

Abstract
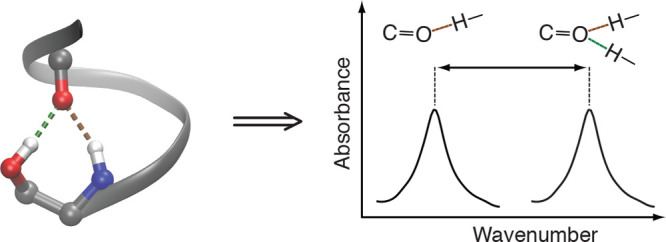
H-bonding is the predominant geometrical determinant of biomolecular structure and interactions. As such, considerable analyses have been undertaken to study its detailed energetics. The focus, however, has been mostly reserved for H-bonds comprising a single donor and a single acceptor. Herein, we measure the prevalence and energetics of multiplex H-bonds that are formed between three or more groups. We show that 92% of all transmembrane helices have at least one non-canonical H-bond formed by a serine or threonine residue whose hydroxyl side chain H-bonds to an over-coordinated carbonyl oxygen at position i–4, i–3, or i in the sequence. Isotope-edited FTIR spectroscopy, coupled with DFT calculations, enables us to determine the bond enthalpies, pointing to values that are up to 127% higher than that of a single canonical H-bond. We propose that these strong H-bonds serve to stabilize serine and threonine residues in hydrophobic environments while concomitantly providing them flexibility between different configurations, which may be necessary for function.
Introduction
H-bonds are relatively weak interactions that are driven by the electrostatic attraction between a positively charged hydrogen and a negatively charged acceptor. Their prevalence is the driving force behind many natural phenomena, perhaps the most notable of which is the flotation of ice on water. Despite their small magnitude, they often amass a considerable impact due to their directional character and their abundance.
Their ability to compound, like Lego bricks, allows H-bonds to achieve a wide range of biological purposes in macromolecules. Complementary H-bonds between the two strands of DNA are responsible for high replication fidelity of genetic information.1 H-bonds between glucose monomers in cellulose provide tremendous physical strength. Finally, as predicted by Pauling and co-workers, specific H-bond patterns in proteins define the secondary structure of helices2 and pleated sheets.3
These secondary structures form during the folding process due to the scarcity of internal water molecules in the hydrophobic core, which requires the protein to self-satisfy its H-bonding potential.4 The lack of water molecules is even more pronounced in the hydrophobic milieu of membrane proteins. This may lead to stronger hydrogen bonding and greater helical uniformity in membrane proteins compared to water-soluble proteins.5,6 Therefore, as one might expect, transmembrane α-helices are frequently more stable than their counterparts in water-soluble proteins and, at times, only unravel when the membrane integrity collapses.7−12
Conventional H-bonds, such as those found in α-helices,2 where the amide H at position i interacts with the i–4 amide carbonyl, have been characterized extensively in terms of geometry and energetics.13 However, these single donor–single acceptor interactions represent only one type of H-bond. More complex H-bonds exist, which are formed with multiple acceptors (multifurcation), multiple donors (over-coordination), or both.
The most common multiplex H-bonds, identified ever since protein structures were first solved,14 involve the over-coordination of a backbone carbonyl with two donors: the backbone amide hydrogen and the hydroxyl side chain of serine or threonine.15 In membrane proteins, such over-coordinated H-bonds have been proposed to accommodate the polarity of serine and threonine in the apolar lipid bilayer.8,16,17
Motivated by the abundance of multiplex H-bonds and their importance to membrane proteins, we have previously measured the strength of one such bond: the over-coordination of the carbonyl of residue i–4 to the hydroxyl and amide hydrogens of serine or threonine residues at position i.18 Our combined experimental and computational study indicated that this bond configuration is about 60% stronger than the single canonical bond.
As shown in Figure 1, however, this is only one of several multiplex H-bonds that serines and threonines may form. In the current study, we provide a comprehensive quantitative analysis of serine and threonine side chains H-bonding to backbone carbonyls in over-coordinated and bifurcated H-bonds. Our results provide a detailed energetic landscape of non-canonical H-bonds in transmembrane helices.
Figure 1.
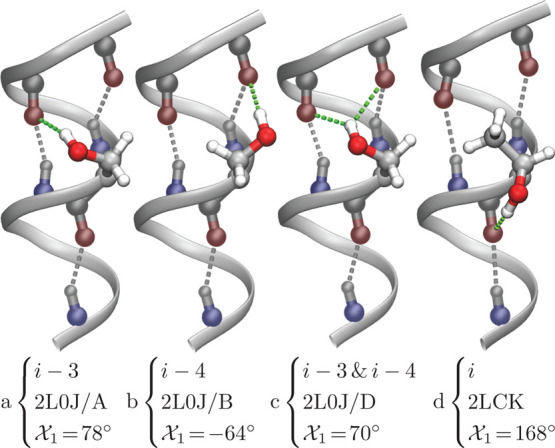
Different H-bond configurations in solved membrane protein structures, as indicated by the PDB ID and chain (if relevant). Structures a, b, and c are from the M2 H+ channel19 and structure d is from the mitochondrial uncoupling protein 2.20 Note that structures a–c were determined by solid state NMR,19 in which the side-chain conformations were obtained by the refinement procedure. The backbone helical H-bonds are colored in gray, while the bonds formed by the hydroxyl side chain are depicted in green. The χ1 rotamer of the hydroxyl group and the particular H-bond acceptor(s) are noted.
Results and Discussion
Prevalence of Polar Residues in TM Helices
Analysis of a non-redundant dataset of transmembrane helices21−23 indicates that residues containing polar side chains that are capable of H-bonding comprise 23% of all amino-acids (16% in bitopic or single-pass proteins and 24% in polytopic or multi-pass proteins, Table S1). Such side chains include serine, threonine, tyrosine, cysteine, histidine, glutamine, aspargine, glutamate, aspartate, lysine, and arginine. Of these polar residues, serines and threonines are the most common, together representing 11% of all transmembrane helical amino acids leading to the fact that 92% of all transmembrane helices contain one or more serine or threonine residues. Finally, the prevalence of serines and threonines in membrane proteins is similar to what is found in water-soluble proteins. However, most other polar/charged residues are more abundant in soluble proteins, as shown in Table S1.
Statistical Analysis of Serine and Threonine H-Bonding
We analyzed each of the serine and threonine residues found in transmembrane helices of solved membrane protein structures for their participation in multiplex H-bonding. H-bonding was determined by a distance of less than 3.5 Å between the hydroxyl O and the carbonyl O. The results indicate that the majority of serines and threonines form such multiplex H-bonds (Table S2). The vast majority of these bonds form with over-coordinated backbone carbonyl groups located at the same residue (i), at three residues prior (i–3), or at four residues prior (i–4) in the sequence.
In order to understand the factors that determine which of these H-bonds is formed, we measured the χ1 rotamer. Residues with χ1 = −60° (Figure 2 red points) are nearly all back-bonded to the i–4 carbonyl group (Figure 2 pink shading). In contrast, at χ1 = +60° (Figure 2 blue points), they back-bond to the i–3 carbonyl group (Figure 2 cyan shading), or simultaneously to both the i–3 and the i–4 carbonyl groups (Figure 2 checkered shading). When the χ1 rotamer is at ±180° (Figure 2 gray points), the side chains H-bond to their own carbonyl group (shown in Figure S1). Similar results are obtained when analyzing threonine residues, as shown in Figure S1.
Figure 2.

Analysis of the distances between the serine Oγ and the oxygen of carbonyl groups located at the i–3 and i–4 positions, as a function of side-chain rotamer (according to the color scale). The cyan shaded region indicates residues whose Oγ is close enough (within 3.5 Å) to H-bond to the i–3 carbonyl group. The pink shaded region indicates residues whose Oγ is close enough (within 3.5 Å) to H-bond to the i–4 carbonyl group. The cyan and pink checkered region indicates residues whose Oγ is close enough to H-bond to both the i–3 and i–4 carbonyl groups simultaneously. The residues are from a dataset of non-redundant transmembrane helices.21−23
The statistical analysis shows that serine and threonine residues are commonly involved in a number of different multiplex H-bonds: to the i–3 carbonyl, to the i–4 carbonyl, to both i–3 and i–4 carbonyls, or to the carbonyl of the same residue (i). Hence, the choice of H-bonding partner depends on the serine or threonine χ1 angles. Previously we have measured the strength of the over-coordinated bond to the i–4 carbonyl.18 In order to obtain a complete understanding of all of these configurations, we now expand our analysis and measure the strength of all of these multiplex H-bonds in a membrane protein solvated in its natural lipid bilayer environment.
FTIR Spectroscopy
As a system to investigate multiplex H-bonds, we chose the 97 amino acid, tetrameric M2 H+ channel from influenza A. Its structure has been extensively characterized by X-ray crystallography,24,25 solution NMR spectroscopy,26 and solid-state NMR spectroscopy.19,27 Moreover, a 25 amino acid peptide that encompasses the protein’s single transmembrane domain exhibits many of the characteristics of the full length protein, such as tetramerization, drug binding, and conductivity.28,29
The M2 channel contains a single serine residue in its transmembrane domain at position 31. Three of the multiplex H-bonding configurations (i–3, i–4, and simultaneous i–3 and i–4) can be observed at this serine location when inspecting the different protein chains and frames of PDB ID 2L0J,19 as depicted in Figure 1. Note that the structure was determined by solid state NMR,19 in which the side-chain conformations were obtained by the refinement procedure. Finally, in all of these configurations, the backbone carbonyl retains its canonical H-bond with the amide H four residues later, and so it does not require the hydroxyl side chain for its own stabilization.
In order to measure the strength of the different multiplex H-bonds, we utilized FTIR spectroscopy, focusing on the vibrational frequency of the carbonyl group. The C=O stretch is the major component of the amide I vibrational mode.30 Consequently, the amide I band is expected to shift to lower frequencies when bound to a single H-bond donor, and even more so when it is over-coordinated to two donors,31 as shown schematically in Figure 3. Hence, FTIR spectroscopy is particularly useful, since the extent of the shift is directly related to the strength of the H-bond in question. Spectroscopic observation of an individual carbonyl group is achieved by 13C=18O labeling, which shifts the labeled carbonyl vibration far from the natural abundance amide I mode.32,33
Figure 3.
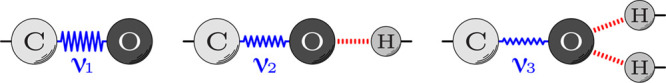
Impact of H-bonding (in red) on the vibrational frequencies of the C=O group (in blue). Left: Non-bonded configuration. Center: Canonical H-bonded configuration composed of a single donor and a single acceptor. Right: Over-coordinated H-bonding configuration with two donors and a single acceptor. Consequently, due to H-bonding the vibrational frequencies are related to one another as follows: ν1 > ν2 > ν3.
In order to analyze the H-bond between Ser31’s hydroxyl to the i–3 carbonyl, we labeled residue Val28 with 13C=18O. Similarly, analysis of the H-bond to the i–4 carbonyl was achieved by labeling Val27. As a control without side-chain over-coordination, we used two additional peptides, once more labeled with 13C=18O at Val27 or Val28, but in these instances Ser31 was replaced with an alanine. Site 31 in the M2 protein has appreciable variability (including S, N, C, G, I, D, K, and R) among currently sequenced naturally circulating viral strains. The overall M2 structure is not altered in any detectable way upon mutation, as can be seen by the FTIR spectra of the amide I and amide II bands that are found at the same frequencies (Figure S2). Finally, an H-bond to the carbonyl of the same residue (i) was not observed in the M2 channel, and hence could not be analyzed experimentally.
The FTIR spectra of the labeled amide I peaks of these four M2 transmembrane peptides in hydrated lipid bilayers are shown in Figure 4. Interestingly, the isotope-edited peaks of Val27 or Val28 change dramatically depending on which residue is located at position 31. In particular, when residue 31 is an alanine, a peak is observed at higher frequencies: 1596 or 1602 cm–1 for the carbonyl stretching mode of residue 27 or 28, respectively. However, an appreciable shift to lower frequencies is obtained when residue 31 is a serine, whose side chain is capable of H-bonding. The carbonyl stretching mode of Val28 (i–3) shifts by 8.4 cm–1, while that of Val27 (i–4) shifts by 14.7 cm–1. These values align with the 7–13 cm–1 downshift reported previously for interactions between cations and an amide carbonyl.34
Figure 4.
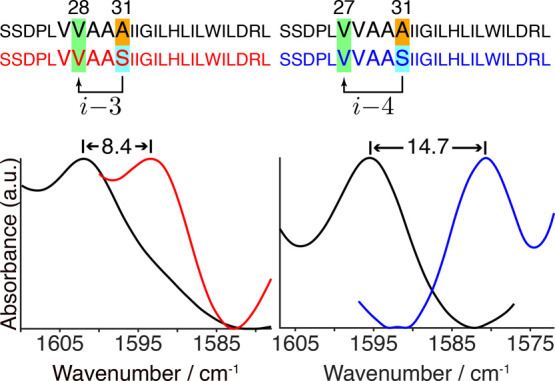
FTIR spectra in the isotope-edited amide I mode region of M2 peptides (Ser22–Leu46, as noted) in hydrated lipid bilayers obtained at room temperature. Amino acids shaded in green are labeled with 13C=18O at position 27 (right panel) or 28 (left panel). The arrows in the sequence depict possible over-coordinated H-bonds by Ser31 (shaded in cyan). Spectra of peptides with an alanine at position 31 (shaded in orange) are depicted in black. The spectra of these peptides with serine at position 31 are depicted in red or blue for peptides labeled at Val28 or Val27, respectively. The spectra were normalized according to each isotope-edited amide I peak.
DFT Calculations
In order to correlate the experimentally measured frequency shifts to bond enthalpies, we undertook DFT calculations. Such calculations yield the frequency of any particular vibrational mode in the system, which can then be compared with the experimental results from FTIR in order to validate the computation.
While a peptide is an exceedingly large system for quantum calculations, it is possible to capture the chemistry and geometry of the relevant H-bonding groups using smaller compounds. For example, two consecutive peptide carbonyls may be effectively mimicked by a 2-acetamido-N-methylacetamide molecule (Figure 5 and Figure S3). Specifically, from the atom coordinates of chains A, B, and D of PDB ID 2L0J,19 we built mimics for the i–3, i–4, and i–3 and i–4 multiplex H-bonding systems (panels d–f, respectively, in Figure 5 and Figure S3). A mimic for the i H-bond system was based on the structure of PDB ID 2LCK(20) (panel g of Figure 5 and Figure S3). Finally, the structures were optimized after assembly, and the resulting minimal deviations can be seen in Figure S4.
Figure 5.

Model compounds used in the DFT calculations in order to calculate the H-bond enthalpies and vibrational frequencies. Middle row models (d–g) are serine side-chain mimics that contain multiplex H-bonds, while the bottom row (h−k) does not, due to the rotation of the hydroxyl group. Similarly, the top row (a–c) are alanine side-chain mimics that represent equivalent systems in which the hydroxyl group is not present, and hence once again, multiplex H-bonds do not exist. Each column differs in the identity of the residue(s) of carbonyl(s) acceptor of the multiplex H-bond, as indicated on top (i–3, i–4, i–3 and i–4, and i). The i, i–3, and i–4 carbonyl groups, and the multiplex H-bonds that they form are colored in purple, red, and blue, respectively. The side-chain hydroxyl group is depicted in green, while canonical H-bonds are depicted in black. Note that these two-dimensional schematic diagrams of the model compounds are intended to clearly show the H-bonds being calculated. For an accurate three-dimensional model of these compounds with the correct bond lengths and angles, see Figure S3.
We then proceeded to calculate the vibrational frequencies of the carbonyls in question (see colored carbonyls in panels d–g of Figure 5 and Figure S3). We followed by calculating the same frequencies for structures in which the hydroxyl group, which participated in the multiplex H-bonding, is absent (panels a–c of Figure 5 and Figure S3). These two calculation series resembled systems with a serine capable of multiplex H-bonding, or conversely, an alanine that is not. Consequently, the vibrational shifts due to multiplex H-bonding could be obtained readily by comparing the two frequencies (top versus middle rows in Figure 5 and Figure S3). The results are very encouraging: The calculated i–4 and i–3 H-bond spectral shifts are 15.7 and 9.01 cm–1, respectively, which are exceptionally close to the 14.7 and 8.4 cm–1 shifts measured experimentally by FTIR (Figure 4).
Following confirmation of the accuracy of the DFT calculations, we proceeded to evaluate the enthalpy of the different multiplex H-bonds. We calculated the overall advantage in stability that serine contributes to the structure. We did so by first calculating the energy of the system with serine back-bonding to a backbone carbonyl (panels d–f in Figure 5 and Figure S3). We then removed any intramolecular influences by calculating the energy of the system again when the two molecules are separated by 100 Å. We did the same for the valine systems (panels a–c in Figure 5 and Figure S3). Subsequently, we subtracted the valine energy values from the serine ones in order to determine the energetic favorability of a serine in this location:
The energetic difference of a serine versus a valine in the i–3, i–4, and multiplex i–3 and i–4 orientations is −4.9 kcal/mol, −5.2 kcal/mol, and −3.0 kcal/mol, respectively.
To determine the particular contribution of the hydroxyl side-chain interaction with the backbone carbonyl, we manipulated each of the above systems to abolish any non-canonical H-bond. This was achieved by rotating the χ1 dihedral such that the hydroxylic side chain is rotated about the Cα–Cβ bond by 180°, thereby breaking the multiplex H-bond (bottom row in Figure 5 and Figure S3). The impact of all other energies, such as any new intramolecular interactions caused by the rotation, can then be accounted for by separating the two molecules apart in both the rotated and non-rotated systems. Hence the enthalpy of the side-chain contribution to each of the multiplex H-bonds (ΔE) is given by
The results listed in Table 1 indicate that the addition of another H-bond donor strengthens the helical H-bond appreciably. In particular, the contribution of a hydroxyl group to the H-bond system involving the i–4 carbonyl increases its stability by 5.8 kcal/mol relative to a canonical (i.e., single donor) H-bond. Similarly, when the hydroxyl group participates in an over-coordinated H-bond with the i–3 or i carbonyl, it results in an H-bonding system that is stronger than a canonical bond by 4.1 and 4.2 kcal/mol, respectively. Finally, when the hydroxyl is simultaneously bound to the i–3 or i–4 carbonyls, the H-bond is strengthened by 1.9 kcal/mol.
Table 1. DFT Calculated Bond Enthalpies of the Different Multiplex H-Bonds Involving Serine Side Chainsa.
| H-bond acceptor(s) | bond enthalpy(kcal/mol) | prevalence (%) |
|---|---|---|
| C=O at i–4 | –5.8 | 63 |
| C=O at i–3 | –4.1 | 56 |
| C=O at i | –4.2 | 32 |
| C=O at i–4 and i–3 | –1.9 | 30 |
The last column is the calculated statistical prevalence of the serine multiplex H-bond in our non-redundant dataset of transmembrane helices (Figure 2). A strict cutoff distance of 3.0 Å between the hydroxyl O and the carbonyl O acceptor was used for classification. Note that due to multifurcation of the hydroxyl group, percentages exceed 100%.
The amide I shift (14.7 cm–1) to hydrogen bond length (1.95 Å) ratio for the i–4 system is 28.4 cm–1/Å, which is very similar to that predicted previously.35 For the i–3 system, however, we receive an amide I shift (8.4 cm–1) to hydrogen bond length (1.93 Å) ratio of 16.4 cm–1/Å, which is nearly half of the value received for the i–4 system. Additional factors, such as environment polarity and geometry, may be responsible for such differences.
Conclusions
We observe that multiplex H-bonds are significantly more stable than canonical H-bonds. Our findings are consistent with their prevalence among serine and threonine residues: over 75% of serines and threonines in TM α-helices form multiplex H-bonds. Moreover, the relative strengths of the different configurations are generally consistent with their prevalence, albeit entropy is not taken into account in the DFT calculations. For example, the strongest bond, in which the hydroxyl is bound to the i–4 carbonyl, is also the most prevalent. Conversely, the weakest bond, in which the hydroxyl is simultaneously bound to both i–3 and i–4 carbonyls, is also the least common.
In analyzing the specific side-chain structure of the M2 peptide, we recall that two isotopomeric peptides were studied, with an identical sequence containing a serine at position 31. The only difference between the two peptides is the location of the 13C=18O label: one at valine 27 (i–4) and the other at valine 28 (i–3). Since we observe a shift in the amide I mode of both carbonyl groups (Figure 4), we can deduce that they both serve as acceptors to an H-bond from the hydroxyl side chain of residue 31. One may speculate that these H-bonding configurations may not behave classically as independent forms but may demonstrate a quantum nature where the proton can tunnel between different proton acceptors and donors. With this view, the position of the proton would not be on any of the donors or acceptors at any given moment, but rather within a potential well somewhere between the acceptors and donors. This has previously been suggested to exist between a proton donor and proton acceptor.36
Polar residues are often necessary for membrane protein function. While most polar or charged residues exist at significantly lower proportions in membrane proteins compared to in water-soluble proteins, approximately equal proportions of serines and threonines exist in both membrane and water-soluble proteins (Table S1). So, while membrane proteins demonstrate a preference for apolar residues over most polar/charged residues, this preference is negated for serine and threonine. The equal ratio of these hydroxylic residues in membrane and water-soluble proteins is due to their ability to form multiplex H-bonds, which provides stability in the hydrophobic membrane environment. The environment-dependent nature of the serine and threonine dihedral preferences that allow the multiplex hydrogen bonding described herein can be applied to statistical and energy-based force fields and scoring functions of bio-computational tools.
The different H-bond configurations may allow serine and threonine residues to form H-bonds at any orientation necessary for protein function. Moreover, the hydroxyl side chain may break and re-form its H-bond to the over-coordinated backbone carbonyl with relative ease since it does not destabilize the carbonyl, which remains H-bonded to the backbone amine hydrogen. This flexible nature of the over-coordinated H-bond of the backbone carbonyl with the hydroxyl side chain makes it uniquely suited for simultaneously stabilizing serine and threonine side chains, while still affording them the versatility needed to function. Moreover, Bowie and co-workers have pointed at the role hydroxyl groups’ over-coordination may have in the pliability of transmembrane helical H-bond patterns.37 Finally, Thiel and co-workers have recently suggested that gating of an ion channel may be controlled by a temporal over-coordinated H-bond.38
We have focused on serine and threonine multiplex hydrogen bonding because these residues are by far the most common polar residues in transmembrane helices. Their behavior highlights the importance of intramolecular hydrogen bonding in the hydrophobic membrane environment. It is quite likely that other residues exhibit equally interesting hydrogen bonding behavior. Similar over-coordination has recently been shown to occur for glutamines in polyglutamine tracts.39 Tyrosines exist with equal prevalence in both membrane and water-soluble proteins. They do not form hydrogen bonds with backbone carbonyls nearly as often as serine and threonine, and they have (together with tryptophan) a preference for the aqueous–lipid interface, where they can interact with the aqueous phase.40 Their specific hydrogen-bonding stabilization mechanism would merit future investigation.
Experimental Section
Statistical Analyses
A list of 27,052 transmembrane α-helices was obtained from PDBTM.21−23 Structures with an X-ray resolution greater than a potential H-bond length (3.5 Å) were pared from the list, resulting in 20,542 transmembrane α-helical segments. Redundancies were removed using CD-HIT41,42 at 80% identity. The representative sequences for each cluster were made into a non-redundant dataset of 2294 transmembrane α-helices. Finally, each of the protein structures was analyzed for the presence of non-canonical H-bonding using in-house written VMD43 TCL scripts.
FTIR Spectroscopy
1-13C=18O isotopic labeling was prepared as described previously.44 Briefly, 4.52 mmol of 3,5-dimethylpyridine hydrobromide45 in 2 mL of anhydrous N,N-dimethylformamide (DMF) (Sigma-Aldrich, MO, USA) was combined with 2.24 mmol of N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (Sigma-Aldrich, MO, USA) and 11.3 mmol of H218O (Sigma-Aldrich, MO, USA) under N2. In order to start the reaction, 225 μmol of l-valine-1-13C-N-FMOC (Cambridge Isotope Laboratories, Inc., MA, USA), dissolved in 3 mL of anhydrous DMF, was added. The reaction mixture was held at room temperature and stirred overnight. After 18 h, another 2.24 mmol of EDC·HCl was added, followed by a third addition of 2.24 mmol of EDC·HCl 8 h later. Sixteen hours after that, the reaction was removed from mixing and N2. Thirty milliliters of ethyl acetate (Gadot-group, Netanya, Israel) was added, and the mixture was transferred to a separatory funnel, where it was washed three times with 0.1 M citric acid and then once with brine. Thirty milliliters of ethyl acetate was then added to the combined citric acid and brine portions and separated. The 60 mL of ethyl acetate containing the labeled amino acid was dried over anhydrous sodium sulfate (Dasit Group, Milan, Italy) and filtered, and finally the ethyl acetate was removed by rotary evaporation, creating an azeotrope with dichloromethane (Gadot-group, Netanya, Israel).
The labeled valine (see above), represented as V̅, was incorporated into four different peptides corresponding to the transmembrane domain of the influenza A M2 channel. The four peptides created include the native sequence with valines 27 or 28 labeled as well as an S31A mutant with valines 27 or 28 labeled (peptide numbering begins at 22):
SSDPLV̅VAASIIGILHLILWILDRL
SSDPLVV̅AASIIGILHLILWILDRL
SSDPLV̅VAAAIIGILHLILWILDRL
SSDPLVV̅AAAIIGILHLILWILDRL
The four peptides were synthesized separately with N-(9-fluorenyl methoxycarbonyl) solid-phase chemistry. Each peptide sample was purified with high performance liquid chromatography on a 20 mL Jupiter 300 Å C4 5 μm high-performance liquid chromatography column (Phenomenex, CA, USA). The column was pre-equilibrated with 80:8:12 (by volume) water:acetonitrile:isopropanol, where all solvents contained 0.1% trifluoroacetic acid (TFA) (Merck, Darmstadt, Germany). Two milligrams of protein sample was dissolved in 2 mL of TFA and injected into the column. The solvent gradient was linearly altered with the VWR Hitachi Chromaster 5160 Pump to remove all water composition while retaining the acetonitrile:isopropanol ratio at 40%:60% with 0.1% TFA. Peptide elution was monitored at 280 nm using the VWR Hitachi Chromaster 5410 UV detector.
All of our experimental measurements were performed on peptides in lipid vesicles. We used organic solvent cosolubilization in order to reconstitute each peptide in a membrane bilayer. Approximately 1 mg of protein and 10 mg of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, AL, USA) were dissolved in 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (Merck, Darmstadt, Germany). The mixture was rotary evaporated at 37 °C until all HFIP evaporated. One milliliter of water was added, and the mixture was rotated at 37 °C to spontaneously form vesicles. The sample was then sonicated to ensure uniformly sized vesicles and no aggregation. The pH of all samples was below 6, and so the M2 protein is in its open conformation.46
For each of the four samples of peptides in a membrane vesicle, separate FTIR spectra were collected. First, 200 μL of sample was deposited on a germanium trapezoid ATR plate (50 mm × 2 mm × 20 mm) with a 45° face angle (Wilmad, NJ, USA). Following removal of bulk solvent, the crystal was incorporated into a 25 reflection variable angle ATR unit (Specac, Orpington, UK), which reflects the incoming FTIR beam 25 times before its exit from the crystal. The ATR unit was incorporated within a Nicolet iS10 FTIR spectrometer, with a mercury cadmium telluride detector (Thermo Scientific, MA, USA), cooled with liquid nitrogen. The FTIR spectrometer was purged with water- and CO2-depleted air, and spectra were collected at room temperature. For each sample, 1000 scans were sampled and averaged at a data spacing of 0.241 cm–1 with two levels of zero filling, N-B strong apodization, and Mertz phase correction. For each of the four samples of peptides in a membrane vesicle, separate FTIR spectra were collected at room temperature. The FTIR spectra that we collected indicate that the DMPC membrane is in the gel phase since the lipid C=O stretch is at 1738 cm–1, the CO–O stretch is at 1177 cm–1, and there are distinct CH2 wag peaks.47
DFT Calculations
The i–3, i–4, and multiplex i–3 and i–4 H-bonding models, were created from chains A, B, and D, respectively, of the solved structure of the influenza A M2 protein with PDB ID 2L0J.19 Each model contains the serine residue, the i–3 and i–4 amide groups, as well as the i + 1 and i amide groups that form canonical H-bonds with the i–3 and i–4 amide groups. Cα’s connecting adjacent amide groups and at the molecule ends were also included, and then H atoms are added with VMD molefacture.43 The models underwent geometric optimization of H atoms and the i–3 and i–4 backbone carbonyls.
The i H-bond model was created from the solved structure with PDB ID 2LCK.20 The model includes the serine residue and the NH group at residue i + 4, involved in a canonical H-bond with the i carbonyl. Cα atoms cap the molecules, and H atoms were added via VMD Molefacture.43 The models underwent geometric optimization of H atoms and the i backbone carbonyl.
All optimization steps, as well as frequency and energy calculations, were conducted with the Q-Chem software package48 using the B3LYP method49,50 and the aug-cc-pVDZ basis set.51,52 The dielectric constant was set to 4 to mimic the hydrophobic membrane environment.
The i–3 and i–4 amide carbonyls of the models were isotopically labeled as 1-13C=18O to imitate the peptides experimentally analyzed by FTIR. The amide I peak shift between the structures in Figure 5d–f and three other structures, where the serine is mutated to an alanine (Figure 5a–c), was calculated. We tested different methods, basis sets, optimization schemes, and even structures until arriving at close correlation between the measured FTIR peak shifts and the calculated DFT peak shifts. The chosen parameters and structures are as described above.
The self-consistent field (SCF) energy calculations were performed on each system in order to derive the energy of the side chain-to-carbonyl H-bond contributions in the different multiplex systems.
The energy of the structures in Figure 5d–f were calculated. The two molecules in each of these systems were separated by 100 Å to remove any influence of H-bonding. By subtracting the far system from the close system, we remove the energy of covalent bonds and atoms from consideration. But we are still left with the energy of all of the H-bonds: the two canonical ones in black and the colored ones (Figure 5).
In order to remove the contribution of the canonical H-bonds, the structures in Figure 5h–j were created, where we rotated the serine side-chain χ1 dihedral by 180°, to break the H-bond. We calculated the energy of these structures, both when the molecules are close together and far apart. We again subtract the far system’s energy from the close system’s energy, giving us the energy of the canonical H-bonds. We deduct this canonical H-bond energy from the energy we calculated previously for all H-bonds, leaving us with the energy of just the colored H-bonds—namely, just the side chain to carbonyl contribution of the different multiplex H-bond schemes.
For the i over-coordinated H-bond, instead of separating the molecules far apart (since that would not break all of the H-bonds, and the side chain to carbonyl H-bond would remain intact), we converted the carbonyl to a methylene group, thereby breaking all H-bonds.
Acknowledgments
The authors thank Prof. A. Senes for invaluable discussions. The authors thank Sophie De-Botton for help synthesizing the isotopically labeled valine. This work was supported in part by grants from the United States-Israel Binational Science Foundation, the Israeli Science Foundation, and the Israeli Ministry of Science and Technology.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04357.
Figures S1–S4, showing H-bonding of serine and threonine in transmembrane α-helices, infrared spectra showing the amide I and II bands of the four M2 peptides, mimetics for DFT simulations showing the correct atomic geometry, and pre- and post-optimization overlays of serine mimetics, and Tables S1 and S2, showing the prevalence of amino acids in transmembrane helices of membrane proteins in the TOPDB53,54 and PDBTM21−23 databases and the H-bonding configuration of serine and threonine residues in a dataset of non-redundant α-helical membrane proteins21−23 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Watson J.; Crick F. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Pauling L.; Corey R.; Branson H. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U. S. A. 1951, 37, 205–211. 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L.; Corey R. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. U. S. A. 1951, 37, 251–256. 10.1073/pnas.37.5.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptitsyn O. B.; Rashin A. A. A model of myoglobin self-organization. Biophys. Chem. 1975, 3, 1–20. 10.1016/0301-4622(75)80033-0. [DOI] [PubMed] [Google Scholar]
- Kim S.; Cross T. A. Uniformity, Ideality, and Hydrogen Bonds in Transmembrane α-Helices. Biophys. J. 2002, 83, 2084–2095. 10.1016/S0006-3495(02)73969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R. C.; Kim S.; Cross T. A. Transmembrane Helix Uniformity Examined by Spectral Mapping of Torsion Angles. Structure 2008, 16, 787–797. 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Membrane protein folding and oligomerization: the two-stage model. Biochemistry 1990, 29, 4031–4037. 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Steitz T. A. The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell 1981, 23, 411–422. 10.1016/0092-8674(81)90136-7. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Steitz T. A.; Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 321–353. 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- Zhang Y. P.; Lewis R. N.; Hodges R. S.; McElhaney R. N. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane protein with phosphatidylcholine bilayers: differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry 1992, 31, 11579–11588. 10.1021/bi00161a042. [DOI] [PubMed] [Google Scholar]
- Zhang Y. P.; Lewis R. N.; Hodges R. S.; McElhaney R. N. FTIR spectroscopic studies of the conformation and amide hydrogen exchange of a peptide model of the hydrophobic transmembrane alpha-helices of membrane proteins. Biochemistry 1992, 31, 11572–11578. 10.1021/bi00161a041. [DOI] [PubMed] [Google Scholar]
- Wang J.; El-Sayed M. A. Temperature jump-induced secondary structural change of the membrane protein bacteriorhodopsin in the premelting temperature region: a nanosecond time-resolved Fourier transform infrared study. Biophys. J. 1999, 76, 2777–2783. 10.1016/S0006-3495(99)77431-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T. The hydrogen bond in the solid state. Angew. Chem., Int. Ed. 2002, 41, 48–76. . [DOI] [PubMed] [Google Scholar]
- Kendrew J. Side-chain interactions in myoglobin. Brookhaven Symp. Biol. 1962, 15, 216–228. [PubMed] [Google Scholar]
- Baker E.; Hubbard R. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984, 44, 97–179. 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- Gray T.; Matthews B. Intrahelical hydrogen bonding of serine, threonine and cysteine residues within alpha-helices and its relevance to membrane-bound proteins. J. Mol. Biol. 1984, 175, 75–81. 10.1016/0022-2836(84)90446-7. [DOI] [PubMed] [Google Scholar]
- Hildebrand P. W.; Preissner R.; Frömmel C. Structural features of transmembrane helices. FEBS Lett. 2004, 559, 145–151. 10.1016/S0014-5793(04)00061-4. [DOI] [PubMed] [Google Scholar]
- Feldblum E. S.; Arkin I. T. Strength of a bifurcated H bond. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 4085–4090. 10.1073/pnas.1319827111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H.-X.; Cross T. A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi M. J.; Shih W. M.; Harrison S. C.; Chou J. J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109–113. 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma D.; Simon I.; Tusnady G. E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2012, 41, D524–D529. 10.1093/nar/gks1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G. E.; Dosztányi Z.; Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2004, 33, D275–D278. 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G. E; Dosztányi Z.; Simon I. Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics 2004, 20, 2964–2972. 10.1093/bioinformatics/bth340. [DOI] [PubMed] [Google Scholar]
- Thomaston J. L.; Alfonso-Prieto M.; Woldeyes R. A.; Fraser J. S.; Klein M. L.; Fiorin G.; DeGrado W. F. High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 14260–14265. 10.1073/pnas.1518493112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer A. L.; Acharya R.; Salom D.; Levine A. S.; Di Costanzo L.; Soto C. S.; Tereshko V.; Nanda V.; Stayrook S.; DeGrado W. F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; Degrado W. F.; Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K. C.; Ashley R. H. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology 1992, 190, 485–489. 10.1016/0042-6822(92)91239-Q. [DOI] [PubMed] [Google Scholar]
- Salom D.; Hill B. R.; Lear J. D.; DeGrado W. F. pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry 2000, 39, 14160–14170. 10.1021/bi001799u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimm S.; Bandekar J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. 10.1016/S0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- Fulara A.; Dzwolak W. Bifurcated hydrogen bonds stabilize fibrils of poly(L-glutamic) acid. J. Phys. Chem. B 2010, 114, 8278–8283. 10.1021/jp102440n. [DOI] [PubMed] [Google Scholar]
- Torres J.; Adams P. D.; Arkin I. T. Use of a new label, 13C=18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 2000, 300, 677–685. 10.1006/jmbi.2000.3885. [DOI] [PubMed] [Google Scholar]
- Torres J.; Kukol A.; Goodman J. M.; Arkin I. T. Site-specific examination of secondary structure and orientation determination in membrane proteins: the peptidic 13C=18O group as a novel infrared probe. Biopolymers 2001, 59, 396–401. . [DOI] [PubMed] [Google Scholar]
- Verbovy D. M.; Smagala T. G.; Brynda M. A.; Fawcett W. R. A FTIR study of ion-solvent interactions in N,N-dimethylacetamide. J. Mol. Liq. 2006, 129, 13–17. 10.1016/j.molliq.2006.08.008. [DOI] [Google Scholar]
- Hamm P.; Lim M.; Hochstrasser R. M. Structure of the Amide I Band of Peptides Measured by Femtosecond Nonlinear-Infrared Spectroscopy. J. Phys. Chem. B 1998, 102, 6123–6138. 10.1021/jp9813286. [DOI] [Google Scholar]
- Tuckerman M. E.; Marx D.; Klein M. L.; Michele P. On the Quantum Nature of the Shared Proton in Hydrogen Bonds. Science 1997, 275, 817–820. 10.1126/science.275.5301.817. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Bowie J. U. Shifting hydrogen bonds may produce flexible transmembrane helices. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8121–8126. 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh O.; Urban M.; Henkes L. M.; Winterstein T.; Greiner T.; Van Etten J. L.; Moroni A.; Kast S. M.; Thiel G.; Schroeder I. Identification of Intrahelical Bifurcated H-Bonds as a New Type of Gate in K+ Channels. J. Am. Chem. Soc. 2017, 139, 7494–7503. 10.1021/jacs.7b01158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobedo A.; Topal B.; Kunze M. B. A.; Aranda J.; Chiesa G.; Mungianu D.; Bernardo-Seisdedos G.; Eftekharzadeh B.; Gairí M.; Pierattelli R.; Felli I. C.; Diercks T.; Millet O.; García J.; Orozco M.; Crehuet R.; Lindorff-Larsen K.; Salvatella X. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. 10.1038/s41467-019-09923-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain A. K.; Lee Y.; Kim S.; Bowie J. U. Snorkeling Preferences Foster an Amino Acid Composition Bias in Transmembrane Helices. J. Mol. Biol. 2004, 339, 471–479. 10.1016/j.jmb.2004.03.072. [DOI] [PubMed] [Google Scholar]
- Li W.; Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Fu L.; Niu B.; Zhu Z.; Wu S.; Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Seyfried M. S.; Lauber B. S.; Luedtke N. W. Multiple-Turnover Isotopic Labeling of Fmoc- and Boc-Protected Amino Acids with Oxygen Isotopes. Org. Lett. 2010, 12, 104–106. 10.1021/ol902519g. [DOI] [PubMed] [Google Scholar]
- Stock 3,5-dimethylpyridine hydrobromide salt was made by dissolving 6 mL of 3,5-lutidine (Sigma Aldrich, MO, USA) in 30 mL of anhydrous diethyl ether (Sigma Aldrich, MO, USA) and adding 12.8 mL of hydrogen bromide solution, 33 wt % in acetic acid (Sigma Aldrich, MO, USA). The mixture was stirred at room temperature under N2 for 1 hour.
- Manor J.; Mukherjee P.; Lin Y.-S.; Leonov H.; Skinner J. L.; Zanni M. T.; Arkin I. T. Gating Mechanism of the Influenza A M2 Channel Revealed by 1D and 2D IR Spectroscopies. Structure 2009, 17, 247–254. 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch W.; Hübner H. H. Orientation of specifically 13C=O labeled phosphatidylcholine multilayers from polarized attenuated total reflection FT-IR spectroscopy. Biophys. J. 1991, 59, 1261–1272. 10.1016/S0006-3495(91)82341-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; DiStasio R. A. Jr; Lochan R. C.; Wang T.; Beran G. J.; Besley N. A.; Herbert J. M.; Yeh Lin C.; Van Voorhis T.; Hung Chien S.; Sodt A.; Steele R. P.; Rassolov V. A.; Maslen P. E.; Korambath P. P.; Adamson R. D.; Austin B.; Baker J.; Byrd E. F. C.; Dachsel H.; Doerksen R. J.; Dreuw A.; Dunietz B. D.; Dutoi A. D.; Furlani T. R.; Gwaltney S. R.; Heyden A.; Hirata S.; Hsu C.-P.; Kedziora G.; Khalliulin R. Z.; Klunzinger P.; Lee A. M.; Lee M. S.; Liang W.; Lotan I.; Nair N.; Peters B.; Proynov E. I.; Pieniazek P. A.; Min Rhee Y.; Ritchie J.; Rosta E.; David Sherrill C.; Simmonett A. C.; Subotnik J. E.; Lee Woodcock H. III; Zhang W.; Bell A. T.; Chakraborty A. K.; Chipman D. M.; Keil F. J.; Warshel A.; Hehre W. J.; Schaefer H. F. III; Kong J.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. 10.1039/B517914A. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H.; Harrison R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
- Dobson L.; Langó T.; Reményi I.; Tusnády G. E. Expediting topology data gathering for the TOPDB database. Nucleic Acids Res. 2015, 43, D283–D289. 10.1093/nar/gku1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G. E.; Kalmár L.; Simon I. TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res. 2007, 36, D234–D239. 10.1093/nar/gkm751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.