

Key Points
In elderly newly diagnosed AMLtosedostat and low-dose cytarabine achieved an ORR of 54.6% with long-term survival in responding patients.
Gene expression profiling predicted sensitivity to the combination with a negative predictive value of 100% validated in an independent set.
Abstract
Tosedostat is an orally administered metalloenzyme inhibitor with antiproliferative and antiangiogenic activity against hematological and solid human cancers. Clinical activity has been demonstrated in relapsed acute myeloid leukemia (AML). Thirty-three elderly patients with AML (median age, 75 years) received 120 mg tosedostat orally once daily combined with subcutaneous low-dose cytarabine (20 mg twice per day for 10 days, up to 8 cycles), until disease progression. Induction mortality was 12%. According to an intention-to-treat analysis, the complete remission (CR) rate was 48.5%, and thus the primary end point of the study was reached (expected CR, 25%). The partial remission rate was 6.1%, with an overall response rate of 54.5%. Furthermore, 4 of 33 patients had stable disease (median: 286 days). The median progression-free survival and overall survival (OS) were 203 days and 222 days, respectively. Responding patients had a longer median OS than nonresponding patients (P = .001). A microarray analysis performed in 29 of 33 patients identified 188 genes associated with clinical response (CR vs no CR). Three of them (CD93, GORASP1, CXCL16) were validated by quantitative polymerase chain reaction, which correctly classified 83% of the patients. Specifically, CR achievement was efficiently predicted by the gene expression patterns, with an overall accuracy exceeding 90%. Finally, a negative predictive value of 100% was validated in an independent series, thus representing the first molecular predictor for clinical response to a specific combination drug treatment for AML. This trial has been registered at the European Medicines Agency and on the European Clinical Trials Database (https://www.clinicaltrialsregister.eu) as #2012-000334-19.
Visual Abstract

Introduction
The treatment of elderly patients with acute myeloid leukemia (AML) is still a major challenge, given the significant increase in median age of the worldwide population. Although several clinical and biological features at diagnosis have been reported as useful predictors of clinical outcome, age remains one of the most relevant prognostic factors for AML, because the prognosis steadily declines with age and is independent from other factors.1
In addition to patient-related factors (ie, comorbidities limiting the usage of aggressive chemotherapy), AML in older patients is associated with an increased incidence of adverse genetics, evolution from myelodysplastic syndromes (MDSs) or myeloproliferative neoplasms, and high rates of expression of multidrug resistance protein-1 in tumor cells, all of which account for adverse clinical outcomes.2 Poor outcomes have been reported in elderly patients, regardless of the well-recognized prognostic factors related to intrinsic adverse features of leukemic cell progenitors.3
Overall, the prognosis of the disease in patients aged >70 years is extremely poor, with most dying within 1 year of diagnosis.4 With currently available chemotherapy regimens, the gold standard is still considered to be the 3+7 daunorubicin/cytarabine combination, and fit patients can achieve complete remission (CR) rates ranging from 45% to 55%; however, fewer than 10% survive at least 5 years. Nonintensive approaches, mainly hypomethylating agents (HMAs), result in CR rates up to 25%, with occasional long-term remissions.5 Recently, 2 new drugs, glasdegib and venetoclax (VEN),6,7 have been approved by the US Food and Drug Administration for the treatment of patients aged >75 years or those considered ineligible for intensive therapies. Despite the approval, results are still generally considered unsatisfactory for glasdegib, which in combination with low-dose cytarabine (LDAC) produced a CR rate and a median overall survival (OS) of 17% and 8.8 months, respectively.8 In contrast, the combination of VEN plus either LDAC or azacitidine (AZA) produced high CR rates (54% and 67%, respectively) and prolonged OS (10.1 and 17.5 months, respectively).9,10 These preliminary observations were subsequently tested in 2 phase 3 clinical trials: VIALE-C and VIALE-A.11,12 VIALE-C was a phase 3 randomized trial of LDAC, with or without VEN.11 Eligibility criteria were similar to those of the phase 1b/2 trial. In total, 211 patients were randomized at a 2:1 ratio to either VEN (n = 143) or placebo (n = 68). The median age was 76 years (range, 36-93), and 38% had secondary AML; half of them had received prior HMA treatment. A third of the patients had poor-risk cytogenetics and TP53, and FLT3 mutations were present in 20% of the VEN group and 18% of the placebo group, respectively, in the VEN arm. The addition of VEN to LDAC resulted in a 25% survival benefit (hazard ratio [HR], 0.75; P = .11), which, surprisingly, was not statistically significant; with a median OS of 7.2 and 4.1 months, respectively.11 VIALE-A was a phase 3 randomized trial of AZA, with or without VEN.12 Patients who were ineligible for intensive induction therapy because of medical comorbidities and/or age ≥75 years (n = 431; median age, 76 years; range 49-91), were randomized in a 2:1 ratio to AZA+VEN (n = 286) or AZA+placebo (PBO; n = 145). Those with prior treatment with HMAs were excluded. With a median follow-up of 20.5 months, the median OS was 14.7 months with AZA+VEN and 9.6 months with AZA+PBO (HR, 0.66; 95% CI, 0.52-0.85; P < .001), representing a 34% reduction in risk of death. Furthermore, certain molecular markers (IDH1, IDH2, NPM1, and TP53) identified patients who were more likely to respond to VEN+HMA.
Even though the results of the VIALE-A were outstanding, all of these therapeutic options, on the one hand, are burdened with significant toxicities, and, on the other hand, offer no predictive biomarkers capable of identifying which patients may benefit more from any of these new agents.13
Tosedostat is a new, orally bioavailable inhibitor of members of the M1 and M17 classes of aminopeptidases that include the zinc-dependent aminopeptidases. Aminopeptidases catalyze the hydrolysis of the terminal amino acids from peptides or polypeptides generated by proteasomal degradation and are involved in the continuous cycle of protein synthesis and degradation.14 The aminopeptidase inhibitor tosedostat is converted in the intracellular compartment into a poorly membrane-permeable, active metabolite (CHR-79888) that inhibits the M1 family of aminopeptidases (particularly puromycin-sensitive aminopeptidase), leukotriene A4 hydrolase, and the M17 family member leucine aminopeptidase. Inhibition of aminopeptidases by tosedostat and its metabolite leads to intracellular accumulation of small peptides, which in transformed cells of the hematopoietic lineage appears to cause a deficiency of free amino acids for new protein synthesis.15 When used as a single agent, tosedostat induced an overall response rate of 27% in elderly patients with AML.16,17 Cytarabine has been widely used in the treatment of AML for several decades, and the safety profile is well characterized. Tosedostat appears to be well tolerated by older patients, and the safety profile does not share features with that of cytarabine. The results of preclinical and early clinical testing suggest that the combination of cytarabine and tosedostat is synergistic with respect to an antidisease effect in AML cells, supporting the idea of a combined treatment.18
Nonetheless, a recent randomized trial (LI-1),19 presented up to now only as an abstract, did not show a survival benefit for the combination of tosedostat and LDAC. In brief, 245 patients (median age, 76 years; range, 60-88) were randomized to receive LDAC+tosedostat vs LDAC alone, with the goal of doubling 2-year survival from 11% to 22% (HR, 0.70). Complete remission was achieved in 22% of the patients treated with LDAC+tosedostat and in 14% of those treated with LDAC alone (odds ratio [OR], 0.59; P = .11). Thirty-day mortality did not increase significantly (17% vs 13%; HR, 1.44; 95% CI, 0.75-2.78; P = .3), and relapse-free survival (HR, 0.93; 95% CI, 0.41-2.16; P = .9) and OS showed no difference (2-year OS, 16% vs 12%; HR, 0.99; 95% CI, 0.74-1.32; P = .9).
We report the final results of a phase 2 study conducted in 33 elderly patients with AML, aged >60 years, whose condition was not suitable for the 3+7 cytarabine+daunorubicin chemotherapy regimen and were treated with tosedostat coupled with LDAC. In this study, we hypothesized that the addition of tosedostat to LDAC would improve the response rate and remission duration over that expected with chemotherapy or tosedostat alone. Furthermore, we performed global gene expression profiling (GEP; Human Gene 2.0 Array; Affymetrix) on purified AML blasts from the peripheral blood or bone marrow of 29 patients at diagnosis, before the initiation of treatment, to verify whether the achievement of CR could be efficiently predicted by 1 or more patterns of gene expression.
Methods
Study design and patients
We designed a prospective, multicenter, single-arm, phase 2 clinical trial that tested the combination of LDAC and tosedostat in previously untreated elderly patients with AML whose conditions were not suitable for receiving intensive chemotherapy. Patients aged ≥60 years with de novo or secondary AML that arose from MDS or MDNs, were considered eligible for the study. Additional inclusion and exclusion criteria are described in the supplemental Data.
The study was approved by an independent research ethics committee and was conducted in accordance with the International Conference on Harmonization Good Clinical Practice Guidelines, the Declaration of Helsinki (1996), and local regulatory requirements and laws. Detailed characteristics of the patients are listed in Table 1.
Table 1.
Patients’ characteristics
| Characteristics | n | % |
|---|---|---|
| Age, median (range), y | 75 (62-85) | — |
| Sex | ||
| Male | 22 | 67 |
| Female | 11 | 33 |
| WHO performance status | ||
| 0-1 | 23 | 70 |
| 2-3 | 10 | 30 |
| Diagnosis | ||
| De novo | 16 | 48 |
| Secondary MDS | 17 | 52 |
| Karyotype | ||
| Intermediate | 18 | 56 |
| Unfavorable | 13 | 39 |
| Not evaluable | 2 | 6 |
| Laboratory median data (range) | ||
| Hb, g/dL | 9.4 (7,6-12.3) | — |
| WBC, ×109/L | 3.05 (0.26-24.53) | — |
| PLT, ×109/L | 69 (6-260) | — |
| Blasts, % | 60 (16-96) | — |
Treatment administration and toxicity assessment
The primary objective of the study was to assess the CR rate of the combination regimen of tosedostat and LDAC in patients aged ≥60 years who had newly diagnosed AML. The secondary objectives were to judge the safety and toxicity of the combination regimen, assess the OS and progression-free survival (PFS), evaluate the response rate, and identify possible biomarker(s) through studying the global GEPs.
Patients received tosedostat 120 mg orally once daily from day 1 to 240 (8 cycles), with LDAC given subcutaneously at 20 mg twice daily from day 1 to 10. Courses of LDAC were repeated every 4 weeks, in the absence of disease progression or unacceptable toxicity, up to 8 cycles. Patients with stable or improving disease conditions (ie, a decrease in blast percentage) received additional courses of tosedostat after the initial 8 cycles until the disease progressed or unacceptable toxicity was identified. To assess treatment efficacy, the patients were evaluated by bone marrow aspiration for morphologic, immunophenotypic, and cytogenetic (fluorescence in situ hybridization) analyses undertaken during the screening, after the second course of therapy, and every 3 months thereafter. In the responding patients who experienced >2 nonhematological toxicity events, according to World Health Organization (WHO) criteria, dose reductions were applied for both tosedostat, from 120 mg daily to 60 mg daily, and LDAC, from 20 mg to 10 mg or 5 mg subcutaneously, once or twice daily. The treatment was intended to be administered in an outpatient setting; however, all patients required hospitalization for the first cycle of therapy because it was required by study protocol as a precaution. Standard antimicrobial prophylaxis and supportive care measures were as reported elsewhere.20
Toxicities were scored according to the National Institutes of Health, National Cancer Institute’s Common Terminology Criteria for Adverse Events (AE), version 4.21 A serious AE resulted in death or immediate risk of death, prolonged hospitalization, or substantial disability. Responses were assessed according to International Working Group criteria,22 and Cytogenetic risk was assessed on the basis of Southwest Oncology Group criteria.23 Patients who completed 1 full cycle of treatment were considered evaluable.
Statistical analysis
This study was designed according to Fleming’s method.24 The primary outcome was complete remission according to International Working Group criteria.22 With the lowest acceptable rate fixed at 10%, the successful rate at 25%, a significance level of α = 0.05, and a 1-β power = 0.80, the necessary sample size was estimated to be 33 patients.
Statistical analyses were performed according to the intention-to-treat approach. OS and PFS were estimated according to the Kaplan-Meier method. The log-rank test was used to assess the significance of differences for each prognostic factor in the univariate analysis. The Cox regression models and the χ2 test were used to assess how patients’ characteristics predicted PFS and OS. The Statistical Package for Social Science (SPSS) was used to analyze the data. A 2-sided test was used in all calculations with the significance level fixed at α = 0.05 for all analyses.
Please see supplemental Data for the methods related to GEP generation and analysis and reverse-transcription and real-time quantitative polymerase chain reaction (qPCR).
Results
Treatment efficacy
All 33 patients received at least 1 course of treatment and were considered evaluable. Overall, the patients received tosedostat for a median of 183 days (range, 12-2094).
According to the intention-to-treat analysis, the CR rate was 48.5% (16 of 33 patients), 2 additional patients obtained a partial response (PR; 6.1%), and the overall response rate was 54.5% (18 of 33 patients). All CRs were full CRs; we did not observe any CR with incomplete or partial hematologic recovery. In responding patients, the median time to best response was 84 days (range, 22-317).
Some patients (11 of 33; 33%) did not show a significant blast reduction. Four of 11 had stable disease for a median of 286 days (range, 145-429), whereas 7 of 11 did not respond and died of progressive disease after having received a median of 2 cycles of LDAC and 50 days of tosedostat.
The overall rate of death during induction was 12%, with 4 of 33 deaths occurring in patients with aplasia (documented by bone marrow biopsy). Among the 18 responding patients (CR+PR), 1 was still in CR at 68 months after the first cycle of therapy, whereas 15 relapsed after a median of 203 days (range, 87-535), and 2 died in CR of heart failure and septicemia.
The median PFS and the median OS were 203 days (95% CI, 18-2062) and 222 days (95% CI, 18-2094), respectively (Figure 1A-B). Responding patients had a longer median OS than nonresponding ones (P = .001; Figure 1C).
Figure 1.
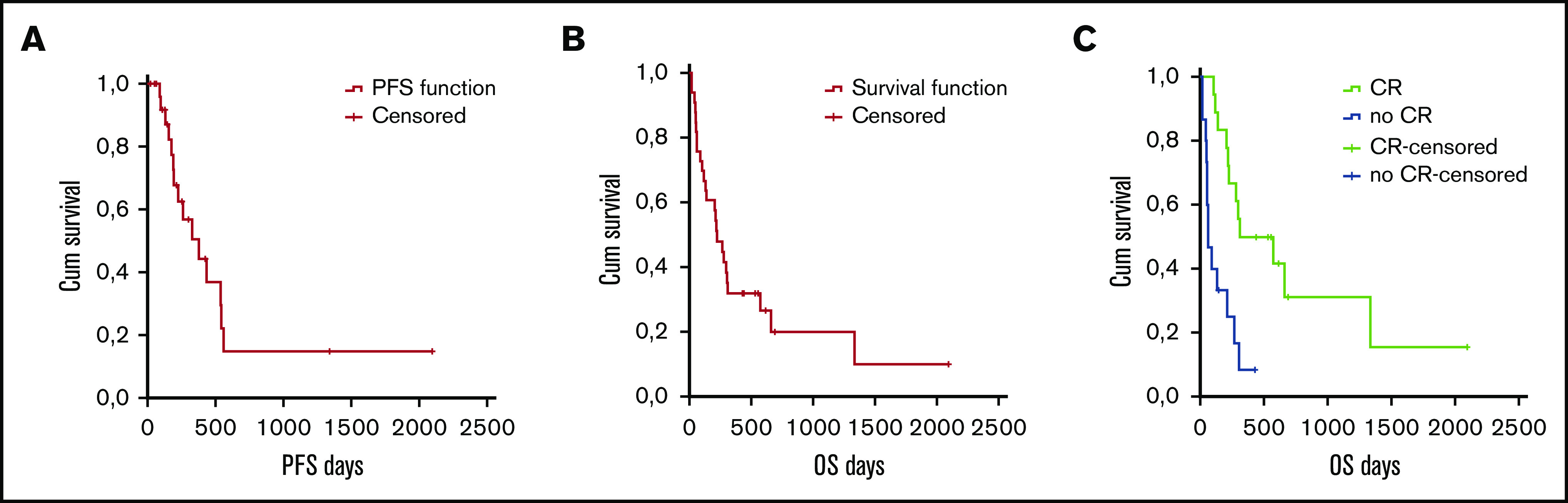
Survival in all patients according to intent to treat. PFS (A) and OS (B) in all patients. (C) OS according to response (CR vs no CR).
Prognostic factors analysis
We then sought to estimate whether additional prognostic factors could be used to stratify patients and to identify potential biomarkers. Age was not associated with OS and PFS when cutoff values of 70 and 75 years were adopted. As far as the disease-related features were concerned, we found no association between PFS/OS and performance status (WHO score of 0, 1, or 2), type of disease (de novo vs secondary), and karyotype (favorable, intermediate, or unfavorable). Finally, we found that achieving a CR was significantly associated with prolonged survival in univariate and multivariate analyses (P < .001).
Treatment toxicity
Overall, the combination of tosedostat and LDAC was well tolerated; no patient had a planned treatment interruption or a dose reduction related to toxicity. As expected, the most common AEs were related to hematological toxicity (Table 2). In particular, of the 33 patients, 3 experienced grade 3-4 anemia; 18, grade 3-4 thrombocytopenia; and 25, grade 3-4 neutropenia. As a consequence of severe neutropenia, infections were observed to occur in 15 patients. The most common infections were pneumonia (8 events) and fever of unknown origin (7 events). Conversely, thrombocytopenia-related severe bleeding was rare, with 2 events occurring, both nonlethal central nervous system hemorrhages.
Table 2.
Adverse events
| Adverse event | Total, n | Grades 1-2, n | Grades 3-4, n |
|---|---|---|---|
| Hematological | 52 | 6 | 46 |
| Infective | 50 | 25 | 25 |
| Hepatic | 33 | 25 | 8 |
| Metabolic abnormalities | 29 | 24 | 5 |
| Other | 28 | 25 | 3 |
| Gastrointestinal | 23 | 22 | 1 |
| Cardiac | 15 | 9 | 6 |
| Bleeding | 6 | 4 | 2 |
| Skin | 6 | 6 | 0 |
| Urinary | 5 | 2 | 3 |
Data are the number of events recorded in the entire population.
Other nonhematological toxicities included hepatic injury (33 events, 8 of which were grades 3-4), metabolic and laboratory result abnormalities (29 events; 5 grade 3 to 4), gastrointestinal events (23 events; 1 grade 3-4), and cardiac events (9 events; 6 grade 3-4). Skin toxicity was relatively rare, occurring in 6 patients (18%) (supplemental Table 2).
Grades 3-4 nonhematological toxicities
Few patients had ≥1 grade 3-4 nonhematological toxicity during or after treatment with tosedostat. One patient had grade 4 acute renal and cardiac failure related to AML and died of progressive disease. AML-related grade 4 central nervous system hemorrhage and atrial fibrillation occurred in the same patient. Eight grade 3-4 liver toxicity events occurred in 3 patients. Two patients developed grade 3-4 increases in alanine transferase and γ-glutamyl transpeptidase and hypoalbuminemia and hypofibrinogenemia during cytarabine+tosedostat treatment. They temporarily discontinued the treatment, received medical therapy, and were successfully rechallenged after the normalization of the blood examination. One additional patient developed a transient grade 4 increase in alanine transferase and decrease in fibrinogen while receiving the combination treatment. Again, he was successfully rechallenged after temporary discontinuation of the drugs. None of the 3 patients experiencing a grade 4 toxicity showed clinical signs of liver toxicity. Furthermore, ultrasonography did not show any sign of hepatic injury. Finally, 2 patients developed grade 4 bronchiolitis obliterans organizing pneumonia after discontinuing the treatment because of disease progression and rapidly died of full-blown disease. Four patients experienced grade 4 respiratory failure leading to death, as a consequence of pneumonia that developed either after treatment (1 patient) or with disease progression after the first cycle of chemotherapy (3 patients).
Molecular profiling discriminated patients according to clinical response
The majority of the patients (29 of 33; 88%) underwent GEP analysis that, in all cases, enabled clinicians to define the clinical outcome (CR vs no CR; no response) while seeking to identify a molecular signature associated with the clinical response. By supervised analysis, 188 probe sets, corresponding to 140 unique genes, were found to be differentially expressed based on the clinical response (CR vs no CR; Mann-Whitney U test; P ≤ .05; fold change, ≥2; Figure 2; supplemental Table 3). The 140 genes differentially expressed were significantly associated with relevant biological functions and pathways, including functions related to the selected oncogenes NPM1, STK33, AKT, and HOXA9, as well as myeloid and hematopoietic stem cell development and immune and inflammatory responses. As far as the latter is concerned, interestingly, a NET-related inflammatory signature, recently shown to be associated with clinical response to lenalidomide-containing regimens for AML,25 was also found to be significantly associated with the response to tosedostat (Figure 3; supplemental Table 4).
Figure 2.
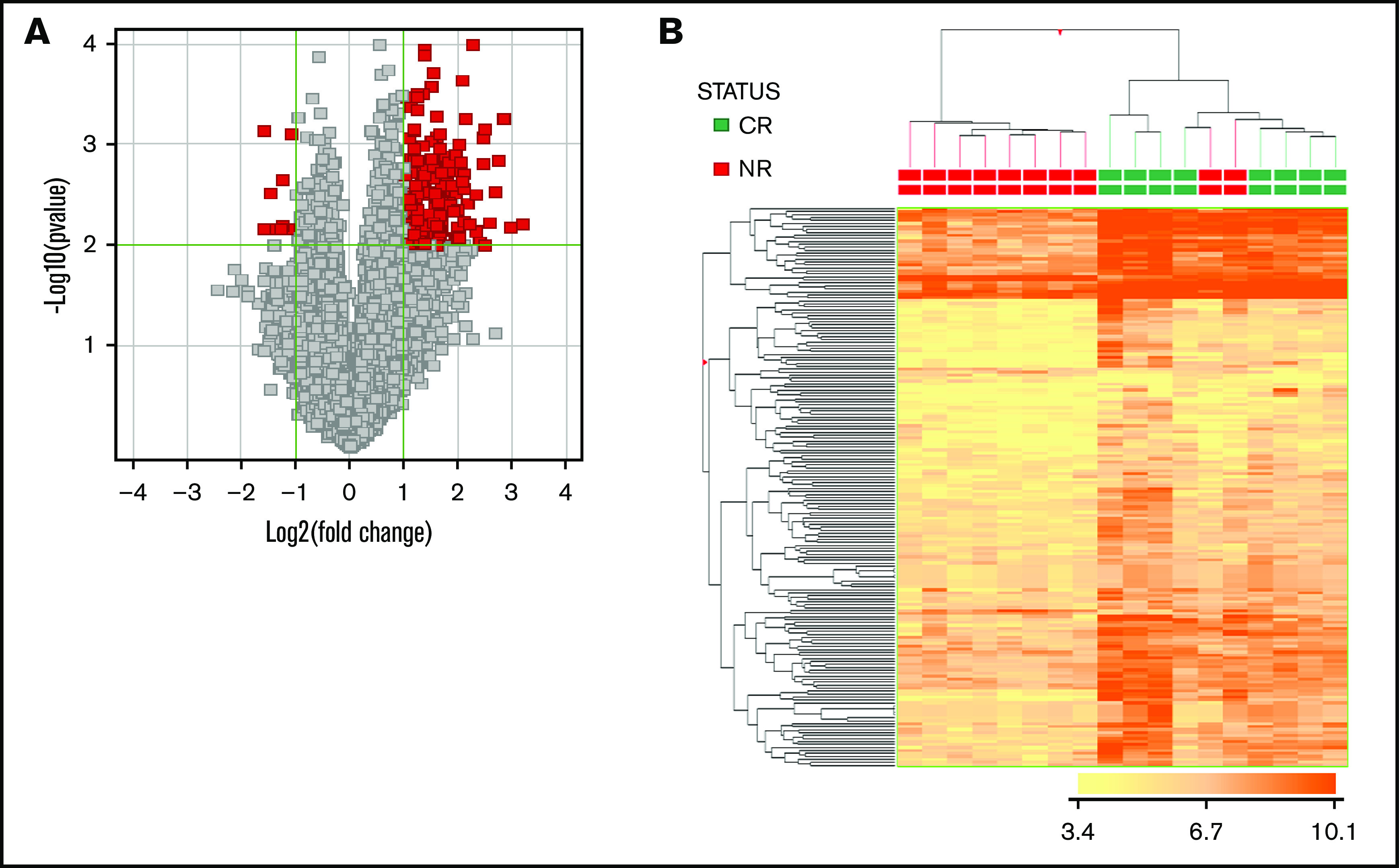
GEP enables a clear distinction of cases based on clinical response. (A) Volcano plot indicating genes differentially expressed in the 2 groups (CR [red] vs NR [gray]). (B) Hierarchical clustering based on the expression of 188 differentially expressed probe sets (corresponding to 140 genes), distinguishing CR vs NR cases.
Figure 3.
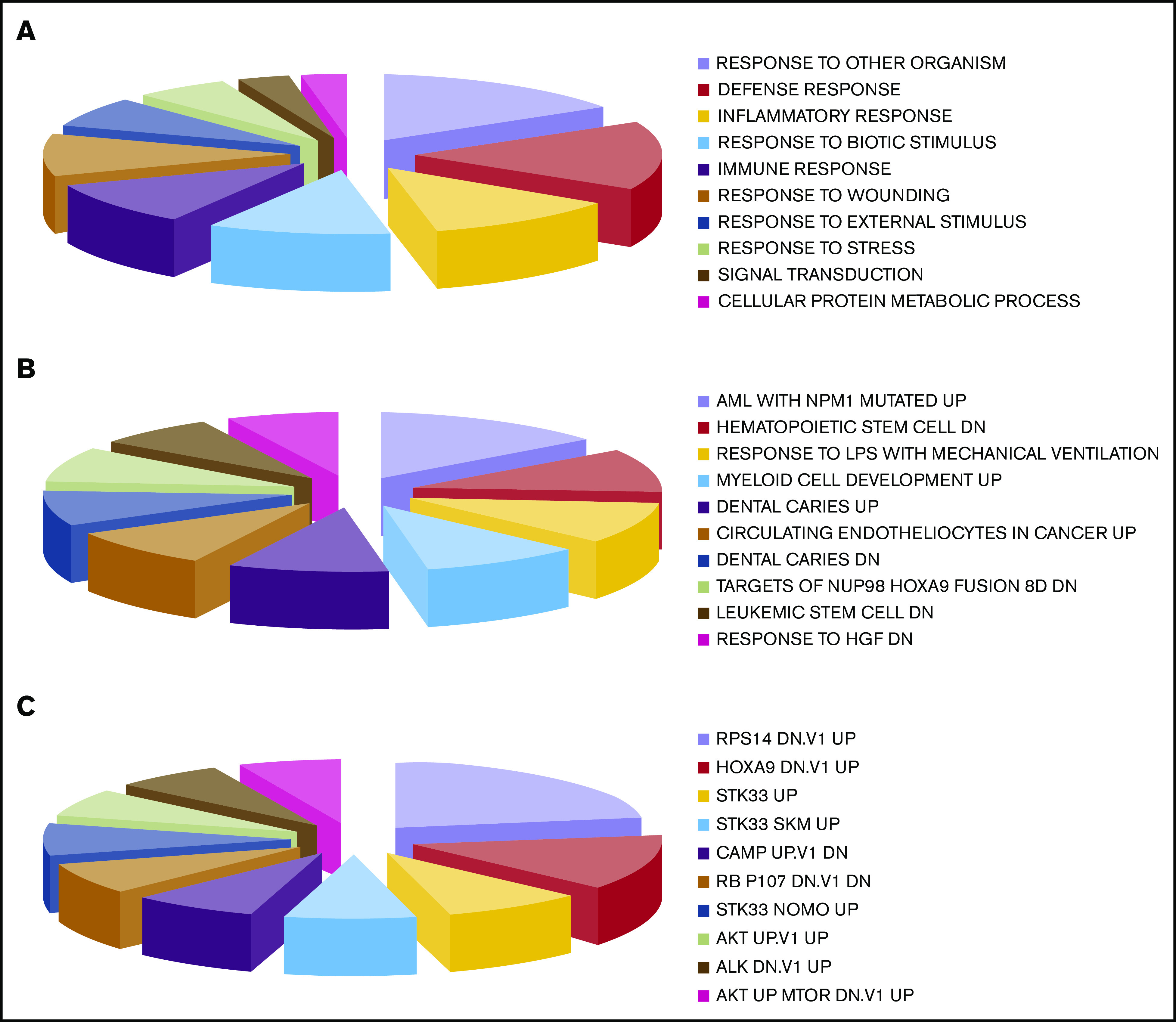
Gene Set Enrichment Analysis of genes differentially expressed in the 2 groups defined by the clinical response, CR vs NR. (A) Gene Ontology biological processes. (B) Curated gene sets. (C) Oncogenic signatures. The size of each group in the pie charts corresponds to the number of genes found in each category.
We also tested whether the expression of a reduced number of genes would be sufficient to predict the clinical response to tosedostat-LDAC. By linear discriminant analysis, we identified 3 genes (CD93, GORASP1, and CXCL16) with predictive capacity. Based on the expression of these 3 genes, cases were classified as CR or NR, with an overall accuracy of 88.9% (16 of 18) and marked sensitivity (88%) and specificity (90%). Seven of 8 responding and 9 of 10 nonresponding patients were correctly identified.
To make these data more robust, we studied the expression of CD93, GORASP1, and CXCL16 by qPCR, obtaining analogue results when compared with microarray analysis. CD93 and CXCL16 were confirmed to be significantly overexpressed in patients who did not achieve a CR (P = .01 and P = .003, respectively). Conversely, GORASP1 showed a nonsignificant trend (P = .095); nonetheless, it was consistent with that observed in the microarray analysis (supplemental Table 5). Based on the qPCR results, remarkably, 15 of 18 (83%) cases were correctly classified according to the clinical response (Figure 4). When the PR cases (n = 2) were removed and the analysis was rerun, no differences were observed (data not shown).
Figure 4.
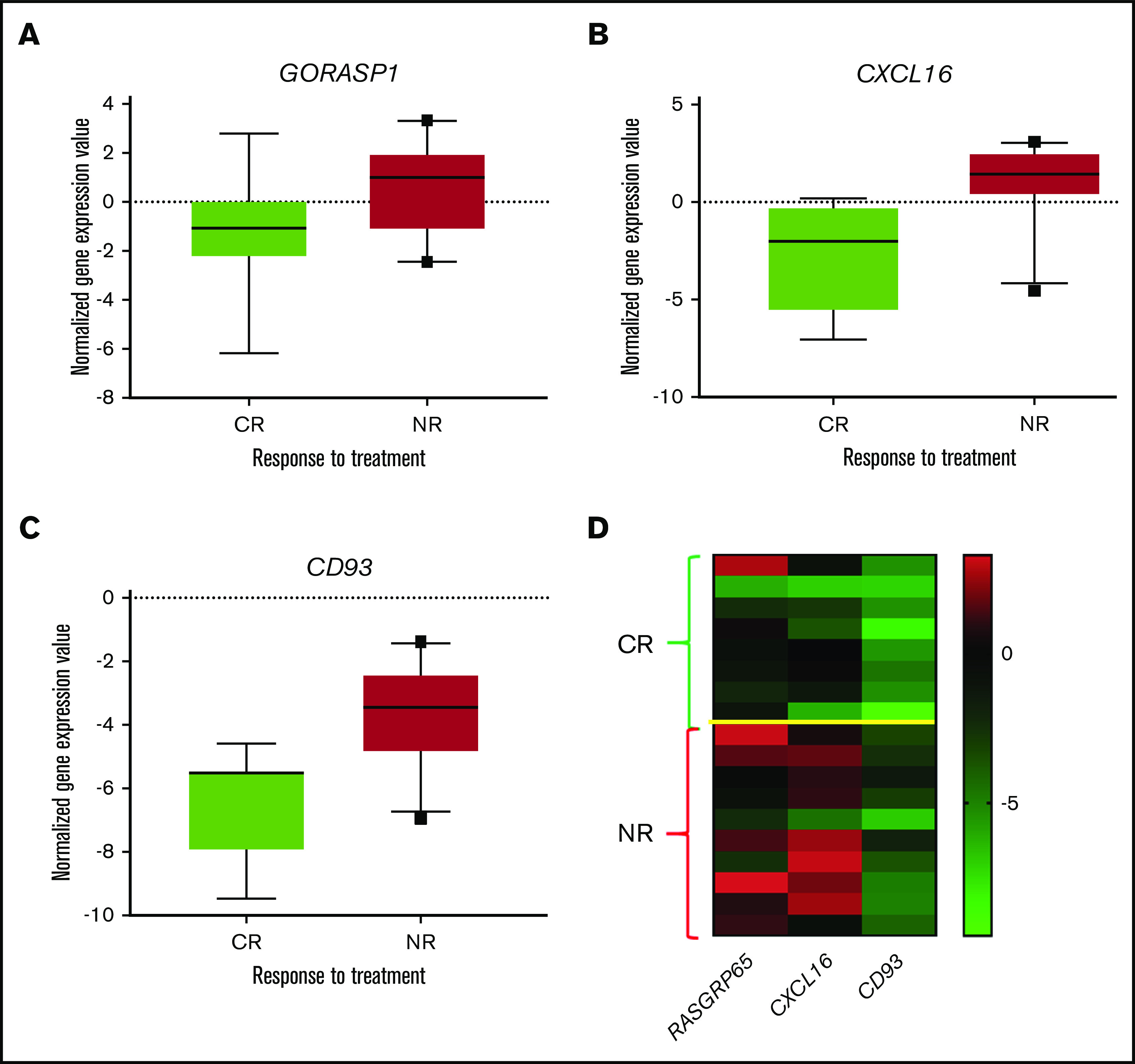
Validation of gene expression analysis by qPCR. (A-C) Single gene expression from the 10th to 90th percentiles plotted in the box. (D) Heat map showing the expression of the 3 genes.
To determine whether the identified molecular signature could also predict the clinical response in patients with AML who were <60 years of age and had received conventional chemotherapy, we tested its ability in a large panel of AML cases (n = 461) for which GEP and clinical data were available in a public database26 (GSE6891). In this case, however, the signature was not associated with the clinical outcome, suggesting specificity for the tosedostat-LDAC combination.
To make our data more robust, we then tested our molecular classifier in an independent cohort of cases. Specifically, RNA samples from 36 patients treated with tosedostat in a distinctive trial in the United Kingdom19 were studied for the expression of CD93, CXCL16, and GORASP1. Remarkably, the overall accuracy of CR vs no CR prediction was 69.4%, with 100% sensitivity and 68% specificity, with a negative predictive value of 100% (95% CI, −4 to 35) (Figure 5).
Figure 5.

Expression of the 3 genes in an independent data set of patients with AML treated with conventional chemotherapy (GSE6891). No significant differences were recorded, suggesting the specificity of the identified signature in discriminating patients likely to respond to the tosedostat-araC combination only.
Overall, these data indicate that GEP may be used to predict clinical sensitivity to the tosedostat-LDAC combination, representing the first molecular predictor for clinical response to a specific drug combination for AML.
Discussion
With the approval of several new drugs in the past 3 years, therapy for AML in elderly patients is finally evolving, with glasdegib,8 VEN,9,12 and CPX-35127 approved by the United States Food and Drug Administration. Additional emerging therapies including FLT-3 inhibitors, ivosidenib, and enasidenib could offer the elderly AML population better prospects of obtaining a CR.11 Nonetheless, some of these drugs, such as VEN, have shown the potential to significantly increase survival in phase 2 trials, even if only the association of VEN+AZA resulted in a statistically significant increase of OS in subsequent phase 3 trials.11,12 Furthermore, in favor of VEN-based therapies, there is increasing knowledge that certain biomarkers (IDH1, IDH2, NPM1, and TP53) indeed identify which patients are more or less likely to respond to VEN regimens.28 Despite the encouraging results recently observed with VEN or other new drugs, relapses are still very common, and it is becoming clear that these combinations will not be curative for most patients with AML.29 Three major hurdles may also explain in part why new drugs and combinations fail to provide CR for many patients. First, primary resistance and clonal evolution leading to adaptive resistance is a recurring theme in AML. Recently, DiNardo et al28 described molecular patterns of response and the failure of the VEN-based combination, with RUNX1, TP53, and FLT3-ITD conferring cross-resistance to both VEN and cytotoxic-based therapies. Second, 30-day mortality with novel drugs is low but not irrelevant. The induction death rate of between 3% and 6%, primarily because of infections such as prolonged neutropenia, increases to >20%, even after only 1 cycle of therapy.28,29 It is well known that at least 25% to 40% of elderly patients with AML receive treatments that not only lack efficacy, but also produce grade 3-4 nonhematological toxicities, such as infections and pneumonia, which are responsible for morbidity and mortality. Last, with the exception of patients bearing IDH-1/IDH-2 or FLT-3 mutations, biomarkers that can predict response and/or long-term survival are still lacking. Accordingly, there is still an urgent need to identify molecular predictors of a clinical response to specific drug combinations for treating AML, to minimize toxicity and maximize efficacy.
The tosedostat and LDAC combination produced a CR rate superior to that predicted (48.6% vs 25%), and the trial thus met its primary end point. Even in an era of successful clinical trials for AML, the primary end point is an important bar for discriminating between failure and success. Remarkably, a specific molecular signature discriminated patients according to treatment response. Our data support30,31 the prospective use of GEP-driven therapy in a cohort of patients with hard-to-treat AML, who are unfit for standard therapy and had an extremely poor prognosis. GEP-based identification of reliable biomarkers that can predict which patients are more likely to achieve clinical responses could be a major achievement of this trial, especially if validated in a phase 3 trial.
The clinical results produced in our trial are in line with the unique previous experience with the combination,18 which reported a 53% overall response rate in a similar setting, as well as poor correlation between response and known prognostic factors. The present study was innovative in that, first, it included only patients with AML and not those with MDS and, second, no alternative regimens were tested. Overall, we evaluated 33 patients with AML who were treated with the combination (in accordance with the study design), whereas Mawad and colleagues studied only 11 patients with AML. Furthermore, their study was set up for evaluating the response rate at a 4-month end point, with long-term follow-up lacking.18 Therefore, the present study offers a clearer evaluation of both efficacy and toxicity of the tosedostat/LDAC combination.
Interestingly, when we reanalyzed the trial with the European LeukemiaNet (ELN) response criteria,32 we found 4 patients with stable disease, without a marrow response, who survived a median of 274 days. This translated into a clear clinical benefit,33 as all of them presented a hematological improvement coupled with a significant decrease in transfusion burden (data not shown) and an increase in OS. In this light, the mechanism of action of tosedostat favors the inhibition of aminopeptidases, resulting in a deficiency of free amino acids for new protein synthesis in hematopoietic cancer cells, which could in part explain these unexpected results.15
The efficacy of currently available conventional chemotherapy is limited by well-characterized patient- and disease-related prognostic factors. Indeed, the use of conventional chemotherapy in some categories (eg, very unfavorable karyotype) is frustrating.34 In our study, we observed that the tosedostat-LDAC combination was equally effective in patients with de novo vs secondary AML, favorable vs unfavorable karyotype, and higher vs lower white blood cell counts. This is noteworthy, in that it suggests that such new agents can overcome classic drug-resistance mechanisms, offering alternatives to high-risk patients, especially elderly ones.
Another relevant point of our trial is the safety of the combination. When elderly patients receive any anticancer treatment, toxicity is a major challenge. The combination of tosedostat-LDAC was well-tolerated overall; no patient had to stop treatment because of toxicity. Besides cytopenias, the commonest severe AEs were infections, hepatic injury, and cardiac events, in line with experience.16,17
The failure to correlate validated clinical-pathological patterns, such as ELN classification,32 in patients treated with tosedostat-LDAC makes it difficult to select patients before treatment, according to validated prognostic factors and guidelines. It should be noted, however, that some molecular prognostic factors, currently included in the ELN classification of AML, were not tested in our study. Those included the mutational status of ASXL1, RUNX1, TP53, CEBPA, NMP1, and FLT3. Nonetheless, the analysis was not significantly affected, because the studied population had already been assessed as very high risk.
With this in mind, we performed dedicated correlative studies with GEP analysis, seeking to identify possible biomarkers predictive of a response to this combo. One hundred eighty-eight unique genes differentially expressed based on the clinical response (CR vs no CR) were identified. When we then tested whether the expression of a reduced number of genes could be sufficient to predict the clinical response to tosedostat-cytarabine, we identified 3 genes (CD93, GORASP1, and CXCL16) with a capacity that could be validated by real-time-qPCR (Figure 5).
Interestingly, all these genes may have a major role in leukemia initiation, progression, and relapse. CD93, a cell surface lectin, has been identified as a marker of the leukemia stem cells (LSCs), which are required for development of MLL-rearranged AML.35 Accordingly, tosedostat should be considered a golden key targeting LSCs through amino acid deprivation response, which occurs selectively in transformed hematopoietic cells, such as LSCs. GORASP1 is involved in Fas/CD95-mediated apoptosis, as well as in Golgi function and cell-cycle progression.36 Finally, CXCL16 is a potent mediator of angiogenesis, and it is upregulated in AML vs normal marrow endothelial cells. Unlike other chemokines, CXCL16 is expressed not only as a membrane-bound molecule but also as a soluble chemokine.34 The CXCL16-CXCR6 (its receptor) axis is involved in TNF-α–induced apoptosis and in the activation of the NF-κB–signaling pathway.37 Nonetheless, the mechanism through which sensitivity and resistance to tosedostat would act is still unclear, certainly warranting further laboratory investigations.
Overall, our data indicate that GEP may be used to predict clinical sensitivity to this new combination. Indeed, the proposed molecular classifier was validated in an independent cohort of patients treated with an analogue regimen in an independent clinical trial. Remarkably, 100% of CRs (maintained at 3 years) were correctly predicted, as well as 100% of no CRs, thus representing the first validated molecular predictor for clinical response to a specific drug combination for treating AML. This finding shows that GEP should still be considered an important tool for predicting response to treatment and opens new scenarios for identifying biomarkers. These data demonstrate that, despite heterogeneous genetics, common GEP patterns may be found in cases characterized by different sensitivity to a given drug. Certainly, similar investigations are warranted in future clinical trials.
The AML landscape has dramatically changed since we started this trial in 2012. New, targeted drugs have been developed and have proven effective. Nonetheless, this abundance translates to a novel approach in patients with AML, for selecting the right drug(s) for the right patients. Our data showed that the combination of tosedostat and LDAC was well tolerated and achieved a higher CR rate than LDAC alone (48.5% vs 25%). Potential biomarkers were identified by GEP. Specifically, the achievement of CR was efficiently predicted by the gene expression patterns with an overall accuracy of ∼70% to 90% in the 2 examined series. Randomized prospective trials are needed to further evaluate the role of the tosedostat-LDAC combination in AML treatment.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank CTI Biopharma for providing tosedostat for the patients free of charge.
The study was supported in part by AIL Pesaro Onlus.
Footnotes
Original data are available by e-mail request to Alessandro Isidori (aisidori@gmail.com).
Authorship
Contribution: G.V. and A.I. conceived and designed the study; F.L., E.Z., A.C., A.S., B.G., G.M., A.M.M., and M.C. acquired the data; G.V., F.L., P.P.P., and A.I. provided critical analysis; G.V., F.L., M.D., E.Z., A.C., A.S., B.G., G.M., A.M.M., M.C., M.R., M.N., A.G., D.G., P.P.P., and A.I. analyzed and interpreted the data; G.V., A.I., F.L., and P.P.P. wrote, edited, and reviewed the manuscript; and G.V. and A.I. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alessandro Isidori, Hematology and Hematopoietic Stem Cell Transplant Center, AORMN, Via Lombroso 1, 61100 Pesaro, Italy; e-mail: aisidori@gmail.com; and Giuseppe Visani, Hematology and Hematopoietic Stem Cell Transplant Center, AORMN, Via Lombroso 1, 61100 Pesaro, Italy; e-mail: giuseppe.visani@ospedalimarchenord.it.
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136-1152. [DOI] [PubMed] [Google Scholar]
- 2.Leith CP, Kopecky KJ, Godwin J, et al. . Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood. 1997;89(9):3323-3329. [PubMed] [Google Scholar]
- 3.Ossenkoppele G, Löwenberg B. How I treat the older patient with acute myeloid leukemia. Blood. 2015;125(5):767-774. [DOI] [PubMed] [Google Scholar]
- 4.Appelbaum FR, Gundacker H, Head DR, et al. . Age and acute myeloid leukemia. Blood. 2006;107(9):3481-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Talati C, Dhulipala VC, Extermann MT, et al. . Comparisons of commonly used front-line regimens on survival outcomes in patients aged 70 years and older with acute myeloid leukemia. Haematologica. 2020;105(2):398-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoy SM. Glasdegib: First Global Approval. Drugs. 2019;79(2):207-213. [DOI] [PubMed] [Google Scholar]
- 7.Deeks ED. Venetoclax: First Global Approval. Drugs. 2016;76(9):979-987. [DOI] [PubMed] [Google Scholar]
- 8.Cortes JE, Heidel FH, Hellmann A, et al. . Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei AH, Strickland SA Jr., Hou JZ, et al. . Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol. 2019;37(15):1277-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiNardo CD, Pratz K, Pullarkat V, et al. . Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei AH, Montesinos P, Ivanov V, et al. . Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiNardo CD, Jonas BA, Pullarkat V, et al. . Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629. [DOI] [PubMed] [Google Scholar]
- 13.DiNardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135(2):85-96. [DOI] [PubMed] [Google Scholar]
- 14.Reid AH, Protheroe A, Attard G, et al. . A first-in-man phase I and pharmacokinetic study on CHR-2797 (Tosedostat), an inhibitor of M1 aminopeptidases, in patients with advanced solid tumors. Clin Cancer Res. 2009;15(15):4978-4985. [DOI] [PubMed] [Google Scholar]
- 15.Krige D, Needham LA, Bawden LJ, et al. . CHR-2797: an antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res. 2008;68(16):6669-6679. [DOI] [PubMed] [Google Scholar]
- 16.Löwenberg B, Morgan G, Ossenkoppele GJ, et al. . Phase I/II clinical study of Tosedostat, an inhibitor of aminopeptidases, in patients with acute myeloid leukemia and myelodysplasia. J Clin Oncol. 2010;28(28):4333-4338. [DOI] [PubMed] [Google Scholar]
- 17.Cortes J, Feldman E, Yee K, et al. . Two dosing regimens of tosedostat in elderly patients with relapsed or refractory acute myeloid leukaemia (OPAL): a randomised open-label phase 2 study. Lancet Oncol. 2013;14(4):354-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mawad R, Becker PS, Hendrie P, et al. . Phase II study of tosedostat with cytarabine or decitabine in newly diagnosed older patients with acute myeloid leukaemia or high-risk MDS. Br J Haematol. 2016;172(2):238-245. [DOI] [PubMed] [Google Scholar]
- 19.Dennis M, Hills R, Thomas I, et al. . A randomised evaluation of low-dose ara-c plus tosedostat versus low dose ara-c in older patients with acute myeloid leukaemia: results of the LI-1 trial [abstract]. EHA Library. 2018;214439. Abstract S117. [Google Scholar]
- 20.Visani G, Ferrara F, Di Raimondo F, et al. . Low-dose lenalidomide plus cytarabine induce complete remission that can be predicted by genetic profiling in elderly acute myeloid leukemia patients. Leukemia. 2014;28(4):967-970. [DOI] [PubMed] [Google Scholar]
- 21.Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 2009. Revised (ver 4.03). 14 June 2010. Available at: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed 14 December 2018.
- 22.Cheson BD, Bennett JM, Kopecky KJ, et al. ; International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia . Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642-4649. [DOI] [PubMed] [Google Scholar]
- 23.Slovak ML, Kopecky KJ, Wolman SR, et al. . Cytogenetic correlation with disease status and treatment outcome in advanced stage leukemia post bone marrow transplantation: a Southwest Oncology Group study (SWOG-8612). Leuk Res. 1995;19(6):381-388. [DOI] [PubMed] [Google Scholar]
- 24.Fleming Thomas, Watelet Luc. Approaches to monitoring clinical trials. J Natl Cancer Inst. 1989;81(3):188-193. [DOI] [PubMed] [Google Scholar]
- 25.Tripodo C, Burocchi A, Piccaluga PP, et al. . Persistent immune stimulation exacerbates genetically driven myeloproliferative disorders via stromal remodeling. Cancer Res. 2017;77(13):3685-3699. [DOI] [PubMed] [Google Scholar]
- 26.Verhaak RG, Wouters BJ, Erpelinck CA, et al. . Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94(1):131-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lancet JE, Uy GL, Cortes JE, et al. . CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol. 2018;36(26):2684-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiNardo CD, Tiong IS, Quaglieri A, et al. . Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Short NJ, Konopleva M, Kadia TM, et al. . Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020;10(4):506-525. [DOI] [PubMed] [Google Scholar]
- 30.Isidori A, Loscocco F, Curti A, Amadori S, Visani G. Genomic profiling and predicting treatment response in acute myeloid leukemia. Pharmacogenomics. 2019;20(7):467-470. [DOI] [PubMed] [Google Scholar]
- 31.Visani G, Loscocco F, Isidori A, Piccaluga PP. Genetic profiling in acute myeloid leukemia: a path to predicting treatment outcome. Expert Rev Hematol. 2018;11(6):455-461. [DOI] [PubMed] [Google Scholar]
- 32.Döhner H, Estey E, Grimwade D, et al. . Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein EM, DiNardo CD, Pollyea DA, Schuh AC. Response Kinetics and Clinical Benefits of Nonintensive AML Therapies in the Absence of Morphologic Response. Clin Lymphoma Myeloma Leuk. 2020;20(2):e66-e75. [DOI] [PubMed] [Google Scholar]
- 34.Isidori A, Venditti A, Maurillo L, et al. . Alternative novel therapies for the treatment of elderly acute myeloid leukemia patients. Expert Rev Hematol. 2013;6(6):767-784. [DOI] [PubMed] [Google Scholar]
- 35.Iwasaki M, Liedtke M, Gentles AJ, Cleary ML. CD93 Marks a Non-Quiescent Human Leukemia Stem Cell Population and Is Required for Development of MLL-Rearranged Acute Myeloid Leukemia. Cell Stem Cell. 2015;17(4):412-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D, Yuan H, Wang Y. The role of GRASP65 in Golgi cisternal stacking and cell cycle progression. Traffic. 2010;11(6):827-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mossanen JC, Kohlhepp M, Wehr A, et al. . CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4+ T-cell–dependent control of senescence. Gastroenterology. 2019;156(6):1877-1889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.