

Abstract
Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) Main protease (Mpro) is one of the vital drug targets amongst all the coronaviruses, as the protein is indispensable for virus replication. The study aimed to identify promising lead molecules against Mpro enzyme through virtual screening of Malaria Venture (MMV) Malaria Box (MB) comprising of 400 experimentally proven compounds. The binding affinities were studied using virtual screening based molecular docking, which revealed five molecules having the highest affinity scores compared to the reference molecules. Utilizing the established 3D structure of Mpro the binding affinity conformations of the docked complexes were studied by Molecular Dynamics (MD) simulations. The MD simulation trajectories were analysed to monitor protein deviation, relative fluctuation, atomic gyration, compactness covariance, residue-residue map and free energy landscapes. Based on the present study outcome, we propose three Malaria_box (MB) compounds, namely, MB_241, MB_250 and MB_266 to be the best lead compounds against Mpro activity. The compounds may be evaluated for their inhibitory activities using experimental techniques.
Keywords: SARS-CoV-2, COVID-19, Mpro, MMV Malaria_box, Virtual screening and MD simulations
Graphical abstract

1. Introduction
COVID-19 pandemic is of great global public health concern triggering worldwide deaths, morbidity, disastrous socio-economic and political consequences too. The causative agent of COVID-19, the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) progresses as phases involving viral translation and replication as an initial phase followed by a second phase comprising of host inflammatory response (Siddiqi and Mehra, 2020). Its successful human-to-human transmission by symptomatic as well as asymptomatic carriers have contributed in the rapid spread of the disease across the world (Ganyani et al., 2020; Mizumoto et al., 2020). Rigorous research studies are being conducted to mitigate the effects of the viral infection by devising novel therapeutic strategies. Many studies on these agents have reported their mechanism of action targeting various aspects of viral life cycle including blocking of viral attachment, entry, hindering viral replication or survival in host cell or reducing exaggerated host immune response (Fukao et al., 2007; Jo et al., 2020; Kruse, 2020; Wang et al., 2020; Yao et al., 2020). The SARS-CoV-2 genome is the largest among all RNA-viruses, ranging from 27 kb to 32 kb, packed with a helical capsid. This structure is formed by a Nucleocapsid protein and RNA surrounded by an envelope. There are three structural proteins associated with this envelope namely, membrane protein (M), envelope protein (E) and spike protein (S protein). The structural proteins, along with several viral proteases, are mainly involved in replication, assembly and fusion. The S protein facilitates the entry of the virus into human cells with the help of surface angiotensin-converting enzyme 2 (ACE2) receptor (Fig. 1 ).
Fig. 1.

Schematic diagram of SARS-CoV-2 surface proteins and Mpro, including ACE2 receptor binding, viral fusion and entry of virus in host cell as well as overall representation of in silico work-flow implemented in the current study.
Owing to its important role in the viral life cycle and initiation of the virus pathophysiology, the main protease (Mpro) or chymotrypsin-like protease (3CLpro) has served as an attractive target for development of drugs directed against coronaviruses. The enzyme is required to cleave replicase polypeptide to generate various viral assembly facilitating proteins (Fig. 1). The proteolytic processing mediated by Mpro involves several cleavage sites, generating various non-structural proteins important for viral replication (Anand et al., 2003; Qamar et al., 2020). It is a cysteine protease composed of three domains (I, II, III) and is conserved among all the coronaviruses (Dai et al., 2020). Along with sharing many common features in different coronaviruses, Mpro caters similar functions involving maturation of viral particles, capsid cleavage, polypeptide release, thereby occurrence of the infection (da Silva Hage-Melim et al., 2020). Use of viral protease inhibitors to block the key proteases to prevent viral replication is one of the most-explored strategies being investigated against coronavirus (Chen et al., 2020). The basic strategy involves identification of the most active compounds which can inhibit the viral protease, preventing the disease progression (Hsu et al., 2005). There are various groups which are conducting extensive research on drug repurposing (Agostini et al., 2018; Guy et al., 2020) or identifying an effective inhibitor against the Mpro (Jin et al., 2020; Mengist et al., 2020; Zhang et al., 2020). Despite the intensive research that has been going on, there are no effective drugs or vaccines for the pandemic till date.
In the present study we have used a comprehensive approach involving virtual screening based molecular docking and Molecular Dynamics (MD) simulations to identify potential Mpro inhibitors from a pool of compounds present in Medicines for Malaria Venture (MMV) Malaria Box (MB) (Spangenberg et al., 2013). The novelty in the present study is the use of MB database, having 400 compounds, that are chemically diverse, pharmacologically active and experimentally proven to inhibit the growth of Plasmodium parasites effectively (Duffy and Avery, 2012; Viswanadhan et al., 1989; Walters and Namchuk, 2003). The 400 compounds, N3 inhibitor and Boceprevir drug (latter two as reference molecules) were subjected to virtual screening to evaluate its effective binding to the active cleft of Mpro. Based on the comparative analysis, we prioritised five compounds, namely MB_183, MB_241, MB_250, MB_266 and MB_380 from the MB dataset. The compounds were further subjected to MD simulations in comparison to reference molecules. The compounds were consequently analysed for ADME/T properties and were found to be potential drug-like candidates that can effectively bind the Mpro enzyme. Furthermore, we analysed the conformational stability of the docked complexes using MD simulations with the help of various parameters such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), Free Energy Landscapes (FEL), Hydrogen bond monitoring, Principle Component Analysis (PCA) and Residue-Residue Contact Map (RRCM). Based on MD simulation results the compounds, MB_241, MB_250 and MB_266 were identified to have high stable confirmations and other favourable properties indicating inhibitory activity towards the active pocket of Mpro, probably hindering the viral replication. Thus, in the current study, we propose three MB compounds to be potential Mpro inhibitors.
2. Material and methods
The work was carried out on High-performance computing (HPC) with 2 x 20 Cores Processors, 256 GB Ram; NVIDIA graphics card (GPU V100 32 GB); installed on an International Business Machines (IBM) server. The server is installed with various Bioinformatics Software such as GROMACS, Xmgrace and other software used in the study. Many offline and online Bioinformatics tools were applied in accomplishing the study.
2.1. Selection of target and standard reference molecules
Two reference molecules were used for all the subsequent analyses in the present study (Fig. 1). The co-crystal structure of Mpro embedded with N3 inhibitor (6LU7) (Liu et al., 2020) and the structure bound with Boceprevir drug (6WNP) were retrieved from PDB database (Anson et al., 2020) and the molecules 3D structures were saved in PDB file format.
2.2. Active site prediction and ligand preparation
The Mpro 4 digit PDB ID was used as input to identify the active site which gives significant insight to recognize surface structural pockets, shape and volume of every pocket, internal cavities of protein and surface areas. The active site and the interactive residues were selected with the help of PDBsum and CASTp online tools (Laskowski et al., 2018; Tian et al., 2018) and also cross-checked with literature sources such as PubMed (Anson et al., 2020; Liu et al., 2020). The ligand 2D/3D structures of the standard reference molecules, i.e. N3 inhibitor and Boceprevir drug were obtained from the PDB. The ligands were prepared using AutoDock Tools (ADT) and the file was saved as pdbqt format (Trott and Olson, 2010).
2.3. Protein preparation and grid generation
The 3D structure of Mpro was prepared using the ADT protein preparation wizard. The polar hydrogen bonds and missing hydrogen atoms were added while water molecules and hetero-atoms were deleted (Forli et al., 2016). The energy minimization was performed with a default constraint of 0.3 Å Root Mean Square (RMS) and charges were assigned. After protein preparation, clean structure was saved as a pdbqt file. The grid box of 126×126×126 Å was generated around the centroid of inhibitor/drug compounds with assigned X, Y and Z axis.
2.4. Virtual screening and binding affinity calculation
The ligand like subset of the MB database was utilized for the virtual screening to identify novel potential antagonist against Mpro receptor. The pdbqt formatted files were provided as input along with standard reference molecules (N3 and Boceprevir) and 400 drug-like compound dataset of MB were screened against Mpro (Duffy and Avery, 2012). The top five compounds were ranked on the basis of the binding energy scores and the docking poses (Forli et al., 2016; Trott and Olson, 2010). The compounds with favourable binding poses were identified with the help of lowest free energy were (ΔG), defined as using the equation as follows,
| ΔG = ΔGcomplex – (ΔGenzyme+ΔGligand) |
where, (ΔGcomplex), (ΔGreceptor) and (ΔGligand) are the average values of Gibbs free energy for the complex, receptor and ligand, respectively.
The stability of the docked complex between the receptor-ligand is better, if it exhibits more negative scores which also indicates a higher potency of inhibitor. All the other docking parameters were kept default and the final visualization of the docked complexes was performed using PyMOL visualization tool (DeLano, 2002). The active pocket of Mpro and docked pose of N3, Boceprevir and the top best MB compounds were compared to find interactive orientations. The detailed workflow followed in the study was presented in Fig. 1.
2.5. Determination of ADME/T properties
The Absorption, Digestion, Metabolism, Excretion/Toxicity (ADME/T) properties of the top five compounds from the virtual screening protocol were determined to evaluate drug-likeness using QikProp module of Schrödinger commercial software (Schrödinger, 2012). The input zone comprises a sketcher that helps the user to draw, edit and import the 2D structure. The output files can be saved as PDB formatted files, which can be enabled in any visualization tool. The analysis provides major physiochemical properties such as flexibility, molecular weight/size, hydrophobicity, bioavailability, permeability and polar solubility (Daina et al., 2017; Lee et al., 2003). The best-screened compounds were further assessed to fulfil the Lipinski's rule of five violations to exhibit drug-likeliness (Lipinski, 2016).
2.6. GROningen MAchine for chemical simulations (GROMACS)
MD simulations were executed for the best docked complexes with maximum binding affinity scores using GROMACS Version 5.18.3. Package (Abraham et al., 2015). The topology of Mpro was generated by using GROMOS9643a1 force field (Van Der Spoel et al., 2005). Due to the lack of suitable force field parameters for drug like molecules in the GROMACS software, PRODRG server was used for generation of molecular topologies and coordinate files (Schüttelkopf and Van Aalten, 2004). All the systems were solvated using simple point charge model (SPC/E) in a cubic box. To neutralize the system 0.15 M counter ions (Na+ and Cl−) were added. The energy minimization of all the neutralized systems was performed using the steepest descent and conjugate gradients (50000 steps for each). The regulation of volume (NVT) and pressure (NPT) was run for system equilibration. Steepest descent, followed by conjugate gradient algorithms was utilized on enzyme-ligand complexes. The NVT ensemble at a constant temperature of 300 K and constant pressure of 1 bar was employed. The SHAKE algorithm was used to confine the H atoms at their equilibrium distances and periodic boundary conditions. Moreover, the Particle Mesh Ewald (PME) method was used to define the long-range electrostatic forces (Abraham et al., 2015). The cut-offs for Van der Waals and columbic interactions were set at 1.0 nm. LINC algorithm was used to constrain the bonds and angles. Using the NPT ensemble, production runs were performed for the period of 100 ns, with time integration. The energy, velocity, and trajectory were updated at the time interval of 10ps. The Cα-atom deviations of the protein were calculated using Root Mean Square Deviations (RMSD). The relative fluctuations of each amino acid were defined with Root Mean Square Fluctuations (RMSF). To measure the compactness of given molecule radius of gyrations (Rg) is implemented and the Solvent Accessible Surface Area (SASA) was employed to know the electrostatic contributions of molecular solvation (Ahamad et al., 2018, 2019; Kanipakam et al., 2020).
The above-mentioned parameters were calculated with the following equations:
| Parameter | Defined equation |
|---|---|
| RMSD |
Where, M = Σi mi and ri(t) is the position of atom i at time t after least square fitting the structure to the reference structure. |
| RMSF |
where, T is the time over which one wants to average and riref is the reference position of particle i. |
| Rg |
Where, mi is the mass of atom i and ri the position of atom i with respect to the center of mass of the molecule. |
| SASA | SASA = 4πR2 *(Nacc/Ntot) Where, *Nacc and Ntot are the number of accessible and total number of points in the shell and where R is the sum of Van der Waal's radius. |
2.7. Principal component analysis (PCA)
PCA is one of the best techniques that help in reduction of complexity to extract the intensive motions in MD simulations analysis (Sang et al., 2017). In this method a matrix was constituted for all the trajectories after excluding rotational and translational movements. Herein, the essential dynamics protocol was implemented to calculate the eigenvectors and eigenvalues as well as their projections along the first two Principal Components (PC). By Diagonalising the matrix eigenvectors and eigenvalues were identified where eigenvalues represent the amplitude of the eigenvector (Abdi and Williams, 2010). The eigenvector of the matrix gives the multidimensional space and the displacement of atoms in the protein along each direction. In this process, the essential subspace is created to understand the movements of the atoms that are plotted by Cartesian trajectory coordinates using GROMACS utilities.
3. Results
3.1. Virtual screening (VS) with Malaria_Box
Based on the structural similarity with the inhibitors N3 and Boceprevir, a total of 400 experimentally proven compounds from the MB database were screened to obtain lead inhibitor. The five hits, namely MB_183, MB_241, MB_250, MB_266 and MB_380 were prioritised based on estimated free binding energies and the docking poses in the active site of Mpro. The five compounds were subjected to further analysis. The VS results yielded potential leads to inhibit Mpro activity, with the highest docking scores in comparison to the reference molecules, suggested as structure-based design reference for coronaviruses. Many studies on coronaviruses have shown that the inhibitor N3 can specifically inhibit Mpro, and has been reported to have potent antiviral activity (Ren et al., 2013; Wang et al., 2016; Xue et al., 2008; Yang et al., 2005). Similarly, another study reported 3D structural similarity with the Hepatitis C virus (HCV) NS3/4 A protease to the SARS-CoV2 Mpro protease particularly at the key active site residues arrangement (Bafna et al., 2020). Boceprevir, a drug to treat HCV, has a broad-spectrum antiviral activity, and its structural scaffolds have the potential to be rapidly optimized into potential anti-SARS-CoV-2 compounds (Anson et al., 2020).
3.2. Docking pose visualization and evaluation
The N3, Boceprevir and the five MB compounds were docked with Mpro with the assistance of AutoDock vina standalone suite. After successful docking experiments, the significant binding and the lowest energy conformations with Mpro were evaluated for further elucidation. The coloured molecular graphics were plotted using PyMOL visualization tool where, the ligands are represented as ball and stick model, whereas Mpro is shown in cartoon mode (Fig. 2 A–G). The amino acid residues interacting with the receptor are shown as line model and the H-bond formation as red dashed lines between the receptor-ligand complexes.
Fig. 2.

Representation of the binding poses, H-bonds and the amino acid residues in the active pocket of Mpro ligand interaction site (A) MB_183, (B) MB_241, (C) MB_250, (D) MB_266, (E) MB_380, (F) N3 inhibitor and (G) Boceprevir drug.
The substrate-binding site of Mpro lies between the domains I and II spanning the residues from 10 to 99 and 100 to 182, respectively, whereas domain III, a globular cluster of five helices starts from residues 198 to 303 is involved in regulating the Mpro dimerization (Zhang et al., 2020). There lies a crucial catalytic dyad formed by the conserved residues His41 and Cys145. In the analysis, it is found that the residues Thr24, Thr25, His41, Thr45, Ser46, Met49, Tyr54, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Glu166, Leu167 and Gln189 interact with all the five MB screened compounds. The interaction details of each complex are mentioned below.
3.2.1. Mpro-N3 and Mpro- Boceprevir complexes
The docking results of Mpro-N3 complex formed 5 H-bonds with Asn142, Gly143, Glu166, Leu167 and Gln189, forming stable confirmations (Fig. 2A). Additionally, the residues Thr24, Thr25, Thr26, Leu27, His41, Met49, Tyr51, Phe140, Leu141, Ser144, Cys145, His163, His164, Pro168, His172, Arg186, Asp187, Thr190, Ala191 and Gln192 provided stability to the Mpro-N3 complex along with several Van der Waal's interactions (Fig. 3 A). The complex Mpro-Boceprevir at the interactive site revealed formation of 5 H-bonds with the residues His41, Met49, Tyr54, Gly143 and His164 (Fig. 2B), and the residues His14, Thr25, Thr26, Leu27, Cys44, Leu50, Pro52, Leu141, Asn142, Ser144, Cys145 Met165, Glu166, Val186, Asp187, Arg188, Gln189, Thr190, Gln192 were involved in Van der Waal interactions (Fig. 3B). The docking scores of both the complexes were estimated to be −8.3 kcal/mol and −6.8 kcal/mol, respectively (Table 1 and Supplementary Table S2).
Fig. 3.

The interactive site between Mpro-ligands after docking studies depicting H-bonds, hydrophobic interactions, Van der Waal's interactions around 4 Å of the binding cavity A) MB_183, (B) MB_241, (C) MB_250 (D) MB_266, (E) MB_380, (F) N3 inhibitor and (G) Boceprevir drug.
Table 1.
The molecular details of five shortlisted MB compounds and the reference compounds, produced by molecular docking.
| S. No. | Ligand | 3D structures | SMILES Format | Binding Score | H-bond residues |
|---|---|---|---|---|---|
| 1 | MB_183 |  |
C1(NC[NH]C1C(=O)NC1[NH]C2CCCCC2N1)C(=O)NC1CCC(C)CC1C | −7.4 kcal/mol | Asn142 |
| 2 | MB_241 | 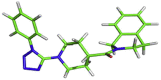 |
[C@H]1(CCN(CC1)[C@@H]1NNNN1C1CCCCC1)C(=O)N1CCC2CCCCC2C1 | −7.6 kcal/mol | Leu141, Ser144 and Glu166 |
| 3 | MB_250 |  |
C1(CCCC(C1NC(=O)C1CC(ON1)C1CCCCC1)O)C1OC2CCCC(C2N1)C(O)O | −8.8 kcal/mol | His41, Leu141 and His163 |
| 4 | MB_266 | 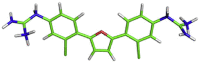 |
C1(OC(CC1)C1CCC(CC1CL)[NH2]C(=[NH2])[NH3])C1CCC(CC1CL)[NH2]C(=[NH2])[NH3] | −8.9 kcal/mol | Thr24, Thr25, Thr45, Ser46, Asn142 and Phe140 |
| 5 | MB_380 | 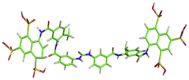 |
N(C(=O)NC1CCCC(C1)C(=O)NC1CC(CCC1C)C(=O)NC1CCC(C2CC(CC(C12)S(=O)(=O)O)S(=O)(=O)O)S(=O)(=O)O)C1CC(CCC1)C(=O)NC1C(C)CCC(C1)C(=O)NC1C2C(CC(CC2C(CC1)S(=O)(=O)O)S(=O)(=O)O)S(=O)(=O)O | −9.2 kcal/mol | Thr24, Thr25, Ser46, Asn142, and Gln189 |
| 6 | N3 inhibitor | 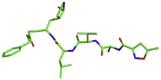 |
CC1=CC(=NO1)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](\C C/C(=O)OCC1=CC CC C1)C[C@@H]1C(NCC1) = O | −8.3 kcal/mol | Asn142, Gly143, Glu166, Leu167, and Gln189 |
| 7 | Boceprevir | 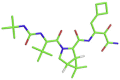 |
[H][C@]12CN([C@H](C(=O)NC(CC3CCC3)C(=O)C(N) O)[C@@]1([H])C2(C)C)C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C | −6.8 kcal/mol | His41, Tyr54, Met49, Gly143, and His164 |
3.2.2. Mpro- MB_183 docked complex
The binding affinity of compound MB_183 towards the Mpro active site is characterized by an H-bond with Asn142 (Fig. 2C). The complex was stabilized with a greater docking score than the standard reference compounds with −7.4 kcal/mol (Table 1). The other amino acid residues like Thr25, Thr26, Leu27, His41, Met49, Leu141, Gly143, Ser144, Cys145, His164, Met165, Glu166, Arg188, Gln189 and Thr190 were involved in hydrophobic interactions (Fig. 3C).
3.2.3. Mpro- MB_241 docked complex
The interaction between the Mpro-MB_241 docked complex shows 3 H-bonds with Leu141, Ser144 and Glu166 (Fig. 2D). Additionally, the residues His41, Cys145, Ser144, Gly143, Asn142, Phe140, His172, His163, His164, Met165, Glu166, Leu167, Pro168, Val186, Arg188, Gln189, Thr190, Ala191 and Gln192 are involved in hydrophobic and Van der Waal's interactions, stabilizing the receptor-ligand complex (Fig. 3D). The binding energy score of the complex was found −7.6 kcal/mol, comparable with N3 and Boceprevir complexes (Table 1).
3.2.4. Mpro-MB_250 docked complex
The molecular docking experiment of MB_250 towards the Mpro active pocket was stabilized with the formation of 3 H-bonds by residues His41, Leu141 and His163, respectively (Fig. 2E). Apart from H-bond formation, the residues Thr25, Thr26, Leu27, Met49, Tyr54, Tyr118, Phe140, Asn142, Gly143, Ser144, Cys145, His164, Glu166, Met165, His172, Val186, Asp187, Arg188, Gln189, Thr190 and Gln192 are seen in the 4 Å region around the active site sharing similar binding with the reference molecules (Fig. 3E). The Mpro-MB_250 docking complex was defined with highest docking energy of −8.8 kcal/mol (Table 1).
3.2.5. Mpro- MB_266 docked complex
The affinity of Mpro- MB_266 complex revealed that it fits in the active site of the reference compounds N3 and Boceprevir. The binding orientation of the docked complex was stabilized with a binding score of −8.9 kcal/mol (Table 1). The stabilization was further strengthened with the formation of 6 H-bonds with the residues Thr24, Thr25, Thr45, Ser46, Phe140 and Asn142 headed for Mpro receptor (Fig. 2F). The residues Cys22, Thr26, Leu27, His41, Cys44, Met49, Leu141, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, His172 and Gln189 were actively involved in the Van der Waal's interaction (Fig. 3F).
3.2.6. Mpro-MB_380 docked complex
The molecular docking results estimated that the complex Mpro-MB_380 interact at the active site cavity, stabilized with 5 H-bonds with the residues Thr24, Thr25, Ser46, Asn142 and Gln189 (Fig. 2G). The additional residues, Cys22, Thr24, Thr26, Leu27, His41, Val42, Ile43, Cys44, Glu47, Met49, Leu50, Asn51, Phe140, Leu141, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, Thr169, Gly170, His172, Ala173, Arg188, Ala191 and Gln192 are involved in hydrophobic interactions, Van der Waal's interactions and other contacts are depicted in the 2D plot (Fig. 3G). The docking energy is −9.2 kcal/mol for the complex, sharing similar binding pose with that of the reference compounds (Table 1).
In the present analysis, it was evident that the shortlisted compounds- MB_183, MB_241, MB_250, MB_266 and MB_380 are mimicking the same binding pattern as the co-crystallized inhibitors of SARS-CoV-2 Mpro. Interestingly, the three complexes MB_241, MB_250, MB_266 are found to have additional hydrogen bonds with the residues Thr24, Thr25, Thr45, Ser46, Phe140 and Ser144 along with the residues His41, Leu141, Asn142, His 163 and Glu166 (common in N3 and Boceprevir). This additional binding indicates a strengthened bond, thus a tighter fit to the binding pocket of Mpro, which may hinder the substrate accessibility and hence the downstream processes. The overall results revealed that the proposed three MB complexes MB_241, MB_250, MB_266 have an edge over the Mpro N3 and Boceprevir complexes attributable to stronger binding abilities.
3.3. Physiochemical and ADME/T properties
ADME/T and pharmacological properties are crucial for selecting and developing the drug candidates. The screened compounds were investigated for the same. The output of QikProp module reported no violation of pharmacological properties for the four compounds, namely MB_183, MB_241, MB_250 and MB_266, with blood-brain barrier penetration (−1.244 to 0.099), logP range from (−0.077 to 3.332), QPlogKhsa (−0.3 to −1.7), SASA (528–610), log Kp. The results revealed that the compounds were well within the acceptable range indicating them to possess high bioavailability and likeliness to be the lead like compounds (Table 2 A and 2B) (Lee et al., 2003; Lipinski, 2016).
Table 2.
ADME/T properties of the ligands in comparison to reference molecules, interpreting pharmacological parameters and Lipinski's rule predicted using QikProp module of Schrödinger.
| Table 2A. ADME/T properties | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | MW | dipole | SASA | FOSA | FISA | PISA | WPSA | volume | DonorHB | AccptHB | % HOA | RO5 | |||
| MB_183 | 396.57 | 0 | 557.49 | 449.09 | 108.40 | 0 | 0 | 1081.89 | 8 | 12.4 | 15.95 | 1 | |||
| MB_241 | 406.61 | 0 | 610.37 | 492.15 | 118.21 | 0 | 0 | 1211.44 | 4 | 11.2 | 58.57 | 0 | |||
| MB_250 | 469.62 | 0 | 528.52 | 413.29 | 115.22 | 0 | 0 | 1130.50 | 7 | 14.2 | 24.37 | 1 | |||
| MB_266 | 423.42 | 0 | 591.19 | 218.28 | 271.33 | 0 | 101.5 | 1103.16 | 10 | 8.7 | 0 | 1 | |||
| MB_380 | 1363.78 | 0 | 1328.07 | 472.39 | 852.84 | 0 | 2.8 | 2976.54 | 29 | 18.8 | 0 | 3 | |||
| Boceprevir | 529.76 | 0 | 809.27 | 639.60 | 169.67 | 0 | s0 | 1634.66 | 10 | 13.8 | 4.47 | 2 | |||
| N3 inhibitor | 694.91 | 0 | 1100.84 | 902.28 | 198.56 | 0 | 0 | 2205.81 | 4.75 | 15.65 | 4.372 | 3 | |||
| Table 2B. ADME/T properties | |||||||||||||||
| Compound | QPlogPo/w | QPlogS | CIQPlogS | QPPCaco | QPlogBB | QPPMDCK | QPlogKp | PSA | QPlogKhsa | ||||||
| MB_183 | −3.206 | 2 | 3.861 | 14.409 | 0.014 | 6.85 | −6.87 | 93.51 | −0.647 | ||||||
| MB_241 | 0.3 | −0.077 | −0.967 | 46.632 | −0.154 | 22.035 | −6.95 | 76.50 | −0.391 | ||||||
| MB_250 | −1.569 | 1.869 | 0.031 | 12.414 | 0.099 | 5.831 | −7 | 104.17 | −0.448 | ||||||
| MB_266 | −3.332 | 2 | 2.743 | 0.411 | −1.244 | 0.527 | −9.87 | 134.99 | −0.676 | ||||||
| MB_380 | −4.811 | 2 | −8.657 | 0 | −11.359 | 0 | −17.042 | 502.64 | −1.735 | ||||||
| Boceprevir | −1.177 | 2 | 1.761 | 3.781 | −1.49 | 1.613 | −6.852 | 132.55 | −0.686 | ||||||
| N3 inhibitor | 1.084 | −3.085 | −2.254 | 3.601 | −2.173 | 3.308 | −7.48 | 220.99 | −0.792 | ||||||
Minimal Ranges: MW = Molecular Weight (130.0/725.0), accPthB = Acceptor - Hydrogen Bonds (2.0/20.0), rotor = No. of Rotatable Bonds (0.0/15.0), logP o/w = log P for octanol/water (−2.0/6.5), dipole = Dipole Moment (1.0/12.5), logS = log S for aqueous solubility (−6.5/0.5), SASA = Total solvent accessible surface area (300.0/1000.0), CIlogS = log S - conformation independent (−6.5/0.5), FOSA =Hydrophobic solvent accessible surface area (0.0/750.0), logBB = log BB for brain/blood (−3.0/1.2), FISA =Hydrophilic solvent accessible surface area (7.0/330.0), log Kp = log Kp for skin permeability (Kp in cm/hr), PISA =Carbon Pi solvent accessible surface area (0.0/450.0), log Khsa = log Khsa Serum Protein Binding (−1.5/1.5), WPSA=Weakly Polar solvent accessible surface area (0.0/175.0), Lipinski Rule of 5 Violations-RO5 (maximum is 4), PSA = vdW Polar surface area (7.0/200.0), % Human Oral Absorption in GI (+-20%) (<25% is poor), volume = Molecular Volume (A∧3) (500.0/2000.0), Apparent Caco-2 Permeability (nm/sec) (<25 poor,>500 great), donorHB = Donor - Hydrogen Bonds (0.0/6.0), Apparent MDCK Permeability (nm/sec) (<25 poor,>500 great).
Lipinski rule violation: The compounds were further examined for the Lipinski rule of five parameters including molecular weight (396–469), Hydrogen bond Acceptor (8.7–14.8), Hydrogen bond donor (4–10), RO5 (0–3) and surface polar atoms (76–134). The values for the mentioned parameters were estimated to look for the presence of drug-like properties (Lipinski, 2016). Out of all, the four compounds namely MB_183, MB_241, MB_250, and MB_266 expect MB_380 fitted with the Lipinski's properties exhibiting drug-likeness there by having good absorption in the biological systems (Table 2A).
3.4. Molecular Dynamics (MD) simulations
To understand the complex stability and interaction profile of the most promising hit compounds inside the active site of SARS-CoV-2 Mpro, MD simulations of Mpro-native, Mpro-reference molecules (N3 and Boceprevir) and Mpro-MB compound complexes were performed for the period of 100ns. Structural parameters including RMSD, RMSF, Rg, SASA, PCA, Intermolecular H-bonds, Residue–Residue Contact Map, were evaluated as a function of time.
3.4.1. RMS-deviation and RMS-Fluctuations
The docked complexes were subjected to RMSD analysis to assess the residual flexibility of Mpro. The native protein, MB_241, MB_250, MB_266 and MB_380 noticeably showed steady RMSD of ~0.4 nm except MB_183, which showed slightly higher RMSD of ~0.5 nm. The N3 and Boceprevir showed RMSD of ~0.3 nm, respectively (Fig. 4 A). The Probability Distribution Function (PDF) analysis revealed that the native protein was stable with a threshold of 0.38 nm of average RMSD (Supplementary Table 1). The docking complexes of Mpro with MB_183, MB_241, MB_250, MB_266 and MB_380 reached equilibrium with average RMSD values of 0.45 nm, 0.32 nm, 0.28 nm, 0.29 nm, 0.29 nm, respectively (Fig. 4B). Subsequently, Mpro-N3 and Mpro-Boceprevir complexes showed stable equilibrium with RMSD-PDF average of 0.28 nm and 0.27 nm. Overall the compounds MB_241, MB_250 and MB_266 revealed similar RMSD values with N3 and Boceprevir, revealing a stable binding with Mpro under given simulation conditions. The RMS deviation of Cα-atoms remained stable with no significant differences in the values of all the three proposed complexes, indicating strong binding between them. The overall results suggested that the prioritised MB compounds reached dynamic equilibriums that were stable and reliable which bolstered the docking results. Further, to identify the flexible and rigid regions of the complex, RMSF analysis was implemented to measure the average atomic flexibility of the Cα-atoms of Mpro-docked complexes. All the complexes, including Mpro-Native N3, Boceprevir, MB_183, MB_241, MB_250, MB_266 and MB_380, revealed similar average RMSF values of ~0.2 nm (Fig. 4C). Even though the average fluctuations of all the complexes were very similar, the compounds MB_241, MB_250 and MB_266 displayed the highest degree of flexibility, exhibiting stable active site residues interaction in comparison to the reference molecules.
Fig. 4.

RMSD and RMSF analysis of the complexes of native protein, complexes of shortlisted ligands and reference complexes (A) RMSD plot (B) PDF of RMSD (C) Combined RMS fluctuations.
3.4.2. Rg and SASA examination
The compactness of the receptor-docked complexes was evaluated by calculation of radius of gyration (Rg). The results showed that the Rg values of the native protein; and the complexes with N3, Boceprevir and five MB compounds remained stable with a range between ~2.4 nm to ~2.13 nm throughout the MD simulation frame of 100ns (Fig. 5 A) (Supplementary Table 1). The PDF analysis confirmed the compactness of the native protein, MB_183, MB_241, MB_250, MB_266 and MB_380, N3 and Boceprevir docked complexes with Rg average of 2.07 nm, 2.04 nm, 2.08 nm, 2.10 nm, 2.12 nm, 2.09 nm, 2.09 nm and 2.13 nm, respectively (Fig. 5B). The comparative Rg results revealed stable folding behaviour of Mpro after binding with MB_241, MB_250, MB_266 compounds indicating high compactness between the complexes than that of reference molecules. This suggests that the MB compounds stayed strongly bound to the active site and they helped to maintain the stability and compactness of the protein structure better than the reference molecules.
Fig. 5.

SASA and radius of gyration (Rg) during MD simulations (A) Rg plot of native, MB_183, MB_241, MB_250, MB_266 and MB_380, N3 and Boceprevir docked complexes (B) The average PDF of Rg for the docked complexes (C) SASA analysis plot of Mpro-docked complexes(D) The PDF plot representing the SASA average values for the docked complexes.
Additionally, we also performed a SASA analysis to understand the solvent behaviour of all the complexes (Supplementary Table 1). SASA profiles revealed that the docked complexes had average values with a range of 170 nm2–173nm2 (Fig. 5C). The average SASA value of the native protein, MB_183, MB_241, MB_250, MB_266, MB_380, N3 and Boceprevir were 173.53 nm2, 170.20 nm2, 172.96 nm2, 173.56 nm2, 172.49 nm2, 173.80 nm2, 172.95 nm2 and 172.51 nm2 respectively (Fig. 5D). The results showed that the compounds MB_241, MB_250 and MB_266 possessed stable hydrophobic contacts between the receptor and ligand complexes, similar to the reference molecules which make the maximum region of Mpro enzyme accessible to the solvent molecules.
3.4.3. Hydrogen bond and eigenvector vetting
To understand the binding affinity of MB compounds towards Mpro, the MD trajectories were analysed for hydrogen bond monitoring to calculate the total number of bonds formed between the receptor-ligand complexes. The MB_183, MB_241, MB_250, MB_266 and MB_380 docked complexes showed the number of hydrogen bonds to be between 0-5, 0–4, 0–5, 0–8 and 0–7, respectively (Fig. 6 A–E). Consequently, the Mpro-N3 and Mpro-Boceprevir docked complexes revealed the number of H-bonds to be between 0-10 and 0–6, respectively (Fig. 6F and 6G). We observed a maximum number of hydrogen bonds in the MB_266-Mpro complex indicating significant interaction lying between the two references. Overall, the hydrogen bonds and hydrophobic interactions revealed that the complexes MB_241, MB_250 and MB_266 were highly stabilizing the active pocket of Mpro (Supplementary Table 2). The results also suggested that the number of hydrogen bonds formed was steady throughout the simulations for all the three docked complexes. The amino acid residues present in the active site are also persistent and contribute to the stability of the overall Mpro geometry throughout the total simulation time, in accordance to that of the complexes of the reference molecules.
Fig. 6.

Hydrogen bonds and Eigen-vector plot of the docked complexes (A) Number of H-bonds in MB_183, (B) MB_241, (C) MB_250, (D) MB_266 (E) MB_380, (F) N3 inhibitor and (G) Boceprevir drug (H). The dynamic energy fluctuation plotted between two eigenvector 1 and 2 generated for the docked complexes showing conformational space of Cα-atoms.
The conformational subspace of the complexes was evaluated using PCA to understand the dynamic behaviour of Mpro. The PCA analysis revealed that the conformational clusters are well defined and the five compounds covered minimum subspaces (Fig. 6H). The graph was plotted between the two eigenvector 1 and 2, which gave the displacement of atomic fluctuation and type of motion obtained between the complexes. The eigenvector values of the docked complexes MB_183, MB_241, MB_250, MB_266 and MB_380 are in the ranges of ~-4 nm2 to ~5 nm2 and ~-8 nm2 to ~5 nm2, respectively, in comparison to the native protein and reference molecules (Supplementary Fig. 1). Among all the five compounds, the complex of MB_241 showed eigenvector 1 in the range -2nm2 to −2.5 nm2, and eigenvector 2 in the range -4nm2–5 nm2 of minimum cluster, MB_250 showed eigenvector 1 and 2 in the range −2.5 nm2 to −3nm2 and -2nm2–3.5 nm2 of range and MB_266 occupied small range occupancy cluster range with eigenvector 1 and 2 in the range of -3nm2–3 nm2 and -4nm2–2 nm2 respectively. The proposed three docked complexes MB_241, MB_250, MB_266 showed restricted space in complex with Mpro, leading to well-defined internal motion behaviour vital for the complex stabilization better than the N3 and Boceprevir inhibitors.
3.4.4. Covariance matrix and free energy landscapes (FEL) analysis
The overall motion of the atoms in complexes was analysed for all the docked complexes by plotting covariance matrices, to calculate the trace values with a maximum number of eigenvectors and amino acid residues. The matrix analysis also helped to understand the positional fluctuations on Cα-atoms and the atomic behaviour of the correlated and anti-correlated motions (Ahamad et al., 2018, 2019). The analysis is illustrated with two intense colour representations, red and blue. The small fluctuations between the atoms are marked with blue and the large fluctuations denoted with red. The covariance predicted that all the complexes were found to be tolerable with the atomic displacement range of 0.183 nm2, 0.184 nm2, 0.255 nm2, 0.218 nm2, 0.207 nm2, 0.193 nm2, 0.403 nm2 and 0.234 nm2 for native protein, MB_183, MB_241, MB_250, MB_266 and MB_380, N3 and Boceprevir, respectively (Supplementary Fig. 2). The overall positional fluctuations on Cα-atoms of all the docked complexes revealed that MB_241, MB_250 and MB_266 were found to be stable with steady residue displacement and amplitude towards Mpro in comparison to reference molecules.
The conformational stabilities of the docked complexes were examined by FEL analysis from the obtained PC1 and PC2 of eigenvectors. The values of FEL ranged from 0 kJ/mol to 11.5 kJ/mol, 13.7 kJ/mol, 14 kJ/mol, 13.3 kJ/mol, 13.9 kJ/mol, 12.6 kJ/mol, 12.9 kJ/mol and 12.1 kJ/mol for the native protein, MB_183, MB_241, MB_250, MB_266, MB_380, N3 and Boceprevir docked complexes (Supplementary Fig. 3). The global free energy minima results showed that the docked complexes revealed steady states on the folding behaviour of Mpro than the reference molecules. The overall analysis revealed that the complexes were persistent with high energy minima suggesting the amino acids between the docking poses are vital for stability and function of Mpro enzyme. Further investigation was performed to know the atomic density distribution of the complexes for atomic orientation towards Mpro. The results elucidated a stable density area values for native protein, MB_183, MB_241, MB_250, MB_266, MB_380, N3 inhibitor and Boceprevir ranging from 2.39 nm−3, 2.52 nm−3, 3.53 nm−3, 3.28 nm−3, 3.78 nm−3, 2.99 nm−3, 3.65 nm−3 and 3.17 nm−3, respectively (Fig. 7 A–H). From the analysis it was shown that the density area of each atom in the complexes MB_241, MB_250, MB_266 were found to be in stable distribution and energy wells showed that all the compounds were sharing similar binding patterns alike N3 and Boceprevir for Mpro, indicating that the complexes are binding to the same active pocket.
Fig. 7.

Density distribution of docked complexes obtained in the MD trajectories during simulation time-frames (A) Native, (B) MB_183, (C) MB_241, (D) MB_250, (E) MB_266 (F) MB_380, (G) N3 inhibitor, (H) Boceprevir drug.
3.4.5. Residue–Residue Contact Map (RRCM)
RRCM (Dorosh and Stepanova, 2017; Shao, 2020) is examined to calculate the smallest distance between Cα-atoms of amino acid residues that conjugate enzyme-ligand docked complexes that influence secondary structure elements to know the allosteric effect on Mpro. The RRCM of native and docked complexes namely MB_183, MB_241, MB_250, MB_266, MB_380, N3 inhibitor and Boceprevir exposed the atomic distance of 5.28 nm, 5.55 nm, 5.57 nm, 5.65 nm, 5.53 nm, 5.76 nm, 5.54 nm and 5.42 nm, respectively (Fig. 8 ). From the above comparative analysis, it is evident that the interacting amino acids of the complexes were in close proximity to tender a strong binding, affirming stable conformations of the ligands towards the active pocket of Mpro in accordance to the reference molecules.
Fig. 8.

Depiction of residue contact map and the mean smallest distance between the Cα-atoms of each amino acid residues of Mpro corresponding to docked compounds (A) Native (B) MB_183, (C) MB_241, (D) MB_250, (E) MB_266 (F) MB_380, (G) N3 inhibitor and (H) Boceprevir drug. (Red, yellow and orange colours indicate strong correlations, whereas longer distance is shown in blue colour).
4. Discussion
It was interesting to conclude from the observations that the five MB_compounds identified in the present study were better than the reported reference molecules N3 and Boceprevir with higher binding affinities. The conformation and strength of interactions were confirmed by screening based molecular docking study validated for all the five docked complexes compared to Mpro-N3 and Mpro-Boceprevir. It was striking to see that out of five compounds, the three MB screened compounds' docking results, namely MB_241, MB_250 and MB_266 were bench-marked with highest docking affinity scores than the standard reference molecules. The results also projected the involvement of important amino acid residues namely, Thr24, Thr25, His41, Thr45, Ser46, Met49, Tyr54, Phe140, Leu141, Asn142, Gly143, Ser144, His163, His164, Glu166, Leu167 and Gln189 that were actively forming hydrogen bonds and thus stabilizing the receptor-ligand complex. It is worthy of highlighting that the three compounds mimicked the similar docking pose and interactive pattern as the reference molecules of Mpro inhibitors used in the present study. From ADME/T properties, the four best candidates MB_183, MB_241, MB_250 and MB_266, found to be orally bioactive, obeying Lipinski's rule of five without any violation of nominal ranges and with no toxicity on productive health. Compound MB_380 was unable to pass the pharmacological screening, owing to its higher molecular weight and larger topological surface area.
The stability of protein-ligand complexes during MD simulation studies was analysed for the protein Cα-atom and ligand structures. RMSD analysis, RMSF showed that the three complexes MB_241, MB_250 and MB_266 amongst the selected five compounds remained stable throughout the simulation stipulated time consequentially than that of N3 and Boceprevir docked complex. Consequently, the three complexes MB_241, MB_250 and MB_266 were found to be more compact with low Rg oscillations than the reference and native, followed by SASA which found to have smaller values in comparison to the N3, Boceprevir and native complexes. This signifies the complexes were having stabilized structural transition and compactness on Mpro and showed high proximity towards the active site residues. The hydrogen bond formed between the complexes showed that the prioritised three compounds MB_241, MB_250 and MB_266 maintained rigidity throughout the MD simulations.
Furthermore, PCA of the covariance matrix between the proposed two system complexes projected well-defined clusters than the reference complexes revealing higher compactness towards the Cα-atomic positional fluctuations of Mpro. The FEL analysis portrayed that the three complexes MB_241, MB_250 and MB_266 have a stable distribution of energy wells headed for Mpro. The residue-residue contacts map was also validated and found that the complexes' interactive amino acids are highly responsible for maintaining conformational stability and proximity, affirming the importance of allosteric effects on Mpro. Therefore, it is evident that the three proposed compounds MB_241, MB_250, MB_266, with greater binding affinity, conformational stability and better pharmacological properties, indicated a strong fit into the active pocket of Mpro, suggesting better ability to block the substrate-binding site, thereby probably hindering the targeted process of viral replication.
5. Conclusion
In the present study, we used pharmacoinformatics to identify potential inhibitors of SARS-CoV-2 Mpro. Summarily, we carried out virtual screening, drug-likeliness analysis and MD simulations to identify best lead compounds amongst the Malaria Box compound library. Virtual screening based molecular docking protocol was carried out to select compounds capable of binding to the Mpro active site, which helped to shortlist five compounds. The five compounds were checked further with various methods like Lipinski's rule of five and ADME/T parameters to filter out the probable drug like candidates. Moreover, the In Silico toxicity evaluation showed the compounds to be of low risk and possess various pharmacological properties. Furthermore, the RMSD, RMSF, hydrogen bonds and free energy wells analysis during MD simulations indicated three MB compounds to be in more stable binding with SARS-CoV-2 Mpro. This indicated that the three compounds, namely MB_241, MB_250 and MB_266 are better than the rest of the two compounds, which may be used for further exploration.
CRediT authorship contribution statement
Shahzaib Ahamad: Project administration, Formal analysis, In silico analysis and, Validation, Data curation, Methodology, Technical parts, Writing - original draft, preparation, Writing - review & editing. Hema Kanipakam: In silico, Validation, Data curation, Methodology, Writing - original draft, preparation, Writing - review & editing. Shweta Birla: Validation, Writing - review & editing. Md Shaukat Ali: Validation, Writing - review & editing. Dinesh Gupta: Supervision, Methodology, Hypothesis, Correspondence, Conceptualization, major revision, Writing - review & editing, All authors revised and reviewed the manuscript.
Declaration of competing interest
None of the authors declared conflict of interests.
Acknowledgment
SA, Research Associate, is thankful for the support and financial assistance (2019–6039 File No. ISRM/11(83)/2019) from Indian Council of Medical Research, India. HK is the recipient of Women Scientist awarded from DBT-BioCARe (No. BT/PR31715/BIC/101/1233/2019), Department of Biotechnology, India. SB is the recipient of National Post-doctoral fellowship from DST-SERB (PDF/2017/001326), Department of Science & Technology, India. DG is endowed by the Department of Biotechnology (Grant No. BT/BI/04/001/2018), Government of India.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejphar.2020.173664.
Contribution of authors
SA contributed to literature mining, Virtual screening based docking, MD simulations, and write-up in the manuscript. HK contributed to write-up and corrections in the manuscript. SB and MSA contributed to write-up in the manuscript. DG contributed in the designing the hypothesis, major inputs, correspondence and entire corrections in the manuscript.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Abdi H., Williams L.J. Principal component analysis. Wiley Interdiscipl. Rev.: Comput. Stat. 2010;2:433–459. [Google Scholar]
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., Smith E.C., Case J.B., Feng J.Y., Jordan R. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9 doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahamad S., Hassan M.I., Dwivedi N. Designing of phenol-based β− carbonic anhydrase1 inhibitors through QSAR, molecular docking, and MD simulation approach. 3 Biotech. 2018;8:256. doi: 10.1007/s13205-018-1278-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahamad S., Islam A., Ahmad F., Dwivedi N., Hassan M.I. 2/3D-QSAR, molecular docking and MD simulation studies of FtsZ protein targeting benzimidazoles derivatives. Comput. Biol. Chem. 2019;78:398–413. doi: 10.1016/j.compbiolchem.2018.12.017. [DOI] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Anson B.J., Chapman M.E., Lendy E.K., Pshenychnyi S., Richard T., Satchell K.J., Mesecar A.D. 2020. Broad-spectrum Inhibition of Coronavirus Main and Papain-like Proteases by HCV Drugs. [Google Scholar]
- Bafna K., Krug R.M., Montelione G.T. 2020. Structural Similarity of SARS-CoV2 Mpro and HCV NS3/4A Proteases Suggests New Approaches for Identifying Existing Drugs Useful as COVID-19 Therapeutics. ChemRxiv. [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Hage-Melim L.I., Federico L.B., de Oliveira N.K.S., Francisco V.C.C., Correa L.C., de Lima H.B., Gomes S.Q., Barcelos M.P., Francischini I.A.G. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020:117963. doi: 10.1016/j.lfs.2020.117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. CCP4 Newsletter on Protein Crystallography. Vol. 40. 2002. Pymol: an open-source molecular graphics tool; pp. 82–92. [Google Scholar]
- Dorosh L., Stepanova M. Probing oligomerization of amyloid beta peptide in silico. Mol. Biosyst. 2017;13:165–182. doi: 10.1039/c6mb00441e. [DOI] [PubMed] [Google Scholar]
- Duffy S., Avery V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012;86:84–92. doi: 10.4269/ajtmh.2012.11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao T., Fukuda Y., Kiga K., Sharif J., Hino K., Enomoto Y., Kawamura A., Nakamura K., Takeuchi T., Tanabe M. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell. 2007;129:617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- Ganyani T., Kremer C., Chen D., Torneri A., Faes C., Wallinga J., Hens N. 2020. Estimating the Generation Interval for COVID-19 Based on Symptom Onset Data. MedRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy R.K., DiPaola R.S., Romanelli F., Dutch R.E. Rapid repurposing of drugs for COVID-19. Science. 2020;368:829–830. doi: 10.1126/science.abb9332. [DOI] [PubMed] [Google Scholar]
- Hsu M.-F., Kuo C.-J., Chang K.-T., Chang H.-C., Chou C.-C., Ko T.-P., Shr H.-L., Chang G.-G., Wang A.H.-J., Liang P.-H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jo S., Kim S., Shin D.H., Kim M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanipakam H., Sharma K., Thinlas T., Mohammad G., Pasha M.Q. Structural and functional alterations of nitric oxide synthase 3 due to missense variants associate with high-altitude pulmonary edema through dynamic study. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2019.1711190. [DOI] [PubMed] [Google Scholar]
- Kruse R.L. 2020. Therapeutic Strategies in an Outbreak Scenario to Treat the Novel Coronavirus Originating in Wuhan, China. F1000Research 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Jabłońska J., Pravda L., Vařeková R.S., Thornton J.M. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27:129–134. doi: 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Lee I., Kim H., Chang G., Chung J., No K. 2003. The PreADME Approach: Web-Based Program for Rapid Prediction of Physico-Chemical, Drug Absorption and Drug-like Properties. EuroQSAR Designing Drugs and Crop Protectants: Processes, Problems and Solutions; pp. 418–420. [Google Scholar]
- Lipinski C.A. Rule of five in 2015 and beyond: target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016;101:34–41. doi: 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- Liu X., Zhang B., Jin Z., Yang H., Rao Z. Protein DataBank; 2020. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3. [Google Scholar]
- Mengist H.M., Fan X., Jin T. Signal Transduction and Targeted Therapy. Vol. 5. 2020. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease M pro; pp. 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumoto K., Kagaya K., Zarebski A., Chowell G. Estimating the asymptomatic proportion of coronavirus disease 2019 (COVID-19) cases on board the Diamond Princess cruise ship, Yokohama, Japan. Euro Surveill. 2020;25:2000180. doi: 10.2807/1560-7917.ES.2020.25.10.2000180. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar T.U.M., Alqahtani S., Alamri M., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharmaceut. Anal. 2020 doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an" Achilles' heel": current effective inhibitor targeting a 3C-like protease. Protein cell. 2013;4:248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang P., Wang L., Cao J. Parametric functional principal component analysis. Biometrics. 2017;73:802–810. doi: 10.1111/biom.12641. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. 2012. QikProp. version 3.5. New York, NY. [Google Scholar]
- Schüttelkopf A.W., Van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Shao Q. Effect of conjugated (EK) 10 peptide on structural and dynamic properties of ubiquitin protein: a molecular dynamics simulation study. J. Mater. Chem. B. 2020 doi: 10.1039/d0tb00664e. [DOI] [PubMed] [Google Scholar]
- Siddiqi H.K., Mehra M.R. COVID-19 illness in native and immunosuppressed states: a clinical–therapeutic staging proposal. J. Heart Lung Transplant. 2020;39:405. doi: 10.1016/j.healun.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T., Burrows J.N., Kowalczyk P., McDonald S., Wells T.N., Willis P. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One. 2013;8 doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W., Chen C., Lei X., Zhao J., Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Viswanadhan V.N., Ghose A.K., Revankar G.R., Robins R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 1989;29:163–172. [Google Scholar]
- Walters W.P., Namchuk M. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discov. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- Wang F., Chen C., Tan W., Yang K., Yang H. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016;6:22677. doi: 10.1038/srep22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.