
Figure 1.
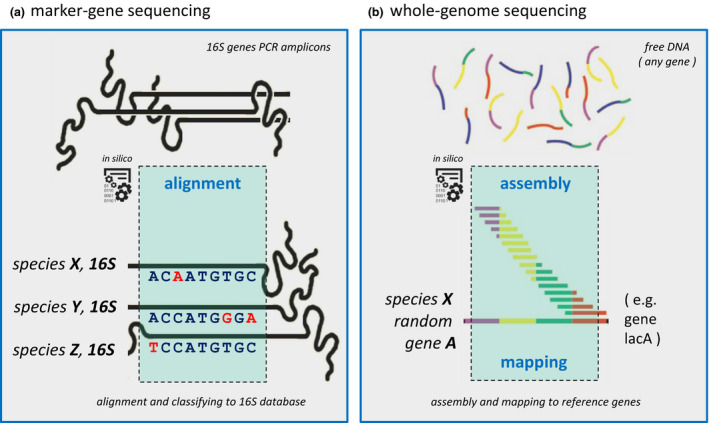
Principal microbiota sequencing approaches. We roughly distinguish two main applications for next‐generation sequencing (NGS) of microbial communities: marker‐gene sequencing (MGS) for metataxonomics and whole‐genome sequencing (WGS) for metagenomics. (a) In the MGS example, 16S is selected as marker gene, which is extracted from a mixed microbial population by polymerase chain reaction (PCR, not shown), and sequenced by NGS techniques. After MGS sequencing, reads (±500 bp) are aligned, and based on informative positional differences in the 16S gene, known reference microbiota can be assigned, or novel taxonomies can be inferred. With WGS, one can extract genomic potential and function information, in contrast with MGS, as with the latter, one can only extract taxonomic information. (b) In the WGS example, typically small sequences (100–150 bp), derived randomly from the full genomic content (i.e. all genes present, not focusing only on 16S) of a mixed microbial population, are assembled into genes of all microbiota present.