

Abstract
How early-life colonization and subsequent exposure to the microbiota impact long-term tissue immunity remains poorly understood. Here we show that the development of mucosal-associated invariant T (MAIT) cells relies on a specific temporal window, after which MAIT cell development is permanently impaired. This imprinting depends on early-life exposure to defined microbes that synthesize riboflavin-derived antigens. In adults, cutaneous MAIT cells are a dominant population of IL-17A-producing lymphocytes, which display a unique transcriptional signature and can subsequently respond to skin commensals in an IL-1-, IL-18-, and antigen-dependent manner. Consequently, local activation of cutaneous MAIT cells promotes wound healing. Together, our work uncovers a privileged interaction between defined members of the microbiota and MAIT cells, which sequentially controls both tissue-imprinting and subsequent responses to injury.
One-Sentence Summary
Early-life microbial exposure imprints the abundance of mucosal-associated invariant T cells and subsequent interactions with the microbiota modulate their ability to promote tissue repair.
Introduction
Every barrier site harbors a unique community of commensal microbes, known as the microbiota, that controls host physiology (1). These microbes promote the maturation and homeostasis of the immune system, in part through the release of microbial products, including metabolites such as short-chain fatty acids, aryl hydrocarbon receptor (AhR) ligands, and polyamines (2). In turn, the immune system modulates the composition of the microbiota and maintains the segregation of commensals by sustaining barrier tissue function (1).
Early-life microbial colonization has been shown to play a fundamental role in the development of the immune system and imparts long-lasting effects on host fitness. For instance, neonatal colonization of the lungs promotes tolerance to allergens in adult mice through the induction of regulatory T (Treg) cells (3), whereas colonization of neonatal skin similarly induces Treg cell-mediated tolerance to commensal microbes (4). During weaning, the microbiota induces a transient upregulation of phosphorylated STAT3 in epithelial cells and innate lymphoid cells (ILCs) (5), as well as an increase in colonic Treg cells (6), which can impact host metabolism and susceptibility to inflammatory disorders. High microbial diversity within the intestine during early life is necessary to impede IgE class switching of mucosal B cells, reducing susceptibility to orally induced anaphylaxis (7). Furthermore, early colonization with the intestinal commensal Bacteroides fragilis inhibits the proliferation of natural killer T (NKT) cells, which prevents oxazolone-induced colitis in adult mice (8, 9). However, despite the importance of this early host-microbiota dialogue, little is known about the microbially derived signals and antigens involved. This question is of particular importance for unconventional T cells, which are predominantly located in tissues colonized by the microbiota and characterized by their recognition of conserved antigenic motifs.
Exhibiting characteristics of both innate and adaptive immunity, innate-like lymphocytes such as mucosal-associated invariant T (MAIT) cells, NKT cells, and γδ T cells acquire their effector functions during development, which direct their localization to tissues and enable them to respond immediately upon primary antigen recognition (10). Consequently, within barrier sites, innate-like lymphocytes are likely poised to respond to the microbiota and play a dominant role in mediating host-commensal interactions. Indeed, both γδ T cells and CD8+ T cells restricted by the non-classical MHC-Ib molecule H2-M3 respond to skin commensals (11, 12), whereas γδ T cells can mediate both beneficial and deleterious effects of the microbiota in the intestines and lungs (13, 14). MAIT cells express semi-invariant αβ T cell receptors that recognize microbial-derived intermediates of vitamin B2 (riboflavin) synthesis presented by the MHC-Ib molecule MR1 (15). Because riboflavin synthesis is broadly conserved among bacteria and fungi (16), MAIT cells are thought to be particularly dependent on the microbiota. Indeed, germ-free (GF) mice exhibit fewer MAIT cells than animals housed in specific-pathogen-free (SPF) conditions (17, 18). Although these cells are the predominant innate-like lymphocyte subset in humans, where they comprise up to 45% of hepatic lymphocytes (19), surprisingly little is known about them. Though the function of MAIT cells remains largely unclear, in humans, these cells can be altered in defined inflammatory or infectious settings (20) and their frequencies show remarkable variability among individuals (21, 22). Although the paucity of MAIT cells in mice has rendered the exploration of their function challenging, MAIT cells have been proposed to promote inflammation and microbial defense (23–25). How commensal-derived antigens contribute to the abundance and function of MAIT cells has not been established. Furthermore, the extent to which MAIT cells promote tissue physiology remains to be determined.
Here we show that the development of MAIT cells and their long-term frequencies within tissues depend on a very specific developmental window and, in particular, early-life exposure to defined microbial communities enriched in riboflavin-synthesizing bacteria, such as Enterobacteriaceae. This defined microbial exposure must occur during the first few weeks of life, after which MAIT cell development is permanently impaired. Following their development, MAIT cells become a dominant, tissue-resident population of IL-17A-producing lymphocytes within the skin and display a transcriptional profile that is distinct from MAIT cells in other tissues. Cutaneous MAIT cells can subsequently respond to defined skin commensals or commensal-derived metabolites in an antigen-dependent manner. Consequently, the local activation of cutaneous MAIT cells promotes wound healing. This work therefore uncovers a privileged interaction between defined members of the microbiota and MAIT cells, resulting in a sequential dialogue that controls both tissue-imprinting and subsequent responses to injury.
Early-life exposure to riboflavin-synthesizing commensals is required for MAIT cell development
Although MAIT cells are abundant in human tissues (19), they are comparatively infrequent in murine organs, where they typically represent less than 1% of αβ T cells (26). Since the skin is characterized by an unusually high frequency of unconventional T cells (Fig. 1A), we assessed the frequency of MAIT cells at this barrier site using an MR1 tetramer loaded with the riboflavin derivative 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU) (27). Though non-specific MR1 tetramer staining was minimal, as demonstrated using an MR1 tetramer loaded with 6-formylpterin (6-FP) (Fig. S1A), we utilized 5-OP-RU-loaded MR1 tetramers conjugated to two different fluorochromes for increased specificity, when possible (Fig. S1B). This analysis revealed that MAIT cells were highly enriched in the skin (Fig. 1B–C), where they comprised up to 40% of αβ T cells (Fig. 1D–E). Importantly, the frequency of MAIT cells in the skin correlated highly with their frequency in other organs (Fig. S1C). Furthermore, MAIT cells were also enriched in human skin compared to the reported ~1% in peripheral blood (20), averaging ~2% of CD3+ lymphocytes (Fig. 1F). Thus, MAIT cells represent a substantial population of skin lymphocytes, both in mice and humans.
Fig. 1. Early-life exposure to riboflavin-synthesizing commensals is required for MAIT cell development.
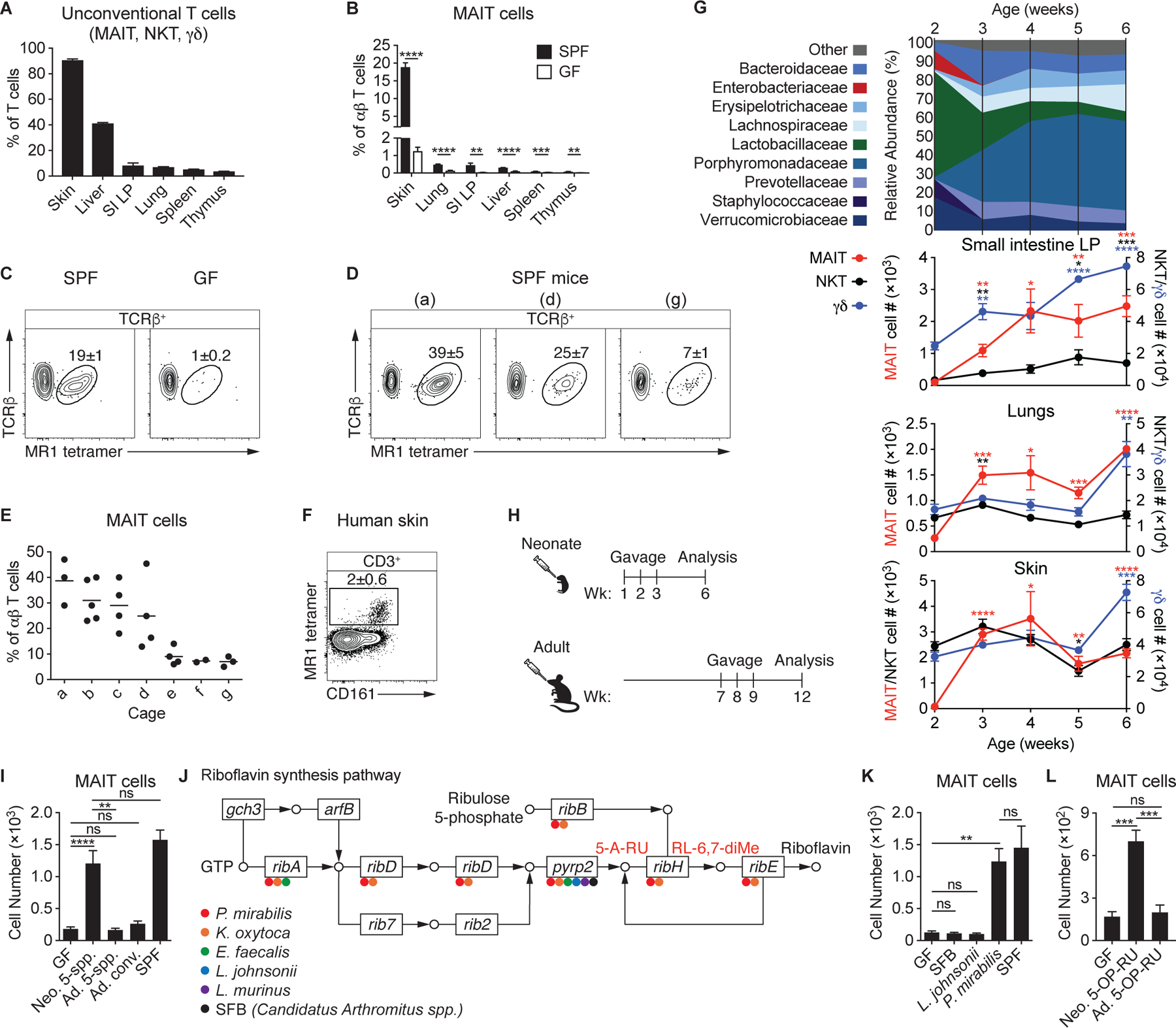
(A) Percentage of T cells (TCRβ+ or TCRγδ+) that are MAIT, NKT, or γδ T cells in wild-type (WT) mice. “SI LP” denotes small intestine lamina propria. (B) Percentage of MAIT cells among αβ T cells in specific-pathogen-free (SPF) and germ-free (GF) WT mice. (C) Flow cytometry of TCRβ+ lymphocytes from the skin of SPF and GF WT mice. (D-E) Analysis of SPF mice housed in different cages (denoted a-g). (D) Flow cytometry of TCRβ+ lymphocytes from the skin of mice housed in the indicated cages and (E) the percentage of MAIT cells among αβ T cells, with each dot representing an individual mouse. (F) Flow cytometry of CD3+ lymphocytes from a human skin biopsy. (G) 16S ribosomal RNA (rRNA) gene sequencing of feces from WT SPF mice longitudinally sampled from 2–6 weeks of age and the number of MAIT, NKT, and γδ T cells present in WT SPF mice at the corresponding ages. Asterisks denote statistically significant changes in cell number from 2 weeks of age. (H) Neonatal GF mice received oral gavages at 1, 2, and 3 weeks after birth, while adult GF mice received oral gavages at 7, 8, and 9 weeks of age. Both were analyzed 5 weeks after the initial gavage. (I) Number of MAIT cells in the skin of GF mice administered oral gavages of the 5-species (5-spp.) community (Proteus mirabilis, Klebsiella oxytoca, Enterococcus faecalis, Lactobacillus johnsonii, and Lactobacillus murinus) either as neonates (Neo.) or adults (Ad.) as depicted in (H) or conventionalized (conv.) by cohousing with SPF mice for 3 weeks as adults. (J) Presence of riboflavin synthesis genes in bacteria of the 5-species community and SFB denoted with the appropriate color. 5-amino-6-D-ribitylaminouracil (5-A-RU), which reacts with methylglyoxal to form the MAIT cell antigen 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU), and 6,7-dimethyl-8-(1-D-ribityl)lumazine (RL-6,7-diMe), are denoted in red. (K) Number of MAIT cells in the skin of GF mice monocolonized as neonates with the indicated bacterial species compared to GF and SPF controls. (L) PBS solution of 1 mM 5-A-RU and 25 mM methylglyoxal (referred to as 5-OP-RU) was topically applied once per week to the skin of GF mice beginning either at 1 week of age (Neo.) or 7 weeks of age (Ad.). Number of MAIT cells in the skin was assessed 5 weeks after the initial application. Flow cytometry gate frequencies and graphs indicate means ± SEM. Data represent at least two experiments with four or more mice per group. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 as calculated by Student’s t-test. “ns” denotes that comparison was not significant.
A comparison of mice housed in conventional specific-pathogen-free (SPF) conditions to germ-free (GF) animals confirmed previous observations that the microbiota is necessary for the presence of MAIT cells in the thymus, spleen, and gut (17, 18), and extended this requirement for MAIT cells in the skin, lungs, and liver (Fig. 1B). Although MAIT cells were highly abundant within the skin, WT SPF mice housed in distinct cages showed striking variability in the proportion of MAIT cells among genetically identical animals, ranging from <5%−40% of αβ T cells (Fig. 1D–E). In contrast, animals in the same cage had similar frequencies of MAIT cells in the skin and other organs (Fig. 1E), supporting the hypothesis that these differences were associated with distinct microbiota.
To establish how the microbiota could modulate MAIT cell development and/or accumulation, we first assessed the kinetics of unconventional T cell accumulation within barrier tissues. Although γδ T and NKT cells were present in barrier tissues of 2-week-old mice, there was a dearth of MAIT cells in the skin, lungs, and intestines (Fig. 1G). MAIT cells accumulated in all barrier tissues assessed by 3 weeks of age, supporting the idea that MAIT cell development occurs during a very specific developmental window and in response to defined microbial exposure. Therefore, we longitudinally assessed the intestinal microbiota of WT mice during early life using 16S ribosomal RNA (rRNA) gene sequencing (Fig. 1G). At 2 weeks of age, the intestinal microbiota was predominantly composed of Lactobacillaceae, with a transient enrichment of Enterobacteriaceae (Fig. 1G), consistent with previous work that has identified an abundance of these two bacterial families in early life (28). Isolation of several bacterial strains from 2-weekold mice, which developed MAIT cells as adults (Fig. S1D–E), allowed us to generate an early-life microbial community composed of two Lactobacillaceae members (Lactobacillus murinus and Lactobacillus johnsonii), two Enterobacteriaceae members (Proteus mirabilis and Klebsiella oxytoca), and the common intestinal commensal Enterococcus faecalis. To determine whether this 5-species community was sufficient to promote MAIT cell development, we colonized GF mice by administering the bacteria at weeks 1, 2, and 3 via oral gavage (Fig. 1H). The colonization of neonatal GF mice with our early-life microbial community induced MAIT cell development to the same extent observed in SPF mice harboring high MAIT cell frequencies (Fig. 1I). Thus, this limited community was sufficient to provide the necessary developmental and/or survival signals. Conversely, exposure later in life failed to promote MAIT cell development and accumulation within tissues (Fig. 1H–I), while still inducing Th17 and IL-17A+ γδ T cells (Fig. S1F). Similarly, conventionalized mice (adult GF mice cohoused with SPF animals) remained deficient in MAIT cells (Fig. 1I). Thus, exposure to the microbiota during an early-life window has a profound impact on MAIT cell development and exposure later in life cannot compensate for a lack of neonatal colonization.
Whole-genome sequencing of the 5-species community indicated that P. mirabilis and K. oxytoca have the genes ribA, ribD, and pyrp2, which encode the riboflavin synthesis enzymes necessary to generate the MAIT cell antigen 5-OP-RU, whereas the Lactobacillus species and E. faecalis do not (Fig. 1J), suggesting that, within our defined community, early colonization with Enterobacteriaceae were responsible for the emergence of the MAIT cell lineage. Indeed, the monocolonization of neonatal GF mice with P. mirabilis, but not L. johnsonii, was sufficient to drive MAIT cell development (Fig. 1K). Segmented filamentous bacteria (SFB), a known inducer of Th17 cells that lacks riboflavin synthesis enzymes (Figs. 1J & S1G) (29, 30), also failed to induce MAIT cells (Fig. 1K). As we observed with the 5-species early-life community, the ability of P. mirabilis to promote MAIT cell development was dependent on neonatal colonization or exposure from birth, both in the skin and other tissues (Fig. S1H). Interestingly, P. mirabilis colonization of both GF neonates and adults was sufficient to induce mature CD24− CD44+ MAIT cells in the thymus (Fig. S1I), indicating that microbial exposure in early life is required for the development of tissue MAIT cells, but not the thymic developmental stages. This corroborates recent work that demonstrates microbially derived 5-OP-RU can promote the thymic development of MAIT cells in GF mice (31). Of note, Enterobacteriaceae, including both Proteus and Klebsiella species, are enriched in the gut microbiota of both human and murine neonates and decrease over time (28, 32, 33). Thus, the early abundance of Enterobacteriaceae may play an essential role in the long-term imprinting of MAIT cells.
We next assessed whether exposure to microbial antigens alone during early life was sufficient to induce MAIT cell development and if such exposure could occur at other barrier sites, such as the skin. To this end, we administered a mixture of 5-amino-6-D-ribitylaminouracil (5-A-RU) and methylglyoxal, which react to form the unstable MAIT cell ligand 5-OP-RU (34), to the skin of GF mice on a weekly basis, beginning at 1 week of age. Topical application of 5-OP-RU was sufficient to induce a significant increase in the number of MAIT cells within the skin (Fig. 1L), without promoting the accumulation of other T cell subsets (Fig. S1J). This demonstrated that antigen recognition in the absence of microbial-induced proinflammatory cytokines was adequate for MAIT cell development. In agreement with our observation that adult microbial colonization was insufficient to promote MAIT cell development (Fig. 1I), topical application of 5-OP-RU to the skin of adult GF mice failed to induce MAIT cells (Fig. 1L). Thus, early-life exposure to commensals that produce defined metabolites, or to microbially derived metabolites alone, is required for MAIT cell development and their presence later in life. Although this exposure to microbial antigens can occur at multiple barrier sites, it must take place during a defined period in early life. If not, MAIT cell development in tissues is permanently impaired.
Cutaneous MAIT cells express a type-17 transcriptional program and require homeostatic IL-23
MAIT cells have previously been shown to produce either type-1- or type-17-associated cytokines (18, 26). Following stimulation, skin MAIT cells robustly produced IL-17A, but not IFNγ (Fig. 2A), suggesting that, within the skin, these cells are exclusively type-17 lymphocytes. As such, in mice with high MAIT cell frequencies, MAIT cells accounted for the largest proportion of IL-17A+ αβ T cells (Fig. 2B), indicating that these innate-like lymphocytes can represent a dominant type-17 effector subset in the skin.
Fig. 2. Cutaneous MAIT cells express a type-17 transcriptional program and require homeostatic IL-23.
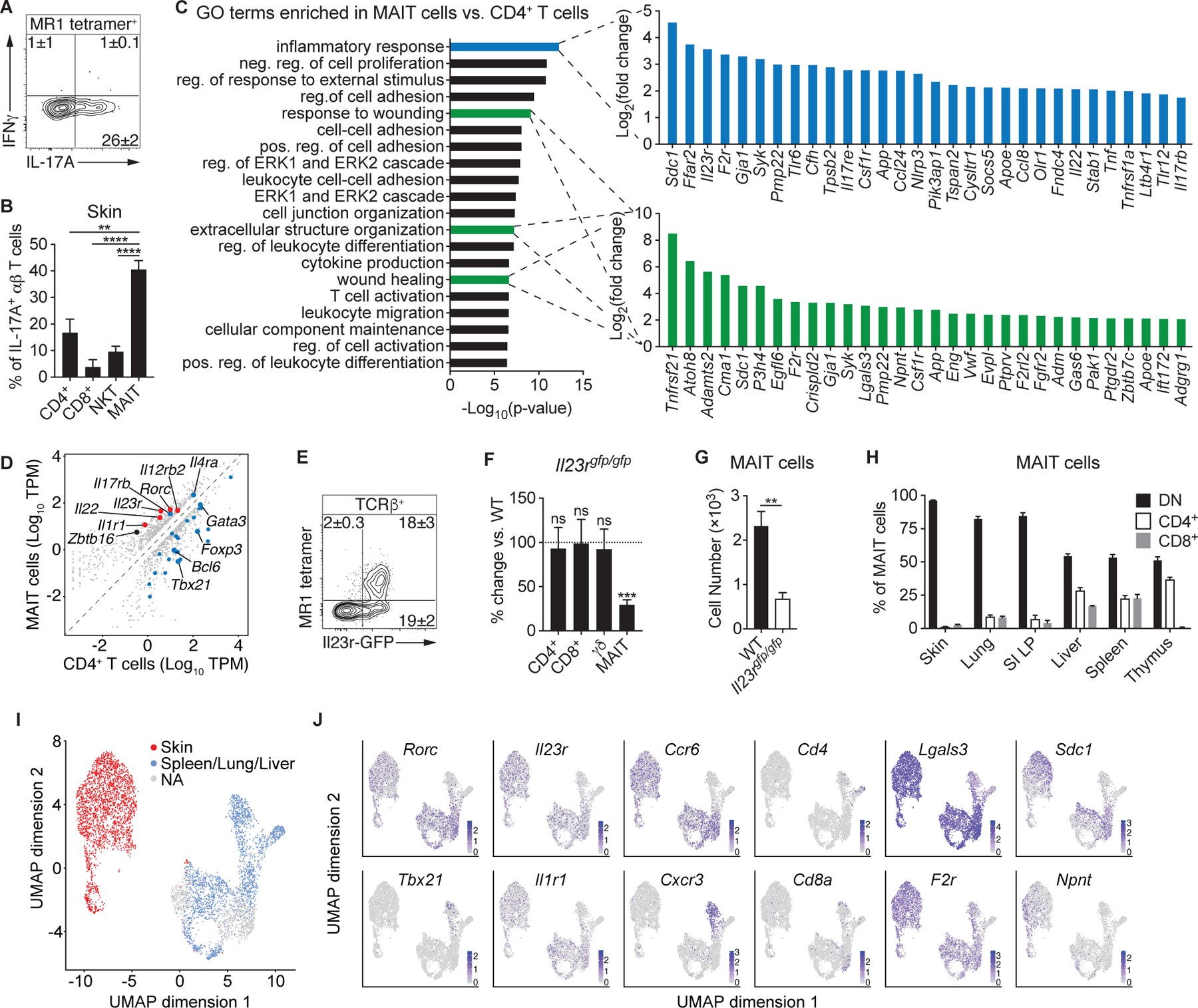
(A) Flow cytometry of cytokine production by MAIT cells from the skin of WT mice. (B) Percentage of IL-17A+ αβ T cells within WT murine skin that were CD4+, CD8+, NKT, or MAIT cells. (C-D) RNA-sequencing (RNA-seq) data of cutaneous MAIT cells and CD4+ CD25− T cells. (C) Top 20 gene ontology (GO) terms that were enriched in MAIT cells and, for the indicated GO terms, the top 30 genes upregulated in MAIT cells. Positive (pos.), negative (neg.), and regulation (reg.) are abbreviated. (D) Expression plot normalized to transcripts per million (TPM), with a minimum fold change of 2 and FDR < 5%. Type-17 genes are denoted in red and genes associated with Type-1, Type-2, T regulatory, and T helper programs are highlighted in blue. (E) Representative flow cytometry plot of TCRβ+ lymphocytes from the skin of Il23rgfp/+ mice. (F) Percentage change of the indicated T cells in the skin of Il23rgfp/gfp mice compared to WT controls. Statistics denote whether the percentage differs significantly from the WT mean (100%). (G) Number of cutaneous MAIT cells in Il23rgfp/gfp mice and WT controls. (H) Percentage of MAIT cells in the indicated organs that expressed CD4, CD8, or neither coreceptor (double negative; DN). (I-J) Single-cell RNA-seq (scRNA-seq) data of MAIT cells sorted from murine skin, spleen, lung, and liver. Clusters were assigned to “Skin” or “Spleen/Lung/Liver” based on the presence of hashtag oligonucleotides (HTOs) from those tissues. Clusters that did not have a predominance of HTOs from either skin or the other tissues were not assigned (“NA”). (I) UMAP plot displaying the distribution of MAIT cell clusters assigned to skin and the other tissues. (J) UMAP plots depicting expression of the indicated transcripts. Flow cytometry gate frequencies and graphs indicate means ± SEM. Data represent at least two experiments with four or more mice per group. **p<0.01, ***p<0.001, and ****p<0.0001 as calculated by Student’s t-test. “ns” denotes that comparison was not significant.
To establish the gene expression profile of cutaneous MAIT cells, we compared them to conventional CD4+ T cells from the skin using RNA-sequencing (RNA-seq; Fig. S2A). Gene ontology (GO) enrichment analysis revealed that cutaneous MAIT cells express a transcriptional profile consistent with T cell activation, including leukocyte differentiation, migration and cell adhesion, cytokine production, and the ERK1/2 cascade, which is downstream of TCR signaling (Fig. 2C). Intriguingly, MAIT cells were also enriched in GO terms associated with tissue repair (Fig. 2C), suggesting that these lymphocytes may contribute to the maintenance or restoration of tissue homeostasis.
As expected, cutaneous MAIT cells expressed the gene Zbtb16 (Fig. 2D), which encodes the transcription factor PLZF and is necessary for their development (18, 26). Consistent with their IL-17A potential, MAIT cells expressed type-17-associated transcripts, including Il22 and Rorc, the gene for the transcription factor RORγt, as well as Il1r1 and Il23r, which encode receptors for interleukins IL-1 and IL-23, respectively (Fig. 2D). Analysis of Il1r1−/− mice indicated that cutaneous MAIT cells do not require IL-1 signaling for their development (Fig. S2B–C). MAIT cells expressed the highest levels of IL-23R among αβ T cells (Fig. 2E) and IL-23 receptor-deficient mice (Il23rgfp/gfp) exhibited fewer MAIT cells than WT animals (Figs. 2F–G & S2D–E), supporting the idea that homeostatic IL-23 contributes to MAIT cell development and/or accumulation within tissues. Although SFB is known to promote IL-23 expression (35), SFB notably did not induce MAIT cells in GF mice (Fig. 1K), indicating that IL-23 is insufficient to promote MAIT cell development in the absence of microbial-derived metabolites. Thus, MAIT cells constitute a substantial IL-17A-producing population within the skin that requires IL-23 signaling for development and/or survival.
Though the MAIT cell TCR does not require the CD4 or CD8 coreceptors to facilitate recognition of antigens presented by MR1, CD4+, CD8+, and CD4− CD8− double-negative (DN) MAIT cells have been described in mice (26), whereas CD8+ and DN subsets exist in humans (36). We found that MAIT cells from the skin and other barrier sites, such as the lungs and gut, were almost exclusively DN (Fig. 2H). Conversely, a large proportion of MAIT cells in the thymus, spleen, and liver expressed either CD4 or CD8. Interestingly, in humans, DN MAIT cells produce more IL-17 and can arise from CD8+ MAIT cells in vitro (36), suggesting that the murine DN MAIT cells at barrier tissues may represent the most mature developmental stage.
We next compared the transcriptional profile of MAIT cells from the skin to other tissues by single-cell RNA-sequencing (scRNA-seq). Cutaneous MAIT cells displayed a markedly different transcriptional profile (Figs. 2I–J & S2F–G). In concordance with our previous data (Figs. 2D & H), skin MAIT cells expressed Rorc, Il1r1, and Il23r, and predominantly lacked expression of Cd4 and Cd8a (Fig. 2J). Type-1 MAIT cells expressing Tbx21, which encodes T-bet, and Cxcr3 were present in non-skin tissues, whereas the Rorc+ MAIT cells expressed Ccr6 (Fig. 2J). Additionally, scRNA-seq confirmed that cutaneous MAIT cells express multiple genes associated with tissue repair, including Lgals3, F2r, Sdc1, and Npnt (Fig. 2J). Intriguingly, MAIT cells expressing either coreceptor did not cluster distinctly from DN MAIT cells from the same tissue (Fig. 2J), suggesting that the transcriptional differences between the CD4+, CD8+, and DN subsets did not outweigh their tissue-specific programs.
Skin-resident MAIT cells respond to cutaneous microbes in an IL-1- and IL-18-dependent manner
The skin is readily exposed to the environment and colonized by a diverse microbial community that is second in size only to the intestinal microbiome (37). We have previously shown that topical application of the skin commensal Staphylococcus epidermidis promotes the accumulation of conventional CD4+ T cells, H2-M3-restricted CD8+ T cells, and γδ T cells within the skin without causing inflammation (11, 12, 38). Although the relative abundance of MAIT cells within tissues is imprinted by early exposure to defined microbes, MAIT cells may also subsequently respond to alterations in the abundance or composition of tissue-resident microbiota, specifically those capable of synthesizing riboflavin. Utilizing strains of S. epidermidis that either induce non-classical CD8+ T cells or not (Fig. S3A) (12), we observed that in both cases, topical association expanded the cutaneous DN MAIT cell population and increased their production of IL-17A (Figs. 3A–D & S3B–D). Furthermore, there was a strong positive correlation between the abundance of MAIT cells prior to and after association with S. epidermidis (Fig. S3E–F), indicating that SPF animals developmentally imprinted with a low MAIT cell frequency will retain a lower level of MAIT cells following subsequent interactions with the microbiota.
Fig. 3. Skin-resident MAIT cells respond to cutaneous microbes in an IL-1and IL-18-dependent manner.
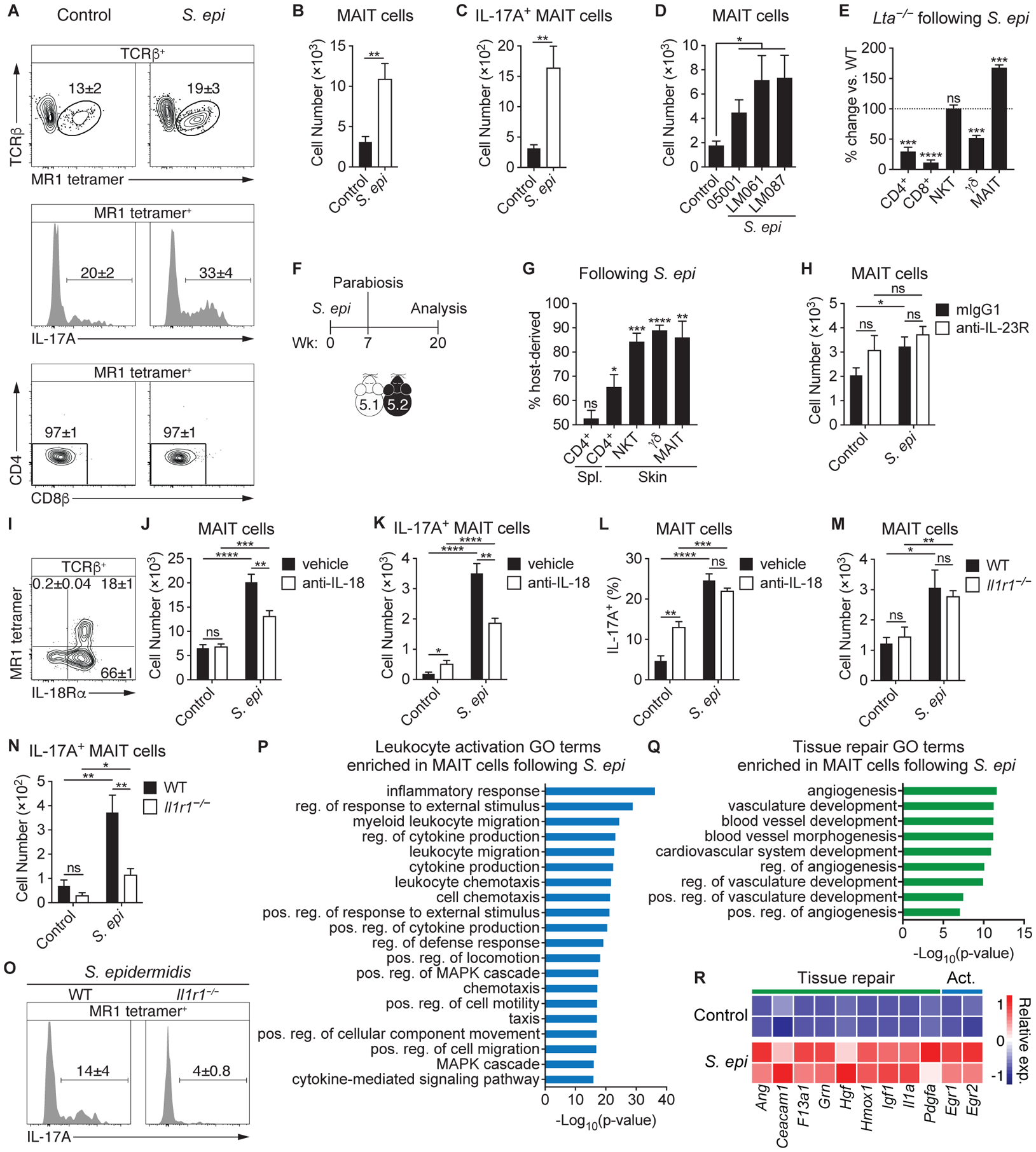
(A-C) S. epidermidis LM061 (S. epi) was topically applied to the skin of WT mice on days 0, 2, 4, and 6. Fourteen days after the initial application, animals were compared to unassociated (control) mice. (A) Flow cytometry of TCRβ+ lymphocytes (top), IL-17A production by MAIT cells (middle), and coreceptor expression of MAIT cells (bottom) within the skin. (B) Number of cutaneous MAIT cells and (C) IL-17A+ MAIT cells. (D) Number of cutaneous MAIT cells following topical association with CD8+ T cell-inducing (LM087) and non-inducing (05001 and LM061) strains of S. epidermidis. (E) Both Lta−/− and WT mice were topically associated with S. epidermidis LM087 and the percentage change of T cells in the skin of Lta−/− mice compared to WT controls is depicted. Statistics denote whether the percentage differs significantly from the WT mean (100%). (F-G) (F) CD45.1 (5.1) and CD45.2 (5.2) mice were topically associated with S. epidermidis LM087, conjoined 7 weeks later, and analyzed 13 weeks following parabiosis. (G) Frequency of T cells in the indicated tissues of parabiotic mice that are host-derived. (H) WT mice were injected subcutaneously with 1 mg of either anti-IL-23R antibody or mIgG1 isotype control 2 days before the initial application of S. epidermidis LM061 on day 0 and again on day 6. Number of cutaneous MAIT cells is depicted. (I) Flow cytometry of TCRβ+ lymphocytes from the skin of WT mice. (J-L) WT mice were injected intraperitoneally with either 1 mg of anti-IL-18 antibody or saline (vehicle) 2 days before the initial application of S. epidermidis LM061 on day 0 and again on days 1, 5, 8, and 11. (J) Number of cutaneous MAIT cells and (K) IL-17A+ MAIT cells and (L) percentage of MAIT cells that are IL-17A+ in anti-IL-18-treated and control mice (vehicle) that were associated with S. epidermidis LM061. (M-O) (M) Number of cutaneous MAIT cells and (N) IL-17A+ MAIT cells in Il1r1−/− mice and WT controls associated with S. epidermidis LM061. (O) Flow cytometry of IL-17A production by MAIT cells within the skin of S. epidermidis-associated mice. (P-R) RNA-sequencing data of cutaneous MAIT cells from mice associated with S. epidermidis LM061 and unassociated controls. GO terms enriched in MAIT cells from S. epidermidis-associated mice that are related to (P) leukocyte activation and (Q) tissue repair. Positive (pos.), negative (neg.), and regulation (reg.) are abbreviated. (R) Heatmap depicting relative expression (exp.) of genes associated with tissue repair and leukocyte activation (Act.). Flow cytometry gate frequencies and graphs indicate means ± SEM. Data represent at least two experiments with four or more mice per group. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 as calculated by Student’s t-test. “ns” denotes that comparison was not significant.
To determine whether the MAIT cell response to S. epidermidis resulted from local expansion or required priming in the lymph node, we associated Lta−/− mice (39). While the number of CD4+, CD8+, and γδ T cells following S. epidermidis association were significantly decreased in the absence of lymph nodes as expected (Fig. 3E), S. epidermidis still promoted an expansion of MAIT cells in the skin of LTα-deficient mice. This suggested that MAIT cells respond to skin commensals locally, either via antigen recognition and/or cytokine stimulation. To further interrogate whether MAIT cells remain within the skin tissue following stimulation, we associated congenically labeled mice with S. epidermidis and performed parabiosis (Fig. 3F). As previously described for NKT cells, subsets of γδ T cells, and MAIT cells in other tissues (40–43), unconventional T cells in the skin, including MAIT cells, were host-derived (Fig. 3G). Thus, MAIT cells persist in the skin as a tissue-resident population and respond locally to skin commensals.
MAIT cells have been shown to respond to cytokine stimulation in an antigen-independent manner (44). More generally, the relative contribution of antigen versus cytokines in MAIT cell activation in vivo remains poorly understood. Though homeostatic IL-23 contributed to early MAIT cell development (Figs. 2F–G & S2E), blocking the IL-23 receptor during S. epidermidis association did not reduce MAIT cell numbers (Fig. 3H). Since MAIT cells are known to upregulate IL-18Rα during development (18), we confirmed that cutaneous MAIT cells expressed IL-18Rα (Fig. 3I). Neutralization of IL-18 significantly reduced the MAIT cell response to S. epidermidis association (Fig. 3J–L), indicating that IL-18 signaling was required for optimal MAIT cell expansion. We have previously shown that topical application of S. epidermidis elicits the release of IL-1α, which promotes IL-17 responses by γδ and conventional αβ T cells (45). Although IL-1 receptor-deficient animals (Il1r1−/−) had equivalent numbers of cutaneous MAIT cells as wildtype mice at steady state and upon S. epidermidis application (Fig. 3M), their ability to produce IL-17A was significantly decreased (Fig. 3N–O). Thus, IL-18 was necessary for the expansion of MAIT cells in response to S. epidermidis and local IL-1 signaling was required for the licensing of IL-17A production.
We next assessed how S. epidermidis association impacts the transcriptional profile of tissue-resident MAIT cells. RNA-seq analysis revealed that application of S. epidermidis led to an enrichment in GO terms associated with leukocyte activation and tissue repair (Fig. 3P–Q), indicating that skin commensals may reinforce the functional capabilities of cutaneous MAIT cells. In addition to inducing the expression of genes downstream of TCR signaling, such as Egr1 and Egr2 (46), S. epidermidis association promoted the expression of genes associated specifically with angiogenesis, including angiogenin (Ang) and Hgf (47, 48), as well as genes that more broadly promote tissue repair, like Ceacam1, Grn, Hmox1, Igf1, and Pdgfa (49–53) (Fig. 3R). Thus, metabolites produced by defined skin commensals may promote the local expansion and activation of cutaneous MAIT cells and enhance their tissue repair program.
MR1-mediated presentation of riboflavin metabolites is necessary and sufficient for MAIT cell recognition of skin commensals
Although the intestine harbors multiple bacterial families that are unable to generate riboflavin, riboflavin synthesis is highly conserved among skin commensals (Fig. 4A). Nearly all Staphylococcaceae species, including our S. epidermidis isolates, express riboflavin synthesis genes (Fig. 4A–B). To assess whether recognition of riboflavin metabolites was necessary for MAIT cell responses within the skin, we generated a mutant strain of S. epidermidis with a deletion of the ribD gene, which is required for the second step of the riboflavin synthesis pathway, preceding generation of 5-OP-RU (27). While this mutation prevented riboflavin synthesis from guanosine-5′-triphosphate (GTP), the S. epidermidis ΔribD mutant retained the ribB, ribH, and ribE genes necessary to produce MAIT cell antigens from ribulose 5-phosphate (Fig. 4B). WT and ΔribD mutant strains colonized the skin equivalently (Fig. S4A). However, in contrast with the WT S. epidermidis strain, association with the S. epidermidis ΔribD mutant failed to induce a MAIT cell response (Fig. 4C), even though the CD8+ and γδ T cell responses were conserved (Fig. S4B–C). Thus, these results support the idea that production of riboflavin intermediates by S. epidermidis is required for local MAIT cell activation and that Staphylococcus species generate riboflavin exclusively from GTP. Finally, to assess whether exposure to microbial antigens was sufficient to activate MAIT cells within the skin, we topically applied 5-OP-RU to SPF mice. Application of antigen alone resulted in a significant increase of cutaneous IL-17A+ MAIT cells, with minimal effects on other T cells subsets (Figs. 4D–H & S4D–E). As with S. epidermidis, the MAIT cell response to 5-OP-RU resulted from local expansion (Fig. S4F–G), without an accompanying expansion of MAIT cells in other tissues (Figs. S4H–M).
Fig. 4. MR1-mediated presentation of riboflavin metabolites is necessary and sufficient for MAIT cell recognition of skin commensals.
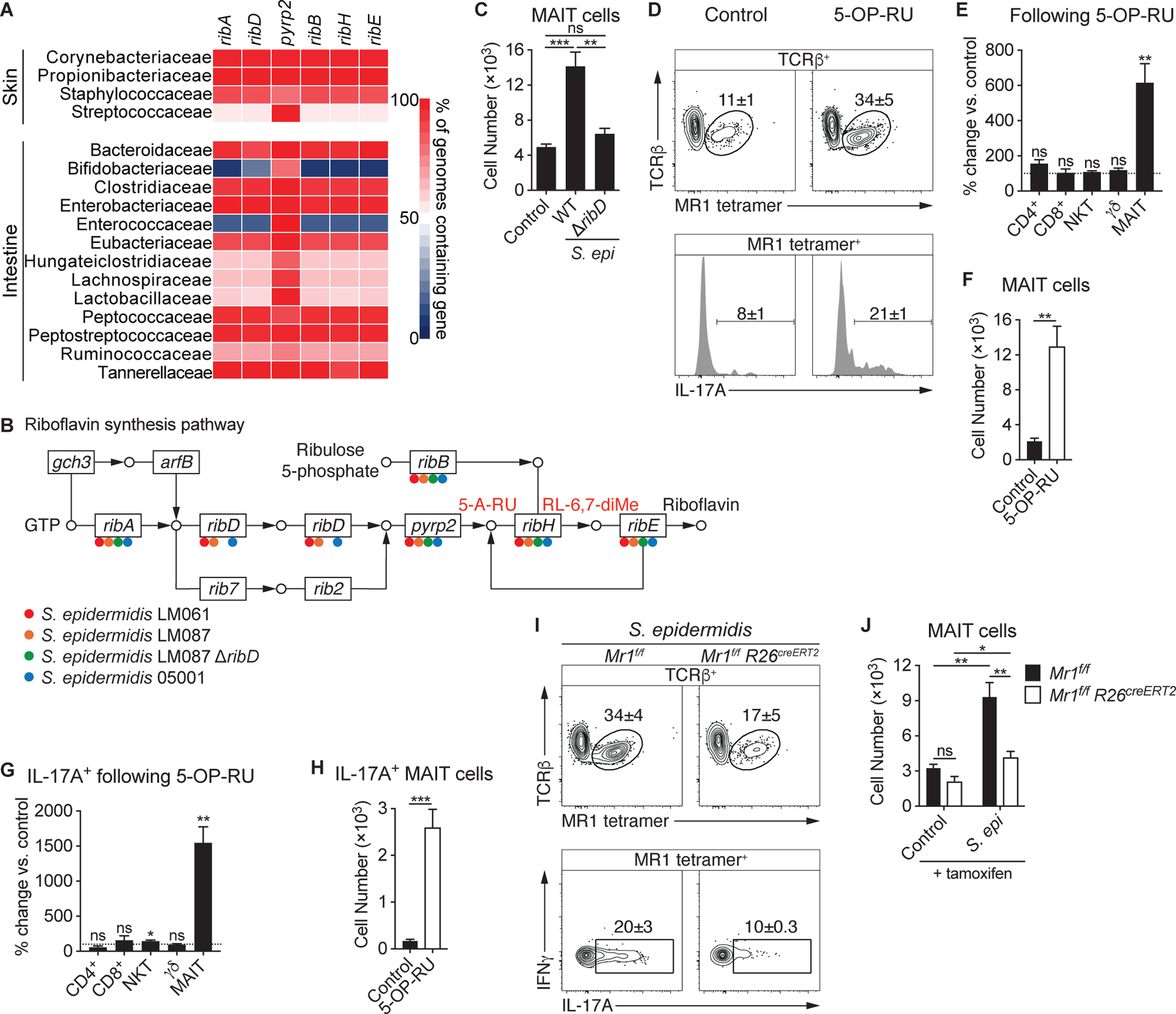
(A) Heatmap displaying the abundance of riboflavin synthesis genes among species in the indicated bacterial families. (B) Presence of riboflavin synthesis genes in strains of S. epidermidis denoted with the appropriate color. (C) Number of cutaneous MAIT cells in WT mice associated with either S. epidermidis LM087 or mutant S. epidermidis LM087 ΔribD. (D-H) 5-OP-RU was topically applied to the skin of WT mice on days 0, 2, 4, and 6. Fourteen days after the initial application, animals were compared to untreated (control) mice. (D) Flow cytometry of TCRβ+ lymphocytes (top) and IL-17A production by MAIT cells (bottom) within the skin of 5-OP-RU-treated and control mice. Percentage change of the indicated (E) T cells and (G) IL-17A+ T cells in the skin of mice treated with 5-OP-RU compared to untreated controls. Statistics denote whether the percentage differs significantly from the control mean (100%). Number of cutaneous (F) MAIT cells and (H) IL-17A+ MAIT cells in 5-OP-RU-treated and untreated control mice. (I-J) Mr1f/f R26creERT2 and Mr1f/f littermate controls were injected intraperitoneally with 3 mg of tamoxifen 8, 6, 4, and 2 days prior to the initial association with S. epidermidis LM061 and were analyzed 14 days later. (I) Flow cytometry of TCRβ+ lymphocytes and cytokine production by MAIT cells. (J) Number of cutaneous MAIT cells. Flow cytometry gate frequencies and graphs indicate means ± SEM. Data represent at least two experiments with four or more mice per group. *p<0.05, **p<0.01, and ***p<0.001 as calculated by Student’s t-test. “ns” denotes that comparison was not significant.
To further interrogate whether antigen presentation is necessary for the MAIT cell response to skin commensals, we generated Mr1f/f conditional knockout mice by inserting LoxP sites flanking exon 3 (Fig. S4N), resulting in the elimination of the α2 helix, which, together with the α1 helix, comprises the ligand-binding cleft of MR1 (15). While previous work has suggested that MAIT cells are selected on double-positive (DP) thymocytes similarly to NKT cells (54), this study utilized TCR transgenic T cells which have been shown to display an unusual PLZF− CD44− phenotype (26). Therefore, whether thymocytes are necessary for the selection of WT MAIT cells remained to be determined. Analysis of Mr1f/f Cd4cre mice revealed a profound reduction in MAIT cells compared to littermate controls while other lineages were unaltered (Fig. S4O–P), indicating that MAIT cells are indeed selected on thymocytes.
Previous studies have suggested that B cells promote MAIT cell development and may present commensal antigens (17, 55). However, Mr1f/f Cd19cre mice and Mr1f/f littermates had similar numbers of MAIT cells prior to and following association with S. epidermidis (Fig. S4Q). To determine if the MAIT cell response to S. epidermidis required antigen recognition, we utilized a tamoxifen-inducible Cre recombinase to enable ablation of MR1 following MAIT cell development (56). Treating the resulting Mr1f/f Rosa26creERT2 mice and Mr1f/f littermates with tamoxifen did not affect MAIT cell numbers 3 weeks later, suggesting that continuous antigen presentation is not necessary to maintain MAIT cells in the skin under steady state conditions (Fig. 4J). Tamoxifen treatment a week prior to the S. epidermidis association revealed that MR1-mediated antigen presentation is required for the MAIT cell response (Figs. 4I–J & S4R–S). Thus, the production of riboflavin metabolites by defined skin commensals and presentation of these antigens promotes local expansion and activation of cutaneous MAIT cells. Although cytokines have been shown to be sufficient to activate MAIT cells (44), our results support the idea that in the context of homeostatic responses to the microbiota, the MAIT cell response is strictly TCR-dependent.
MAIT cells promote tissue repair
To explore a potential role for MAIT cells in skin physiology, we first determined their localization within the skin. Since MAIT cells account for the vast majority of IL-17A-producing αβ T cells following topical application of 5-OP-RU (Fig. S5A), we imaged the skin of Il17acre R26-STOP-YFP fate-mapping mice by confocal microscopy and found that MAIT cells predominantly resided near the interface of the dermis and epidermis, in close proximity to the basal layer (Fig. 5A). Dissociation of the dermis and epidermis confirmed that, while few MAIT cells are present in the epidermal layer, the majority are localized in the dermis (Fig. 5B). This localization contrasted with that of conventional CD4+ or CD8+ T cells, which localized close to hair follicles and within the epidermis, respectively (38). This defined localization of MAIT cells, coupled with the tissue remodeling signature of these cells (Figs. 2C, 2J, & 3Q–R), supported the idea that MAIT cells may regulate tissue responses to injury.
Fig. 5. MAIT cells promote tissue repair.
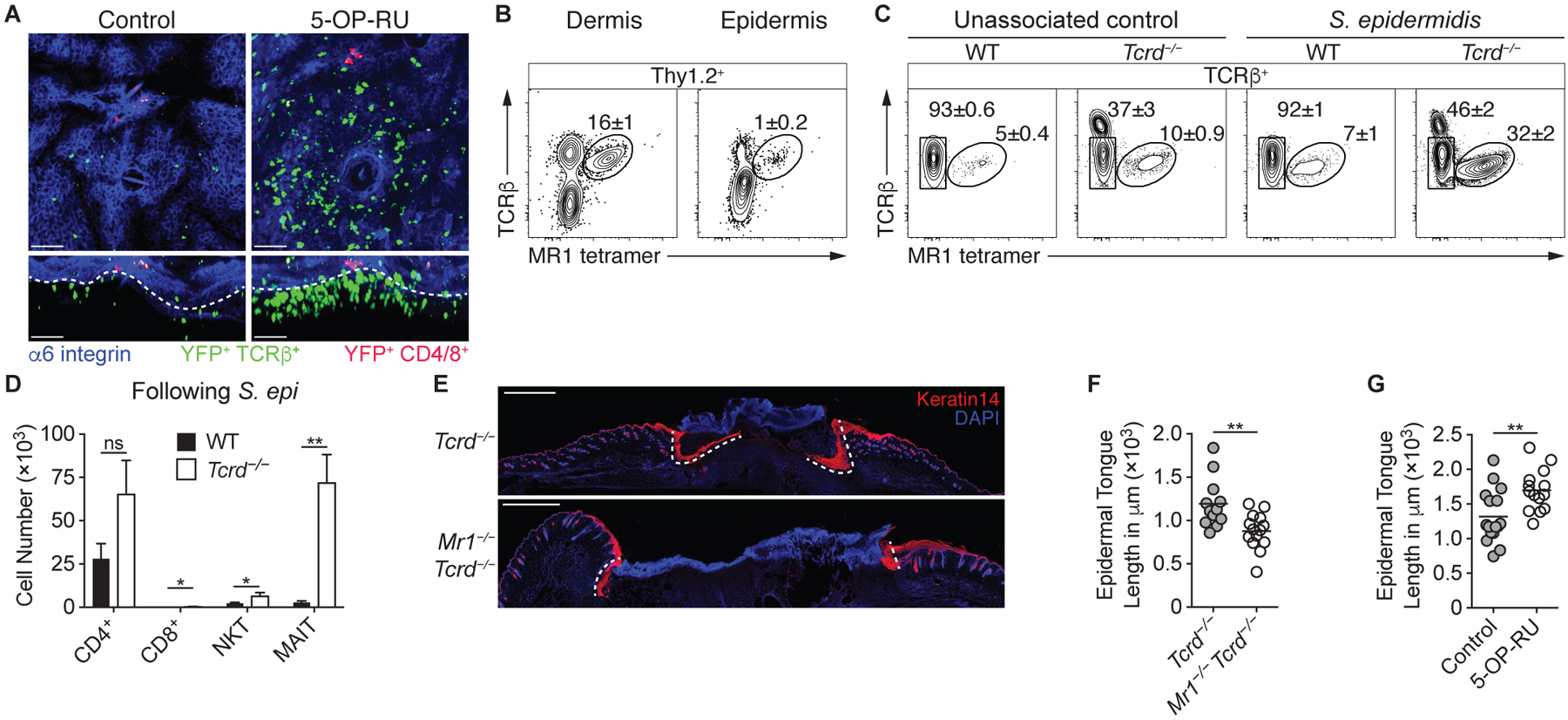
(A) Representative confocal microscopy images of the skin of Il17acre R26-STOP-YFP mice that received topical 5-OP-RU and untreated controls. Projection along the z-axis (top) or x-axis (bottom). Colocalization of YFP and TCRβ is depicted in green, whereas the colocalization of YFP and either CD4 or CD8 is in red. White dashed lines denote the dermal/epidermal interface. Scale bars represent 50 μm. (B) Flow cytometry of Thy1.2+ lymphocytes within the dermis and epidermis of WT mice. (C) Flow cytometry of TCRβ+ lymphocytes from the skin of Tcrd−/− and WT mice either associated with S. epidermidis LM061 or left untreated. Note that the TCRβhi cells in the Tcrd−/− samples are dendritic epidermal T cells (DETCs) that express αβ TCRs. (D) Number of cutaneous T cells in Tcrd−/− and WT mice following association with S. epidermidis LM061. (E-F) S. epidermidis LM061 was topically applied to the backs of MAIT cell-deficient Mr1−/− Tcrd−/− mice and Tcrd−/− littermates on days 0, 2, 4, and 6. Twelve days after the initial application, punch biopsies were taken through the back skin, and animals were assessed 5 days later. (E) Representative immunofluorescence images of back skin wounds 5 days after punch biopsies. Tissue sections were immunolabeled with Keratin 14 (red), which stains the advancing epidermal tongues (demarcated with white dashed lines) during re-epithelialization of the wounds. Scale bars represent 1 mm. (F) Quantification of the epidermal tongue length 5 days after wounding, with each dot representing the measured length of an individual epidermal tongue. (G) 5-OP-RU was topically applied to the backs of WT mice on days 0, 2, 4, and 6. Twelve days after the initial application, punch biopsies were taken through the back skin, and animals were assessed 5 days later, when epidermal tongue lengths were quantified. Flow cytometry gate frequencies and graphs indicate means ± SEM. Data represent at least two experiments with four or more mice per group. *p<0.05 and **p<0.01 as calculated by Student’s t-test. “ns” denotes that comparison was not significant.
Though alterations in MAIT cell frequencies have been associated with various pathologies, including skin conditions (57, 58), the functions of these cells remain largely unclear. The previous difficulty assessing MAIT cell function in mice may have been due to the low frequency of MAIT cells in murine tissues other than skin, as well as the potentially overlapping functions and compensatory responses of unconventional T cell subsets. Indeed, NKT cell-deficient Cd1d−/− mice harbor greater numbers of thymic and splenic MAIT cells (18), suggesting that this subset of innate-like lymphocytes competes with the MAIT cell population. However, such a compensatory response was not observed in the skin of Cd1d−/− mice (Fig. S5B–C), likely due to the relatively small number of NKT cells in the skin compared to γδ T cells. Conversely, in the absence of γδ T cells, MAIT and NKT cell numbers were significantly increased at steady-state and following S. epidermidis association (Figs. 5C–D & S5D–F). Therefore, γδ T cells restrict the MAIT cell population at steady-state and constrain their response to commensals. This observation provided us with an approach to test the function of MAIT cells when competition with other innate-like lymphocytes is limited, a setting that is relevant to the lymphocyte composition of human skin, where γδ T cells are far less abundant than in mice (59).
Previous studies from our laboratory and others have indicated that innate-like lymphocytes can promote tissue repair. For instance, CD8+ T cells that recognize commensal-derived N-formylated peptides presented by the MHC-Ib molecule H2-M3 accelerate wound closure in mice (12), whereas murine dendritic epidermal T cells (DETCs) promote epidermal thickening and keratinocyte proliferation (60). We utilized an experimental model that involved taking a punch biopsy through the murine back skin 12 days after the initial association with S. epidermidis and assessing the re-epithelialization of the wound 5 days later by measuring the epidermal tongue of proliferating keratinocytes (Fig. S5G) (12, 61). To minimize confounding effects of H2-M3-restricted CD8+ T cells, we topically associated Tcrd−/− mice and MAIT cell-deficient Mr1−/− Tcrd−/− littermates with a strain of S. epidermidis that does not induce CD8+ T cells (12). Post wounding, the epidermal tongue lengths of MAIT cell-sufficient Tcrd−/− mice were significantly longer that those observed in MAIT cell-deficient Mr1−/− Tcrd−/− animals (Fig. 5E–F), indicating that MAIT cells promote tissue repair in the absence of compensatory responses by γδ T cells.
To assess whether an increase in MAIT cells was sufficient to impact skin physiology and response to injury, we directly applied the 5-OP-RU antigen to the skin of WT mice prior to wounding to selectively increase MAIT cells (Fig. 4E). The topical application of 5-OP-RU significantly increased the epidermal tongue length (Fig. 5G), demonstrating that the microbially derived antigen 5-OP-RU was sufficient to promote wound healing in an intact host.
Discussion
Post development, the skin harbors tissue-resident IL-17A+ MAIT cells and exposure to defined skin microbes or microbially derived antigens can further MAIT cell accumulation and strengthen the tissue repair functions of these cells in an antigen-dependent manner. The majority of skin commensals were capable of synthesizing riboflavin-derived metabolites (Fig. 4A), suggesting that these antigens contribute to skin health and homeostasis and may represent novel therapeutic approachs for promoting tissue repair.
Here, we also uncover how early microbial encounters have long-term effects on the composition of the immune system. Notably, we show that MAIT cells are induced during a specific early-life window in response to riboflavin-synthesizing commensals, a phenomenon that permanently imprints the abundance of this subset in tissues. Previous work uncovered a role for early-life exposure to epithelial cell-derived butyrophilin-like proteins for Vγ7+ γδ IEL development, gestational colonization for the induction of ILCs, and early colonization with the intestinal commensal B. fragilis to constrain the abundance of NKT cells in adults (8, 62, 63). Together, these findings imply that temporal windows of development and permanent imprinting of tissues may represent a general phenomenon for non-classically restricted T cells. Our work also provides a possible explanation for the remarkable variation in MAIT cell abundance observed in humans (21, 22). In contrast to mice, human T cells develop in utero supporting the hypothesis that such early MAIT cell imprinting may occur prior to birth. Indeed, functionally mature MAIT cells have been found in second trimester human fetuses (64). How, in humans, microbiota-derived riboflavin intermediates prior to and post-birth contribute to the final maturation and long-term tissue imprinting of MAIT cells remains to be addressed. In conclusion, our work proposes that a central strategy utilized by the immune system to interact with the microbiota may be via the recognition of canonical microbially derived antigens by unconventional T cells, including MAIT cells, a dialogue that may play a dominant role in the control of host physiology.
Materials and Methods
Mice
Il17acre (65), Il23rgfp/gfp (66), and Mr1−/− (17) mice have been previously described and were generously provided by B. Stockinger (Francis Crick Institute), M. Oukka, (Seattle Children’s Research Institute), and S. Gilfillan (Washington University School of Medicine), respectively. Wild-type (WT) C57BL/6NTac and BALB/c specific-pathogen-free (SPF) mice were purchased from Taconic Biosciences. CD45.1 (B6.SJL-Ptprca Pepcb/BoyJ) and Il1r1−/− (C57BL/6-[KO]IL1r1) were obtained through the NIAID-Taconic exchange program. Germ-free (GF) C57BL/6 mice were bred and maintained in the NIAID Microbiome Program gnotobiotic animal facility. Cd1d−/− (B6.129S6-Del(3Cd1d2-Cd1d1)1Sbp/J), Cd4cre (Tg(Cd4-cre)1Cwi/BfluJ), Cd19cre (B6.129P2(C)-Cd19tm1(cre)Cgn/J), Lta−/− (B6.129S2-Ltatm1Dch/J), R26-STOP-YFP (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J), R26creERT2 (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J), and Tcrd−/− (B6.129P2-Tcrdtm1Mom/J) mice were purchased from The Jackson Laboratory. All mice were bred and maintained at an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility at the NIAID and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals. All experiments were performed at NIAID under an animal study proposal (LISB-19E or LISB-20E) approved by the NIAID Animal Care and Use Committee. Unless otherwise noted, age- and sex-matched mice between 6 and 12 weeks of age were used for each experiment.
Generation of Mr1f/f mice
CRISPR guide RNAs (gRNAs) were synthesized following a published protocol (67). The following oligos were used to target the second and third introns of Mr1: 5′ -TAGGTTAAAGCCATCCTCCCAT-3′ & 5′ -AAACATGGGAGGATGGCTTTAA-3′ (second) and 5′ -TAGGCTAGGCATGTTAAGAATG-3′ & 5′ -AAACCATTCTTAACATGCCTAG-3′ (third). Briefly, oligos were annealed and ligated into the pT7-gRNA vector (Addgene). These constructs were linearized and used as templates for in vitro transcription (IVT) using the MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific), which yielded the gRNAs. Cas9 mRNA was generated by using linearized pT3TS-nCas9n (Addgene) as the template for IVT with the mMESSAGE mMACHINE T3 Transcription Kit (Thermo Fisher Scientific). Both the Cas9 mRNA and the gRNAs were purified using the MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific) and eluted in RNase-free water. C57BL/6NCr embryos were microinjected with Cas9 mRNA (100 ng/μL), both gRNAs (100 ng/μL), and the following two PAGE-purified ssDNA oligos (300 ng/μL; Integrated DNA Technologies), which contain the LoxP sequences flanked by 75 bases of homology: 5′ -CAAAAGTGCTTTCAGCCATGCTCCAGTTGTACCACTTGAGAAGTCTCTCTCTGTCTCT GTTTAAAGCCATCCTCCATAACTTCGTATAATGTATGCTATACGAAGTTATCATAGG CTGTATATGTTTCAGGGCTTCACACCTACCAGAGAATGATTGGCTGTGAGTTGCTAG AAGATGGCAGCA-3′ & 5′ -TGGCTAAAGAGGTTCTTGGAATATGGAAGAGATACCCTAGAAAGAACAGGTAATGG GAAGAGAGAACACCTCATATAACTTCGTATAATGTATGCTATACGAAGTTATTCTTA ACATGCCTAGGAAAACTGGCTTTGTGTCTGACTTCCTCTTCTGCAGGTGATATTCGCC CTGTGCTGAAAT-3′ and surgically transferred to recipient females. Progeny were screened by PCR amplification with primers 5′ -TTTCTGGCTCCTGCGTATCT-3′ & 5′ -ATTTCAGCACAGGGCGAATA-3′, followed by Sanger sequencing with the primer 5′ -CTAATTCCCCAAGGCAAGC-3′. The founder was bred to C57BL/6NTac mice to propagate the strain.
Inducible deletion of MR1
Mr1f/f R26creERT2 and Mr1f/f littermate controls were injected intraperitoneally with 3 mg of tamoxifen (Sigma-Aldrich) in a corn oil-ethanol (95:5) mixture 8, 6, 4, and 2 d prior to association with S. epidermidis NIHLM061.
Commensal culture and colonization
For the isolation of intestinal commensals, a stool pellet was homogenized in 200 μL of BHI and 10 μL of the homogenate was streaked on MacConkey (BD Biosciences) and de Man, Rogosa, and Sharpe (MRS; Sigma-Aldrich) agar plates and incubated at 37°C under aerobic and microaerophilic conditions, respectively. After 48 h of incubation, colonies bearing unique morphologies were sub-cultured to obtain pure cultures. Additionally, reiterative 1:10 dilutions in Brain Heart Infusion (BHI) broth allowed us to isolated single bacteria in 96-well plates under aerobic conditions. Taxonomic identification of pure isolates was determined by 16S rRNA Sanger sequencing. To prepare bacterial cultures for oral gavage, 10 μL of each frozen strain was propagated on Columbia agar with 5% sheep blood (Thermo Scientific) and incubated under microaerophilic conditions overnight at 36°C. 2–3 colonies of each strain were inoculated in Peptone Yeast Glucose (PYG) broth and incubated at 37°C under microaerophilic conditions for 12 h, except for Lactobacillus strains, which were incubated for 24 h. All cultures were normalized to an optical density (OD) of 1 at 600 nm, mixed in equal volumes when required (5-species community), centrifuged, and resuspended in 10% the volume of PYG broth. Neonates received 20 μL of the bacterial culture by oral gavage, whereas adult animals received 200 μL. For the topical association with S. epidermidis NIH05001, NIHLM061, and NIHLM087 (referred to as 05001, LM061, and LM087 in the text) (68), strains were cultured for 18 h in Tryptic Soy Broth (TSB) at 37°C, to a density of approximately 109 CFU/mL. Mice were colonized by topically applying up to 5 mL of culture across the entire skin surface using a sterile swab. Association was repeated every other day, for a total of 4 times. To quantitate S. epidermidis colonization, the ear skin was swabbed with a sterile cotton swab soaked in TSB. Swabs were streaked on blood agar and plates were then incubated at 37°C under aerobic conditions for 18 h before counting CFUs.
Generation of S. epidermidis mutant
The S. epidermidis NIHLM087 ΔribD mutant was constructed using a recently described method (Chen et al., submitted). Approximately 1 kb up- and downstream of the gene ribD were amplified with the following primers: 5′ -CGGTATCGATAAGCTTGATATCTGGAATAGTTTGAGCGTTAA-3′ & 5′ -GATACCTGTAAACATAGACATTCTACTCAATTGATCACCTC-3′ and 5′ -GAGGTGATCAATTGAGTAGAATGTCTATGTTTACAGGTATC-3′ & 5′ -TATAGGGCGAATTGGAGCTCTTACGTGTTAACACACCATT-3′. These two flanking regions were assembled into the plasmid pIMAY (Addgene) (69) using Gibson Assembly (New England Biolabs) (70). The resulting plasmid was transformed into S. epidermidis NIHLM087. Temperature shift to 37°C and anhydrotetracycline were then used to select for allelic recombination events that resulted in a ribD deletion without a genomic scar. Successful knockouts were confirmed by colony PCR and sequencing.
Parabiosis experiments and surgery
Age-matched CD45.1 and CD45.2 mice were topically associated with S. epidermidis NIHLM087 and 7 weeks after the initial colonization, parabiosis surgery was performed as described (71). Animals were treated with oral antibiotics for 2 weeks and remained conjoined for 13 weeks prior to analysis.
In vivo cytokine blockade
Mice were injected subcutaneously with 1 mg of either anti-IL-23R antibody (21A4; Merck) or mIgG1 isotype control (27F11; Merck) 2 d before the initial application of S. epidermidis NIHLM061 and again on day 6. Anti-IL-18 monoclonal antibody (SK113AE-4) (72) was purified from hybridoma supernatant using Protein G Sepharose beads (Abcam). Mice were injected intraperitoneally with either 1 mg of anti-IL-18 or saline (vehicle) 2 d before the initial application of S. epidermidis NIHLM061 and again on days 1, 5, 8, and 11.
Human skin specimens
Normal adult human skin was obtained from patients at UCSF undergoing elective surgery, in which healthy skin was discarded as a routine procedure. The study was conducted in accordance with the Declaration of Helsinki principles.
Human skin processing
Skin samples were stored at 4°C in a sterile container with PBS and gauze until the time of digestion. Subcutaneous fat was removed, skin was minced finely with dissection scissors, and mixed in a 6-well plate with 3 mL of digestion buffer consisting of 0.8 mg/mL collagenase type 4 (Worthington), 0.02 mg/mL DNase I (Sigma-Aldrich), 10% FBS, 1% HEPES, and 1% penicillin/streptavidin in RPMI medium. Samples were incubated overnight in 5% CO2 and filtered passed twice through a 100-μm filter with wash buffer (2% FBS, 1% penicillin/streptavidin in RPMI medium).
Murine tissue processing
To isolate skin cells, ear pinnae were excised and separated into ventral and dorsal sheets, which were digested by placing them dermal side down in RPMI 1640 media supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 50 mM β-mercaptoethanol, 20 mM HEPES, 100 U/mL penicillin, 100 mg/mL streptomycin, 0.5 mg/mL DNase I (Sigma-Aldrich), and 0.25 mg/ml of Liberase TL purified enzyme blend (Roche), and incubated for an hour and 45 min at 37°C. Digested skin sheets were homogenized using the Medimachine tissue homogenizer system (BD Biosciences) and passed through 50-μm filters. To separate the epidermis from the dermis, ear pinnae were first digested for 45 min with 500 CU Dispase (Corning Life Sciences) and curved forceps were used to remove the epidermis, before proceeding with the Liberase TL digestion as previously described. Lungs were diced and incubated in 2 mL of pre-warmed RPMI 1640 containing 1 mg/mL DNase I (Sigma-Aldrich) and 0.25 mg/ml of Liberase TL (Roche) for 45 min in a 37°C water bath. The digested tissue was passed through a 70-μm filter, centrifuged, resuspended in 5 mL of 40% Percoll (Sigma-Aldrich), and centrifuged again. Thymus, spleen, and liver were dissociated through 70-μm filters, after which 40% Percoll (Sigma-Aldrich) was used to remove hepatocytes from liver samples. Small intestine lamina propria were processed as previously described (73).
In vitro restimulation
Cells were cultured for 2.5 h at 37°C in RPMI complete media (RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 50 mM β-mercaptoethanol, 20 mM HEPES, 100 U/mL penicillin, and 100 mg/mL streptomycin) containing 50 ng/mL of phorbol myristate acetate (PMA; Sigma-Aldrich), 5 mg/mL ionomycin (Sigma-Aldrich), and a 1:1,000 dilution of GolgiPlug (BD Biosciences).
Flow cytometric analysis
Fluorophore-conjugated antibodies that were used are listed in Table S1. 5-OP-RU and 6-FP-loaded mMR1 and hMR1 tetramers and PBS57-loaded mCD1d tetramers were provided by the NIH Tetramer Core Facility and cells were stained in RPMI complete media for 1 hour at room temperature. For intracellular cytokine staining, cells were fixed and permeabilized using the Fixation/Permeabilization Solution Kit (BD Biosciences) and stained with fluorophore-conjugated antibodies for at least 60 min at 4°C. For transcription factor staining, cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stained with fluorophore-conjugated antibodies for at least 60 min at 4°C. All staining was performed in the presence of purified anti-mouse CD16/32 and purified rat gamma globulin (Jackson ImmunoResearch). Dead cells were excluded from live samples using 4′, 6-diamidino-2-phenylindol (DAPI; Sigma-Aldrich), whereas either LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen Life Technologies) or Ghost Dye Violet 510 (Tonbo Biosciences) was used in fixed samples. All antibodies were purchased from BD Biosciences, BioLegend, or eBioscience. Cells were acquired on an LSRII flow cytometer (BD Biosciences) running FACSDiva software (BD Biosciences) and data were analyzed using FlowJo (TreeStar). Unless otherwise indicated, murine MAIT cells were gated as MR1 tetramer+ TCRβ+ TCRγδ− CD3+ CD90.2+ CD45+ (CD44+ as well in tissues other than skin); NKT cells as CD1d tetramer+ TCRβ+ TCRγδ− CD3+ CD90.2+ CD45+; CD4+ T cells as CD4+ CD8β− MR1 tetramer− CD1d tetramer− TCRβ+ TCRγδ− CD3+ CD90.2+ CD45+; CD8+ T cells as CD8β+ CD4− MR1 tetramer− CD1d tetramer− TCRβ+ TCRγδ− CD3+ CD90.2+ CD45+; and γδ T cells as TCRγδ+ TCRβ− CD3+ CD90.2+ CD45+.
Tetramer enrichment
For the enrichment of MAIT cells, samples were stained with phycoerythrin (PE)-conjugated MR1 tetramer, bound to anti-PE MicroBeads (Miltenyi Biotec), and enriched on an autoMACS Pro Separator (Miltenyi Biotec) using the POSSEL_S program.
Confocal microscopy
Ear pinnae were split with forceps, fixed in 1% paraformaldehyde (Electron Microscopy Sciences) overnight at 4°C, and blocked in PBS containing 1% BSA and 0.25% Triton X-100 for 2 h at room temperature. Tissues were first stained with anti-CD4 (RM4–5, eBioscience), anti-CD8α (53–6.7, eBioscience), anti-CD49f (GoH3, eBioscience), anti-GFP (A21311, Life Technologies), and anti-TCRβ (H57–597, eBioscience) antibodies overnight at 4°C, washed three times with PBS, and then stained with DAPI (Sigma-Aldrich) overnight before being mounted with ProLong Gold (Invitrogen). Ear pinnae images were captured on a Leica TCS SP8 confocal microscope equipped with HyD and PMT detectors and a 40X oil objective (HC PL APO 40X/1.3 oil). Images were analyzed using Imaris (Bitplane).
Wounding and epifluorescence microscopy of wound tissue
Wounding and quantitation of wound healing were performed as previously described (61). Male mice in the telogen phase of the hair cycle were anesthetized with ketamine/ xylazine and their back skin was then shaved using a ChroMini Pro (Moser). A 6-mm biopsy punch was first used to partially perforate the skin and then iris scissors were used to cut the epidermis and dermis along the punch outline, resulting in a circular, full-thickness wound. 5 d after wounding, the skin tissue was excised and fixed in 4% paraformaldehyde for 4 h at 4°C, incubated overnight in 30% sucrose at 4°C, embedded in OCT compound (Tissue-Tek), frozen on dry ice, and cryo-sectioned (20-μm-thick sections). Sections were fixed in 4% paraformaldehyde for 10 min at room temperature, rinsed with PBS, permeabilized with 0.1% Triton X-100 (Sigma) for 10 min, and blocked for 1 hour at room temperature in blocking buffer (2.5% normal goat serum, 1% BSA, 0.3% Triton X-100 in PBS). Chicken anti-mouse Keratin 14 antibody (Poly9060, BioLegend) was diluted at 1:400 in blocking buffer containing rat gamma globulin and anti-CD16/32 and incubated overnight at 4°C. After washing with PBS, sections were stained with a polyclonal anti-chicken IgY-AlexaFluor647 secondary antibody (Jackson ImmunoResearch) at a 1:800 dilution for 1 hour at room temperature, washed with PBS, stained with DAPI, washed with PBS, and mounted with ProLong Gold (Invitrogen). Wound images were captured with a Leica DMI 6000 widefield epifluorescence microscope equipped with a Leica DFC360X monochrome camera. Tiled and stitched images of wounds were collected using a 20X/0.4 NA dry objective. Images were analyzed using Imaris (Bitplane).
16S rRNA sequencing and analysis
Fecal DNA was purified using the MagAttract PowerMicrobiome DNA/RNA kit (Qiagen). Amplification of the V4 hypervariable region of the bacterial 16S rRNA gene was performed using the 515f and 806r primers (515F: 5′ -GTGCCAGCMGCCGCGGTAA-3′; 806R: 5′ -GGACTACHVGGGTWTCTAAT-3′), followed by an additional PCR to append unique barcodes to each sample. Amplicons were quantified using Kapa Library Quantification Complete Kit (ROX Low) (Kapa Biosystems) and pooled at equimolar concentrations before being sequenced on a MiSeq (Illumina) using the V2 MiSeq Reagent kit (Illumina). The DADA2 algorithm (74) was used to denoise raw 16S reads after primer trimming and to tabulate sequence variants. Read counts were converted to relative abundance and taxonomy was assigned to each sequence variant using the RDP Training Set 16. Sequence variants were collapsed by family-level classifications, and families present in greater than 5% abundance in at least one time point were graphically represented, whereas remaining families were collapsed into the category “Other”.
Bacterial whole-genome sequencing
For the E. faecalis, K. oxytoca, L. johnsonii, L. murinus, and P. mirabilis isolates, DNA libraries were prepared using the paired-ended Nextera DNA Flex Library Prep Kit (Illumina). Sequencing was performed on a NextSeq 550 (Illumina) using the High Output v2 Kit (Illumina).
Assessing the presence of riboflavin synthesis genes
Protein sequences of representative riboflavin biosynthesis genes were downloaded from Genbank: ribA (NP_764994), ribB (CDO12415), ribD (NP_764996), ribE (NP_764995), ribH (NP_764993), and pyrp2 (NP_764225). Isolates were either sequenced as described above or genomes were downloaded from NCBI: Candidatus Arthromitus (BioProject PRJDA67835), S. epidermidis NIH05001 (BioProject PRJNA62391), S. epidermidis NIHLM061 (BioProject PRJNA62355), and S. epidermidis NIHLM087 (BioProject PRJNA62349). For taxa that are generally present in the intestinal and skin microbiota, we also downloaded genomes of unique strains from NCBI: Corynebacteriaceae (txid 1653), Propionibacteriaceae (txid 31957), Staphylococcaceae (txid 90964), Streptococcaceae (txid 1300), Bacteroidaceae (txid 815), Bifidobacteriaceae (txid 31953), Clostridiaceae (txid 31979), Enterobacteriaceae (txid 543), Enterococcaceae (txid 81852), Eubacteriaceae (txid 186806), Hungateiclostridiaceae (txid 2304686), Lachnospiraceae (txid 186803), Lactobacillaceae (txid 33958), Peptococcaceae (txid 186807), Peptostreptococcaceae (txid 186804), Ruminococcaceae (txid 541000), and Tannerellaceae (txid 2005525). These nucleotide scaffolds were used as subjects in a tBLASTn search using representative riboflavin biosynthesis genes, using an e-value <1×10−10. Alignments that met this threshold were considered as homologs, and thus those genomes were considered to contain that gene.
RNA-sequencing and analysis
The following populations were isolated via FACS from the ear pinnae of C57BL/6NTac mice associated with S. epidermidis NIHLM061 and untreated control animals: MAIT cells (MR1 tetramer+ TCRβ+ TCRγδ− CD90.2+ CD45+ DAPI−) and CD4+ T cells (CD4+ CD25− CD8β− MR1 tetramer− TCRβ+ TCRγδ− CD90.2+ CD45+ DAPI−). RNA was isolated using the Arcturus PicoPure RNA Isolation Kit (Applied Biosystems), cDNA was synthesized using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech Laboratories), libraries were generated using Nextera XT DNA Library Preparation Kit (Illumina), and the samples were sequenced on a NextSeq 500 (Illumina) using the High Output v2 Kit (Illumina). Fastq files were mapped with STAR (10.1093/bioinformatics/bts635) to the mouse mm10 reference genome with default parameters. For analysis, the Hypergeometric Optimization of Motif EnRichment (HOMER) program was used (10.1016/j.molcel.2010.05.004). Tag directories were created with default parameters. To analyze differential gene expression, reads on genes were counted for two replicates per condition with analyzeRepeats rna with parameters -noadj and -condenseGenes. Subsequentially getDiffExpression.pl was used with default parameters (log2 fold change 1, FDR < 5%) using DESeq2 and rlog normalization. For GO term enrichment analysis Metascape (10.1016/j.chom.2015.11.002) was used.
Single-cell RNA-sequencing and analysis
Lymphocytes from the ear pinnae (skin), spleens, lungs, and livers of pooled WT mice were stained with antibodies against surface markers and TotalSeq-A hashtag oligonucleotide antibodies (BioLegend; hashtag 1 for skin, 2 for livers, 3 for lungs, and 4 for spleens). MAIT cells were sorted from each organ as MR1 tetramer+ TCRβ+ TCRγδ− CD3+ CD90.2+ B220− CD11b− CD11c− CD49f− CD45+ (10,000 cells from skin, spleens, and lungs and 3,500 cells from livers). Samples were pooled, encapsulated into droplets using the Chromium Single Cell Controller (10X Genomics), and libraries were prepared using Chromium Single Cell 3′ Reagent Kits v2 (10X Genomics). The mRNA library was prepared following the 10X Genomics user guide, while the HTO library was prepared according to published guidelines (75). Libraries were sequenced on a NextSeq 550 with 10% of the lane occupied by the HTO library and the other 90% by the mRNA library.
Data was demultiplexed using Cell Ranger 3.0.1 resulting in data for 6,463 cells. HTO libraries were processed using CITE-seq-Count (DOI: 10.5281/zenodo.2590196). Subsequently, data was analyzed using Seurat 3.0 (76). Cells with less than 900 or more than 3,000 gene counts, more than 5% mitochondrial contamination, or more than 10,000 RNA counts were filtered out, leaving 5,678 cells for downstream analysis. Data was normalized, scaled, PCA was performed, and neighbors were found using 5 dimensions. UMAP reduction was performed on this dataset with 10 dimensions. HTO were read in and assigned to the single cells and only singlets were used for further analysis leaving 585 cells. To define origins of cells, number of HTOs per cluster were considered and classified as “Skin”, “Spleen/Lung/Liver”, or not assigned (“NA”). To analyze gene expression between tissues, all cells, including cells without HTO from the respective clusters were used.
Statistical analysis
Two-tailed unpaired Student’s t-test was performed using Prism 7 (GraphPad) to determine statistical significance. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. “ns” denotes that the comparison was not significant.
Supplementary Material
Acknowledgements:
We thank National Institute of Allergy and Infectious Diseases (NIAID) animal facility staff, in particular, D. Trageser-Cesler and C. Acevedo (NIAID Microbiome Program Gnotobiotic Animal Facility), J. Chen, N. Modi, and S. Mistry (NIH Center for Human Immunology), M. Smelkinson (NIAID Biological Imaging Section), T. Hawley and E. Stregevsky (NIAID Flow Cytometry Section), R. Awasthi (NCI Transgenic Mouse Model Laboratory), G. Trinchieri, R. Salcedo, J. McCulloch, S. Sen, K. Beacht, J. LeGrand, E. Lewis, and J. Kehr. MR1 and CD1d tetramers were provided by the NIH Tetramer Core Facility. We acknowledge the NIAID Microbiome Program Sequencing Platform for performing the 16S rRNA and genomic sequencing. This study used the Office of Cyber Infrastructure and Computational Biology (OCICB) High Performance Computing (HPC) cluster at NIAID and the high-performance computational capabilities of the Biowulf Linux cluster at NIH.
Funding: This research was supported in part by the Intramural Research Program of NIAID, NIH (ZIA-AI001115 and ZIA-AI001132 to Y.B.); the NIH Extramural Research Program (U19-AI111143 to J.A., R01-DK110174 and DP1-DK113598 to M.A.F., DP2-AI144968 to T.C.S.); the Cancer Research Institute Irvington Postdoctoral Fellowship Program (M.G.C. and I.V.-C.); the European Molecular Biology Organization (EMBO) Long-Term Fellowship Program and the ARC Foundation (S.T.); the National Institute of General Medical Sciences (NIGMS) Postdoctoral Research Associate Training (PRAT) Program (J.L.L.); HHMI Hanna H. Gray Fellows Program (Y.E.C.); and the HHMI-Simons Faculty Scholars Program and the Chan Zuckerberg Biohub (M.A.F.).
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: Reagents are available upon signing a material transfer agreement. Anti-IL-18 (SK113AE-4) is available from I. Förster under a material transfer agreement with the University of Bonn. The RNA-seq data, 16S rRNA sequences, and bacterial genomes were deposited with the NCBI under accession numbers GSE128814 and PRJNA529261. All other data needed to evaluate the conclusions in this paper are present either in the main text or the supplementary materials.
Supplementary Materials:
Figures S1–S5
Table S1
References (65–76)
References and Notes:
- 1.Belkaid Y, Hand TW, Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rooks MG, Garrett WS, Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16, 341–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gollwitzer ES et al. , Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med 20, 642–647 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Scharschmidt TC et al. , A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity 43, 1011–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao K et al. , Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554, 255–259 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Al Nabhani Z et al. , A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity. [DOI] [PubMed] [Google Scholar]
- 7.Cahenzli J, Koller Y, Wyss M, Geuking MB, McCoy KD, Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14, 559–570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olszak T et al. , Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An D et al. , Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Constantinides MG, Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol 103, 409–419 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Ridaura VK et al. , Contextual control of skin immunity and inflammation by Corynebacterium. J Exp Med 215, 785–799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linehan JL et al. , Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 172, 784–796 e718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ismail AS et al. , Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A 108, 8743–8748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin C et al. , Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 176, 998–1013 e1016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kjer-Nielsen L et al. , MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Le Bourhis L et al. , Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol 11, 701–708 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Treiner E et al. , Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422, 164–169 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Koay HF et al. , A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol 17, 1300–1311 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Dusseaux M et al. , Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117, 1250–1259 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Vorkas CK et al. , Mucosal-associated invariant and gammadelta T cell subsets respond to initial Mycobacterium tuberculosis infection. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben Youssef G et al. , Ontogeny of human mucosal-associated invariant T cells and related T cell subsets. J Exp Med 215, 459–479 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen P et al. , Circulating Mucosal-Associated Invariant T Cells in a Large Cohort of Healthy Chinese Individuals From Newborn to Elderly. Front Immunol 10, 260 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ussher JE, Klenerman P, Willberg CB, Mucosal-associated invariant T-cells: new players in anti-bacterial immunity. Front Immunol 5, 450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H et al. , MAIT cells protect against pulmonary Legionella longbeachae infection. Nat Commun 9, 3350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilgenburg BV et al. , MAIT cells contribute to protection against lethal influenza infection in vivo. Nat Commun 9, 4706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rahimpour A et al. , Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med 212, 1095–1108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbett AJ et al. , T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509, 361–365 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Kim YG et al. , Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 356, 315–319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanov II et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaboriau-Routhiau V et al. , The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Legoux F et al. , Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science, eaaw2719 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Chu DM et al. , Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med 23, 314–326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Combellick JL et al. , Differences in the fecal microbiota of neonates born at home or in the hospital. Sci Rep 8, 15660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li K et al. , Synthesis, stabilization, and characterization of the MR1 ligand precursor 5-amino-6-D-ribitylaminouracil (5-A-RU). PLoS One 13, e0191837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sano T et al. , An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dias J et al. , The CD4(−)CD8(−) MAIT cell subpopulation is a functionally distinct subset developmentally related to the main CD8(+) MAIT cell pool. Proc Natl Acad Sci U S A 115, E11513–E11522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byrd AL, Belkaid Y, Segre JA, The human skin microbiome. Nat Rev Microbiol 16, 143–155 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Naik S et al. , Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520, 104–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Togni P et al. , Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 264, 703–707 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Jiang X et al. , Dermal gammadelta T Cells Do Not Freely Re-Circulate Out of Skin and Produce IL-17 to Promote Neutrophil Infiltration during Primary Contact Hypersensitivity. PLoS One 12, e0169397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas SY et al. , PLZF induces an intravascular surveillance program mediated by long-lived LFA-1-ICAM-1 interactions. J Exp Med 208, 1179–1188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salou M et al. , A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J Exp Med 216, 133–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobkowiak MJ et al. , Tissue-resident MAIT cell populations in human oral mucosa exhibit an activated profile and produce IL-17. Eur J Immunol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Wilgenburg B et al. , MAIT cells are activated during human viral infections. Nat Commun 7, 11653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik S et al. , Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seiler MP et al. , Elevated and sustained expression of the transcription factors Egr1 and Egr2 controls NKT lineage differentiation in response to TCR signaling. Nat Immunol 13, 264–271 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bussolino F et al. , Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 119, 629–641 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyake M, Goodison S, Lawton A, Gomes-Giacoia E, Rosser CJ, Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene 34, 890–901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toulon A et al. , A role for human skin-resident T cells in wound healing. J Exp Med 206, 743–750 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He Z, Ong CH, Halper J, Bateman A, Progranulin is a mediator of the wound response. Nat Med 9, 225–229 (2003). [DOI] [PubMed] [Google Scholar]
- 51.LeBlanc S et al. , CEACAM1 deficiency delays important wound healing processes. Wound Repair Regen 19, 745–752 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Demaria M et al. , An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grochot-Przeczek A et al. , Heme oxygenase-1 accelerates cutaneous wound healing in mice. PLoS One 4, e5803 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seach N et al. , Double-positive thymocytes select mucosal-associated invariant T cells. J Immunol 191, 6002–6009 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Martin E et al. , Stepwise development of MAIT cells in mouse and human. PLoS Biol 7, e54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ventura A et al. , Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Teunissen MBM et al. , The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol 134, 2898–2907 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Li J et al. , The frequency of mucosal-associated invariant T cells is selectively increased in dermatitis herpetiformis. Australas J Dermatol 58, 200–204 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Suwanpradid J, Holcomb ZE, MacLeod AS, Emerging Skin T-Cell Functions in Response to Environmental Insults. J Invest Dermatol 137, 288–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jameson J et al. , A role for skin gammadelta T cells in wound repair. Science 296, 747–749 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Keyes BE et al. , Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 167, 1323–1338 e1314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Marco Barros R et al. , Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific gammadelta T Cell Compartments. Cell 167, 203–218 e217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomez de Aguero M et al. , The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Leeansyah E, Loh L, Nixon DF, Sandberg JK, Acquisition of innate-like microbial reactivity in mucosal tissues during human fetal MAIT-cell development. Nat Commun 5, 3143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.