;10:e0137851. doi: 10.1371/journal.pone.0137851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lorenz C, et al. Geometric morphometrics in mosquitoes: What has been measured? Infect. Genet. Evol. 2017;54:205–215. doi: 10.1016/j.meegid.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 67.Ayala D, et al. Chromosomal and environmental determinants of morphometric variation in natural populations of the malaria vector Anopheles funestus in Cameroon. Infect. Genet. Evol. 2011;11:940–947. doi: 10.1016/j.meegid.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gardner TA, et al. Prospects for tropical forest biodiversity in a human-modified world. Ecol. Lett. 2009;12:561–582. doi: 10.1111/j.1461-0248.2009.01294.x. [DOI] [PubMed] [Google Scholar]
- 69.Ceretti-Junior W, et al. Species composition and ecological aspects of immature mosquitoes (Diptera: Culicidae) in bromeliads in urban parks in the City of São Paulo, Brazil. J. Arthropod. Borne. Dis. 2016;10:102–112. [PMC free article] [PubMed] [Google Scholar]
- 70.Forattini OP, Kakitani I, Massad E, Marucci D. Studies on mosquitoes (Diptera: Culicidae) and anthropic environment: 11—Biting activity and blood-seeking parity of Anopheles (Kerteszia) in South-Eastern Brazil. Rev. Saude Publica. 1996;30:107–114. doi: 10.1590/S0034-89101996000200001. [DOI] [PubMed] [Google Scholar]
- 71.Forattini OP, O de Souza Lopes, E. X. R. Investigações sobre o comportamento de formas adulta de mosquitos silvestres no estado de São Paulo, Brasil. Rev Saude Publ 2, 111–173 (1968). [PubMed]
- 72.Deane LM. Simian malaria in Brazil. Mem. Inst. Oswaldo Cruz. 1992;87(Suppl 3):1–20. doi: 10.1590/S0074-02761992000700001. [DOI] [PubMed] [Google Scholar]
- 73.Deane, L. M., Deane, M. P., Ferreira Neto, J. A. & Barbosa de Almeida, F. On the transmission of simian malaria in Brazil. Revista do Instituto de Medicina Tropical de São Paulo 13, 311–319 (1971). [PubMed]
- 74.Lorenz C, Marques TC, Sallum MAM, Suesdek L. Altitudinal population structure and microevolution of the malaria vector Anopheles cruzii (Diptera: Culicidae) Parasit. Vectors. 2014;7:1–12. doi: 10.1186/s13071-014-0581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Rezende Dias G, et al. Cryptic diversity in an Atlantic Forest malaria vector from the mountains of South-East Brazil. Parasit. Vectors. 2018;11:36. doi: 10.1186/s13071-018-2615-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rona LD, Carvalho-Pinto CJ, Peixoto A. Evidence for the occurrence of two sympatric sibling species within the Anopheles (Kerteszia) cruzii complex in southeast Brazil and the detection of asymmetric introgression between them using a multilocus analysis. BMC Evol. Biol. 2013;13:207. doi: 10.1186/1471-2148-13-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rona LDP, Carvalho-Pinto CJ, Mazzoni CJ, Peixoto AA. Estimation of divergence time between two sibling species of the Anopheles (Kerteszia) cruzii complex using a multilocus approach. BMC Evol. Biol. 2010;10:91. doi: 10.1186/1471-2148-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forattini OP, et al. The synanthropic potential of Kerteszia and Culex mosquitoes (Diptera: Culicidae) in Southeastern Brazil. Rev. Saude Publica. 2000;34:565–569. doi: 10.1590/S0034-89102000000600001. [DOI] [PubMed] [Google Scholar]
- 79.Deane LM. Malaria Studies and Control in Brazil. Am. J. Trop. Med. Hyg. 1988;38:223–230. doi: 10.4269/ajtmh.1988.38.223. [DOI] [PubMed] [Google Scholar]
- 80.Gomes AD, Rabello EX, Natal D. Uma nova câmara coletora para armadilha CDC-miniatura. Rev. Saude Publica. 1985;19:190–191. doi: 10.1590/S0034-89101985000200009. [DOI] [PubMed] [Google Scholar]
- 81.Hale ML, Burg TM, Steeves TE. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS ONE. 2012;7:e45170. doi: 10.1371/journal.pone.0045170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernández ME, et al. Comparison of the effectiveness of microsatellites and SNP panels for genetic identification, traceability and assessment of parentage in an inbred Angus herd. Genet. Mol. Biol. 2013;36:185–191. doi: 10.1590/S1415-47572013000200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russello MA, Waterhouse MD, Etter PD, Johnson EA. From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ. 2015;3:e1106. doi: 10.7717/peerj.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alvarado-Serrano DF, Van Etten ML, Chang SM, Baucom RS. The relative contribution of natural landscapes and human-mediated factors on the connectivity of a noxious invasive weed. Heredity (Edinb). 2019;122:29–40. doi: 10.1038/s41437-018-0106-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Narasimhan V, et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32:1749–1751. doi: 10.1093/bioinformatics/btw044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lanes ÉC, et al. Landscape genomic conservation assessment of a narrow-endemic and a widespread morning glory from Amazonian Savannas. Front. Plant Sci. 2018;9:1–13. doi: 10.3389/fpls.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol. Ecol. Notes. 2007;7:574–578. doi: 10.1111/j.1471-8286.2007.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinforma. 1, 117693430500100 (2005). [PMC free article] [PubMed]
- 91.R Core Team. R: A Language and Environment for Statistical Computing. 2, 1–12 (2017).
- 92.Archer FI, Adams PE, Schneiders BB. stratag: an r package for manipulating, summarizing and analysing population genetic data. Mol. Ecol. Resour. 2017;17:5–11. doi: 10.1111/1755-0998.12559. [DOI] [PubMed] [Google Scholar]
- 93.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 94.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 95.Dray S, Dufour A-B. The ade4 Package: implementing the duality diagram for ecologists. J. Stat. Softw. 2007;22:1–20. doi: 10.18637/jss.v022.i04. [DOI] [Google Scholar]
- 96.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 97.Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the conclusions are included within the article. The dataset analyzed during the current study are available in the Mendeley Data repository, https://doi.org/10.17632/jd72b5v8vv.1.