

Abstract
Mitochondrial damage is a critical contributor to cardiac ischemia/reperfusion (I/R) injury. Mitochondrial quality control (MQC) mechanisms, a series of adaptive responses that preserve mitochondrial structure and function, ensure cardiomyocyte survival and cardiac function after I/R injury. MQC includes mitochondrial fission, mitochondrial fusion, mitophagy and mitochondria-dependent cell death. The interplay among these responses is linked to pathological changes such as redox imbalance, calcium overload, energy metabolism disorder, signal transduction arrest, the mitochondrial unfolded protein response and endoplasmic reticulum stress. Excessive mitochondrial fission is an early marker of mitochondrial damage and cardiomyocyte death. Reduced mitochondrial fusion has been observed in stressed cardiomyocytes and correlates with mitochondrial dysfunction and cardiac depression. Mitophagy allows autophagosomes to selectively degrade poorly structured mitochondria, thus maintaining mitochondrial network fitness. Nevertheless, abnormal mitophagy is maladaptive and has been linked to cell death. Although mitochondria serve as the fuel source of the heart by continuously producing adenosine triphosphate, they also stimulate cardiomyocyte death by inducing apoptosis or necroptosis in the reperfused myocardium. Therefore, defects in MQC may determine the fate of cardiomyocytes. In this review, we summarize the regulatory mechanisms and pathological effects of MQC in myocardial I/R injury, highlighting potential targets for the clinical management of reperfusion.
Key words: Mitochondrial quality control, Mitochondrial fission, Fusion, Mitophagy, Mitochondrial death, Cardiomyocyte I/R injury, Apoptosis, Necroptosis
Graphical abstract
Mitochondrial quality control contributes to acute cardiac I/R injury. The mitochondrial network is constantly reshaped by the antagonistic activities between mitochondrial fission and fusion. Mitophagy allows autophagosomes to selectively degrade damaged mitochondria. When these adaptive responses fail, programmed cell death by apoptosis or necroptosis is activated.
1. Introduction
The heart is a strong muscular pump that enables tissue and organ perfusion. Therefore, a continuous supply of fresh blood is vital for cardiac function. In coronary artery disease, plaques or thrombi induce rapid occlusion, which restricts blood flow to the heart1, 2, 3. The primary effect of coronary artery disease is substantial cardiomyocyte death, which prevents the heart from effectively pumping blood to vital organs4,5. Emergency coronary recanalization through a coronary artery bypass graft or percutaneous transluminal coronary intervention can limit cardiomyocyte death6. However, laboratory experiments have revealed that a significant proportion of cardiomyocyte death occurs during the first few minutes of reperfusion, in what is known as myocardial ischemia/reperfusion (I/R) injury7,8.
Several molecular mechanisms have been proposed to explain the pathological alterations in cardiac I/R injury, including rapid reactive oxygen species (ROS) release, calcium overloading, energy depletion, mitochondrial dysfunction and programmed cell death activation9, 10, 11, 12. Mitochondria have been recognized as key triggers of cardiac I/R injury13,14. First, mitochondria are abundant in cardiomyocytes and determine more than 90% of their energy supply15. Second, mitochondria can promote cardiomyocyte death by inducing apoptosis or necroptosis in the reperfused myocardium16. Third, other pathological situations such as calcium overload, oxidative stress, endoplasmic reticulum stress and immune responses are triggered by, integrated with or augmented by mitochondrial dysfunction17. Therefore, it is highly important to understand the regulatory mechanisms and biochemical contributions of mitochondrial dysfunction in cardiac I/R injury.
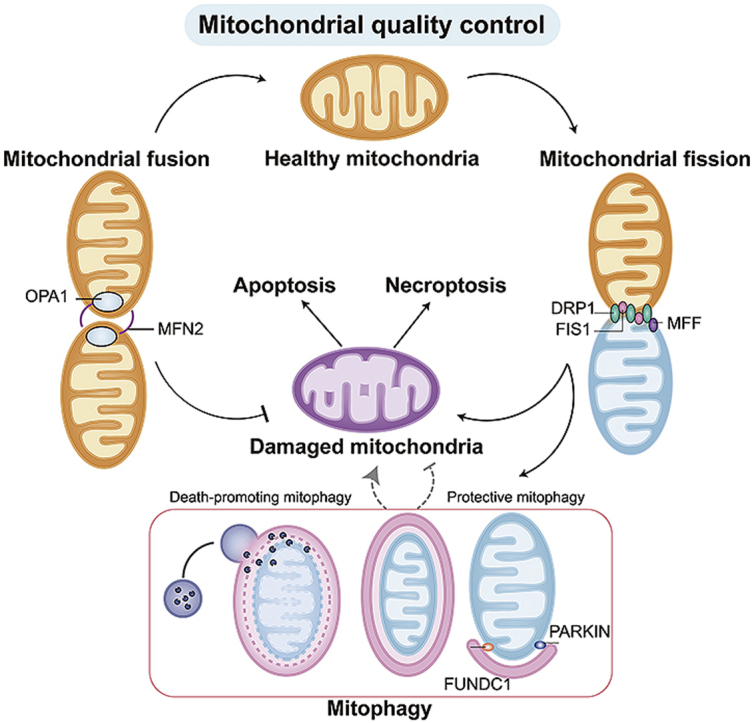
In response to stressful conditions, the mitochondria can activate mitochondrial quality control (MQC) to preserve mitochondrial structure and function18. MQC is a group of adaptive responses that regulate mitochondrial protein turnover, mitochondrial fusion, mitochondrial fission and mitophagy19 (Fig. 1). The main consequences of MQC are the rapid removal of defective mitochondrial debris and the timely replenishment of the mitochondrial network20,21. These biophysical processes protect the mitochondria from damage and therefore attenuate the vulnerability of cardiomyocytes to I/R injury.
Figure 1.

Overview of mitochondrial quality control (MQC) under physiological conditions and I/R injury. MQC coordinates various processes (fission, fusion, mitophagy and mitochondria-controlled cell death) to ensure cellular homeostasis. Mitochondrial dysfunction, amplified by failing quality-control processes, is believed to be a major mechanism of cardiac I/R injury. Several potential targets of MQC could be harnessed to treat cardiac I/R injury by inhibiting mitochondrial fission, promoting mitochondrial fusion, moderately activating mitophagy and inhibiting mitochondria-dependent cell death.
This review summarizes how MQC protects the myocardium from I/R injury, focusing on mitochondrial fission, fusion, mitophagy and mitochondria-dependent programmed cell death (Fig. 1). Recent findings on the contribution of mitochondrial fission to mitochondrial damage and cardiomyocyte death (apoptosis and/or necroptosis) are discussed, along with receptor-dependent and -independent mitophagy-based mechanisms of cardiomyocyte mitochondrial protection. The debate over whether mitochondrial fusion induces or reduces I/R-related mitochondrial dysfunction is also analyzed22,23. A special emphasis is given to mitochondria-induced cell death, especially necroptosis, a novel mechanism that contributes to MQC and determines cardiomyocyte viability during cardiac I/R injury. We hope the information presented here will provide new insights into the molecular pathways underlying mitochondria-related myocardial damage in I/R, and will offer useful targets for cardioprotection.
2. Mitochondrial fission
Although mitochondria were originally believed to be static, it is now well accepted that mitochondria are dynamic organelles that are constantly reshaped by fusion and fission. Mitochondrial fission can remove dysfunctional mitochondria from cardiomyocytes, and the extent of mitochondrial fission is largely determined by the metabolic needs of the cells. Proper mitochondrial fission generates many offspring and thus provides the boost in cardiomyocyte oxidative phosphorylation necessary for myocardial development and performance24,25. Fission also enables mitochondria to separate damaged fractions from reticular mitochondria26, and thus is indispensable for cardiomyocyte mitochondrial homeostasis.
Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) and its receptors (Fig. 2), including mitochondrial fission factor (MFF), mitochondrial fission one protein (FIS1), mitochondrial dynamics protein of 49 kDa (MID49) and mitochondrial dynamics protein of 51 kDa (MID51)27. Under physiological conditions, DRP1 is primarily free in the cytoplasm in an inactive form that cannot bind to its receptors, which are anchored to the outer mitochondrial membrane (OMM). Therefore, mitochondrial fission is relatively low under normal conditions. Interestingly, under stressful conditions, DRP1 undergoes conformational changes through post-transcriptional modifications including ubiquitination, acetylation and phosphorylation28,29. These structural alterations expose the binding site on DRP1 and increase its likelihood of binding to its receptors on the OMM, enabling cytoplasmic DRP1 to translocate to the surface of the mitochondria. On the other hand, the transcriptional upregulation of DRP1 is not a reliable measure of DRP1-induced mitochondrial fission30.
Figure 2.

Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) and its receptors, including mitochondrial fission factor (MFF), mitochondrial fission one protein (FIS1). Increased mitochondrial fission is associated with oxidative stress, mitochondrial DNA (mtDNA) damage, mitochondrial membrane potential reduction and mitochondrial apoptosis activation.
Multiple post-transcriptional modifications of DRP1 have been discovered through mass spectrometry-based proteomics, as shown in the online open database PhosphoSitePlus31 and a thorough recent review by Jhun et al.32. Two post-transcriptional modification sites on DRP1 have been well explored: the phosphorylation sites at Ser616 and Ser637. Ser616 phosphorylation promotes DRP1 oligomerization around the OMM, a prerequisite for the formation of a potential mitochondrial fission ring33. Phosphorylation at Ser637 has the opposite effect of impairing DRP1 oligomerization and therefore preventing mitochondrial fission34,35.
Post-transcriptional modification also occurs on DRP1 receptors, including MFF36, FIS137, MID4938 and MID5139. Phosphorylation of MFF at Ser146 enhances its affinity for DRP1, and this alteration has been reported in cardiac microvascular I/R injury36. Interestingly, the N-terminal arm of FIS1 auto-inhibits its access to DRP1, whereas phosphorylation of this N-terminal arm enhances the binding of FIS1 to DRP137. The effects of MID49/MID51 phosphorylation on DRP1-induced mitochondrial fission in cardiac I/R injury have not been described.
In the context of cardiac I/R injury, mitochondrial fission is associated with mitochondrial damage and cardiomyocyte death (Fig. 2). Following cardiac I/R injury, DRP1 phosphorylation at Ser637 decreases, so the mitochondrial localization of DRP1 increases34. Consequently, excessive mitochondrial fission occurs, which induces cytosolic calcium overload and thus promotes cardiomyocyte death and myocardial contractile dysfunction. In contrast, DRP1 phosphorylation at Ser616 increases after myocardial I/R injury40, and ROS production and cardiomyocyte oxidative stress are elevated. The expression of MFF41 and its post-transcriptional phosphorylation at Ser14636 are found to be augmented in a mouse model of cardiac microvascular I/R injury, and genetic ablation of Mff is reported to attenuate mitochondrial DNA (mtDNA) breaks, restore mtDNA copying and transcription, improve mitochondrial respiration and enhance endothelial viability.
The increased fission under cardiac I/R injury is known to induce other pathological alterations, including the reduction of ATP levels, the translocation of cytochrome c (Cyt-c) from the mitochondria to the cytoplasm, the opening of the mitochondrial permeability transition pore (mPTP) and the dissipation of the mitochondrial membrane potential; these effects are coupled with caspase-3 activation and cardiomyocyte apoptosis42, 43, 44, 45. Of note, the cardiomyocyte antioxidant capacity, as reflected by the levels of superoxide dismutase two and heme oxygenase 1, is also found to be altered by mitochondrial fission, although the mechanism is unknown46. Moreover, autophagy, a procedure that degrades damaged intracellular components, is reported to be drastically repressed by mitochondrial fission, as demonstrated by the reduced LC3II/I ratio, beclin-1 expression and ATG5/7 expression46. In vivo, the extent of mitochondrial fission is found to correlate positively with the size of the myocardial infarction and negatively with cardiac function measures such as the left ventricular ejection fraction and left ventricular fractional shortening46,47. These results illustrate the sufficiency of mitochondrial fission to promote myocardial I/R injury.
On the other hand, genetic or pharmacologic blockades of mitochondrial fission can protect the reperfused heart48,49. Mdivi-1 pharmacologically inhibits mitochondrial fission by preventing DRP1 from binding to its receptors. Administration of Mdivi-1 to mice before myocardial I/R injury markedly inhibits DRP1 translocation to the mitochondria, and thus reduces serum cardiac troponin I levels and lactate dehydrogenase activity48. Mdivi-1 treatment primarily improves mitochondrial function by blocking mPTP opening and stabilizing the mitochondrial membrane potential49,50. Mdivi-1 treatment can also partly reverse mitochondria-induced apoptosis by suppressing Cyt-c release and caspase-9 activation41. Ding and coworkers48 observed that Mdivi-1 treatment increases the activity of the antioxidant enzyme manganese superoxide dismutase and reduces the content of malondialdehyde, indicating that mitochondrial fission is also associated with the redox status.
Mitochondrial fission influences a variety of cardiac protective pathways, including the protein kinase B (PKB), extracellular-signal-regulated kinase (ERK), 5′-adenosine monophosphate-activated protein kinase (AMPK) and nitric oxide pathways51, 52, 53. Several of these proteins have been identified as upstream regulators of the post-transcriptional modifications of DRP1 and its receptors. For instance, ERK and AMPK can attenuate DRP1 phosphorylation at Ser616, whereas PKB can promote DRP1 phosphorylation at Ser63740.
The above data indicate that mitochondrial fission is a complex and progressive process involving either positive or negative feedback signals between various signaling pathways. However, several critical events should be emphasized. First, Mdivi-1 can improve cardiac function when it is given during ischemia and at the onset of reperfusion, but to a lesser extent than when it is administered before ischemia54. One possibility is that inhibiting physiological mitochondrial fission impairs cardiac function, whereas pathological mitochondrial fission primarily takes place after ischemia or reperfusion. Second, the inhibition of fission attenuates apoptosis but exacerbates necroptosis in cardiomyocytes55. This unexpected phenomenon seems to be present in Mdivi-1-injected mice, but not in Drp1-deleted56 or Mff-depleted mice41. Thus, it is possible that Mdivi-1 is not a specific blocker of mitochondrial fission. Actually, several findings have suggested that Mdivi-1 can repress mitophagy51,57, a protective pathway that prevents cell death by impeding apoptosis or necroptosis. Lastly, Mdivi-1 treatment following ischemia primarily seems to reverse cardiac diastolic dysfunction, as evidenced by the improved left ventricular developed pressure and lower left ventricular end diastolic pressure after such treatment34. Thus, careful attention is needed when interpreting studies in which Mdivi-1 has been used to inhibit mitochondrial fission in myocardial I/R injury.
3. Mitochondrial fusion
In contrast to mitochondrial fission, fusion is a process that integrates several mitochondrial fractions into long filamentous mitochondria. Mitochondrial fusion can be divided into three distinct steps: tethering, outer membrane fusion and inner membrane fusion58,59. Like mitochondrial fission, mitochondrial fusion is regulated by large guanosine triphosphatases (GTPases). The transmembrane GTPases mitofusin one and 2 (MFN1/2) are OMM-localized proteins, whereas the dynamin-like GTPase optic atrophy 1 (OPA1) promotes IMM intermingling60 (Fig. 3). Structurally, MFN1 and MFN2 on two physically contacting mitochondria promote homotypic or heterotypic coordination to stimulate OMM fusion61. The long isoform of OPA1 (L-OPA1) triggers IMM interactions between two mitochondria, resulting in the formation of the short isoform of Opa1 (S-OPA1) with the help of the proteases yeast mitochondrial escape one like one ATPase (YME1L1) and OMA1 zinc metallopeptidase (OMA1)62.
Figure 3.

Mitochondrial fusion is controlled by outer mitochondrial membrane (OMM)-localized mitofusin 2 (MFN2) and inner mitochondrial membrane (IMM)-localized optic atrophy 1 (OPA1). Increased mitochondrial fusion inhibits mitochondrial fission, sustains mitochondrial potential, promotes mitochondrial bioenergetics and suppresses mitochondrial apoptosis.
Most experimental evidence indicates that mitochondrial fusion protects cells during stress by two independent mechanisms. First, fusion offsets the effects of excessive mitochondrial fission and thus limits fission-initiated mitochondrial apoptosis63. Second, fusion generates a long, shared electrochemical potential within the mitochondrial network, enhancing the timely detection of damaged parts in the mitochondrial mass64. Fusion also equilibrates mitochondrial proteins, lipids, metabolites and mtDNA, which is thought to alleviate the local stress response and restore mitochondrial homeostasis65.
While fused mitochondria may be protective under physiological conditions, the involvement of fusion-related factors in cardiac I/R injury is the subject of hot debate. First, Mfn1-null mice are healthy and fertile, whereas Mfn2-null mice die soon after birth66. Mfn1 deletion seems to have little influence on cardiac function under either physiological or pathological conditions67, whereas Mfn2-deficient hearts exhibit extensive mtDNA breaks and mitochondrial damage68. More surprisingly, cardiomyocyte-specific Mfn1-knockout mice display a normal respiratory repertoire and are protected from mitochondrial depolarization69. In addition, Mfn1-knockout cardiomyocytes exhibit improved viability in a hydrogen-peroxide-induced oxidative stress microenvironment due to their reduced mPTP opening rate69, suggesting that Mfn1 deletion may protect cardiomyocytes from oxidative-stress-induced injury. In contrast, Mfn2 deficiency in cardiomyocytes promots mPTP opening, augments ROS production and triggers cell death70,71. In a hypoxia/reoxygenation-mimicked I/R injury model in vitro, Mfn2 silencing sensitizes H9C2 cells to apoptosis, and this process could be partly reversed through the inhibition of caspase-9 or the overexpression of BCL-x(L)72. In accordance with the effects of cardiomyocyte-specific Mfn2 ablation, Mfn1/Mfn2 double depletion causes defective mitochondria to accumulate and exhibit an unfolded protein response73,74. These data may reflect an additional function of MFN1 that has not yet been documented. However, relatively low levels of MFN1 have been detected in many tissues75, 76, 77, especially the brain, which may explain why MFN1 loss is not so detrimental to cardiac function. This concept requires additional studies for verification. MFN2 and MFN1 may exert completely different effects on cardiomyocyte viability upon cardiac I/R injury, despite their similar functionality in promoting mitochondrial fusion.
Unlike the effects of MFN1 and MFN2, the effects of OPA1 on cardiomyocyte fate and mitochondrial function have been well established. The heart-specific knockdown of Opa1 increases mitochondrial morphometric heterogeneity and ultimately induces ventricular dilation with irreversible contractile dysfunction78. In myocardial I/R injury, OPA1 expression is found to be reduced, while the genetic activation of OPA1 suppresses mitochondrial fission and cardiomyocyte death63. Knocking out Opa1 expands the infarction size and induces cardiac dysfunction in reperfused hearts79. Reperfusion induces the self-cleavage and activation of OMA1, and cleaved OMA1 accelerates the conversion of L-OPA1 to S-OPA1, leading to mitochondrial fragmentation, Cyt-c release and apoptosis80. OPA1 dysregulation impairs mitochondrial bioenergetics and exacerbates oxidative stress81. Of note, other molecular mechanisms may account for OPA1-induced cardioprotection. For example, OPA1 is found to enhance myocardial fatty acid utilization and thus attenuate ROS generation and sustain the mitochondrial morphology in failing hearts82,83. However, this finding has not been validated in the process of cardiac I/R injury.
Like DRP1, MFN1/2 can be post-transcriptionally phosphorylated by a number of kinases. However, the phosphorylation of MFN1/2 partially reduces their GTPase activities and thus abolishes their ability to induce mitochondrial fusion. MFN1 phosphorylation at Ser86 by beta II protein kinase C (bIIPKC) leads to a buildup of mitochondrial fragments in heart failure84. In addition, mitogen-activated protein kinase/ERK phosphorylates MFN1 at T56285, thereby reducing its efficiency in oligomerization and mitochondrial tethering but increasing the susceptibility to BAX-induced mitochondrial apoptosis. Similarly, MFN2 phosphorylation by PTEN-induced putative kinase protein 1 (PINK1) facilitates depolarization-induced PARKIN translocation onto mitochondria, thus promoting mitophagy and reducing the accumulation of morphologically and functionally abnormal mitochondria86. In contrast, after exposure to stress, MFN2 is primarily phosphorylated and then degraded by c-Jun N-terminal kinase, which impairs mitochondrial fusion and enhances cell death87. In the context of cardiac I/R injury, although post-transcriptional modifications of MFN1/2 have not been confirmed, MFN1/2 protein levels are significantly downregulated88.
Of note, unlike MFN1/2, OPA1 is primarily regulated at the protein level by two mechanisms: redox status and mitochondrial proteolytic enzyme activity. OMA1 and YME1L1, which are mainly upregulated by stress or the mitochondrial unfolded protein response, have been acknowledged as upstream inducers of OPA1 degradation during cardiac I/R injury82. In addition, mitochondrial ROS levels are found to correlate with the extent of proteolytic processing of OPA1, while the scavenging of mitochondrial ROS is reported to prolong the protein stability of OPA1 in cardiomyocytes89. OPA1 transcription in cardiomyocytes is activated by STAT3 and RelA, which form a supercomplex that binds to the promoter region of OPA190. Although STAT3 has not been observed to transcriptionally modify OPA1 in cardiac I/R injury, STAT3 activity is significantly downregulated in the reperfused heart91, 92, 93. This downregulation, together with OMA1/YME1L1-induced OPA1 degradation, may further reduce OPA1 expression during myocardial I/R injury. Additionally, in hearts under pressure overload or hyperglycemic conditions, OPA1 is found to be hyperacetylated94 and O-GlcNAcylated95, respectively. These structural modifications reduce the GTPase activity of OPA1, leading to mitochondrial morphological disorder and cardiomyocyte death. It would be interesting to explore the post-transcriptional hyperacetylation and O-GlcNAcylation of OPA1 in myocardial I/R injury.
Several drugs and gene-modifying technologies have been created to restore mitochondrial fusion96, 97, 98. Sevoflurane-induced anesthetic postconditioning has been demonstrated to reduce cardiac I/R injury in basic research and clinical surgery99. Interestingly, the benefits of sevoflurane are attributed to the upregulation of OPA1 and MFN2 in hypoxia/reoxygenation-treated neonatal rat cardiomyocytes99,100. OPA1 and MFN2 levels are also found to be induced by vagal nerve stimulation, which improves mitochondrial dynamics in the ischemic myocardium52. Epigallocatechin gallate effectively inhibits OPA1 degradation by OMA1, and therefore maintains mitochondrial morphological homeostasis in reperfused hearts80,101. Melatonin transcriptionally upregulates OPA1 expression through the AMPK pathway and thus increases the resistance of mitochondria and cardiomyocytes to I/R injury79. Based on this information, preserving mitochondrial fusion through MFN2 activation or OPA1 stabilization is critical when designing cardioprotective therapies for myocardial I/R injury.
4. Mitophagy
Mitochondrial components are eventually recycled through a specialized autophagic pathway known as mitophagy. Mitophagy is a kind of selective organelle autophagy that prevents the accumulation of abnormal mitochondria that might otherwise trigger cardiomyocyte dysfunction or death102. Proper mitophagy also recycles metabolic substrates that are vital for cardiomyocyte metabolism under stressful conditions103,104. BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), FUN14 domain containing 1 (FUNDC1) and NIX are expressed on the OMM and therefore elicit receptor-dependent mitophagy (Fig. 4). However, PARKIN is mainly localized in the cytoplasm and translocates onto the mitochondria with lower membrane potential to initiate receptor-independent mitophagy after stimulation. Mechanismically, the targeted mitochondria are then engulfed by the pre-autophagosome, forming an autophagosome. Subsequently, microtubule-associated protein 1A/1B-light chain 3 (LC3) binds to phosphatidylethanolamine, generating the LC3-phosphatidylethanolamine conjugate (LC3II). Finally, the lysosome induces the proteolytic degradation of the autophagosomal proteins, nucleic acids, carbohydrates and lipids, which are recycled by the cell to restore homeostasis105,106.
Figure 4.

The most recognized mitophagy pathway in mammalian cells is mediated by PARKIN, a receptor-independent pathway. In addition to PARKIN-mediated mitophagy, receptor-dependent pathway for mitophagy induction includes Fun14 domain-containing protein 1 (FUNDC1). Mechanismically, the targeted mitochondria are engulfed by the pre-autophagosome, forming an autophagosome. Subsequently, microtubule-associated protein 1A/1B-light chain 3 (LC3) binds to phosphatidylethanolamine, generating the LC3-phosphatidylethanolamine conjugate (LC3II). Finally, the lysosome induces the proteolytic degradation of the autophagosomal proteins, nucleic acids, carbohydrates and lipids, which are recycled by the cell to restore homeostasis.
Since mitophagy is a “self-eating” process, excessive mitophagy is maladaptive and has been linked to cell death. Accordingly, genetically or pharmacologically blocking mitophagy can attenuate cell death107,108. Three molecular mechanisms have been proposed to explain mitophagic cell death. First, many stimuli can trigger both mitophagy and cell death, so the cell fate is mainly determined by the degree and duration of stress. Under mild stress, when parts of the mitochondria are damaged, the selective removal of mitochondria via mitophagy is protective for the cell. However, once stress becomes severe, the number of damaged mitochondria increases and may overwhelm the capacity of mitophagy, leading to cell death. Thus, mitophagy is pro-survival, and cell death occurs when mitophagy cannot preserve mitochondrial homeostasis109,110. Second, excessive mitophagy significantly reduces the mitochondrial mass and therefore causes ATP exhaustion. Cells with low ATP levels have a pronounced susceptibility to stress-induced death via necroptosis rather than apoptosis, because apoptosis is ATP-dependent programmed cell death. Therefore, mitophagy is fatal when it “eats” too many mitochondria111. Third, mitophagy is stimulated by many proteins that also induce apoptosis, such as the BCL2-family proteins NIX and BNIP3112. Although BNIP3 initiates mitophagy by promoting the binding between LC3 and mitochondria, BNIP3 overexpression sensitizes cells to the intrinsic apoptotic cell death pathway113. Thus, the death-suppressing or -promoting actions of mitophagy are determined by upstream adaptors. Lastly, we must emphasize that cell fate management via mitophagy may also depend on the tissue and cell type.
In the setting of cardiac I/R injury, numerous experiments have investigated how mitophagy influences myocardial function and cardiomyocyte viability. The effects of mitophagy (i.e., suppressing or promoting cell death) mainly seem to depend on the adaptors involved. For example, reperfusion-induced cardiomyocyte death through calcium overload is followed by OPA1-induced mitophagy; however, the pharmacologic activation of OPA1-induced mitophagy is found to protect the heart against I/R injury63. Similarly, an early study indicated that the genetic ablation of Opa1 impaires mitophagy and augments I/R-induced myocardial damage79. In addition, Saito et al.114 demonstrated that protective mitophagy during myocardial ischemia is mediated by RAB9 for the first time. Phosphorylated RAB9 at Ser179 promotes the assembly of the ULK1–RAB9–RIP1–DRP1 complex, and then activates mitophagy to protect myocardium against ischemia. In contrast, PARKIN-induced mitophagy is found to be fatal for reperfused hearts because it enhances cyclophilin D (CypD)-induced mPTP opening, a feature of necroptosis115. In a cardiac microvascular I/R injury model, PARKIN-induced mitophagy causes excessive mitochondrial elimination and an ATP undersupply, thus conveying death-promoting signals to cardiac microvascular endothelial cells116. BNIP3-induced mitophagy is also demonstrated to be lethal in cardiac I/R injury117. Abrogating BNIP3 activity not only prevents mitophagy, but also suppresses necrotic cell death in cardiomyocytes117.
Unlike PARKIN- and BNIP3-induced mitophagy, cardiolipin-induced mitophagy is a cardioprotective process that attenuates mitochondrial oxidative stress, reduces calcium overload and promotes cardiomyocyte survival during I/R injury118,119. Protective mitophagy can also be triggered by FUNDC1, an OMM protein that is regulated through post-transcriptional modification120. At the stage of ischemia, FUNDC1 is found to be activated (dephosphorylated) and to foster mitophagy, thus reducing reperfusion-induced myocardial damage121. FUNDC1-induced mitophagy has been reported to reverse the mitochondrial membrane potential, reduce mitochondrial ROS production and prevent mitochondria-induced apoptosis36,122. TNF-receptor-associated factor 2 (TRAF2), an E3 ubiquitin ligase, also has been found to trigger protective mitophagy and reduce mitochondrial fragmentation in reperfused hearts123,124.
Although the induction of mitophagy by different adaptors can have distinct effects on cell fate, ranging from survival to death, little is known about the molecular crosstalk among these adaptors. Thus, the net effect of mitophagy on cardiac I/R injury remains unclear. Of note, some studies have found that mitophagy is activated during I/R injury113,115, whereas others have found that it is inhibited79,125. This may be due to the different time points of reperfusion evaluated after ischemia. There is no doubt that ischemic/hypoxic stress induces autophagy (mitophagy)126,127. During reperfusion, autophagy flux is reduced in the early phase (0–24 h after I/R injury), but is augmented at the later recovery stage (1–3 days after I/R injury)128,129. In a careful recent study130, autophagy reporter (CAG-RFP-EGFP-LC3) mice are generated and subjected to renal I/R injury. Autophagy is unaltered in the first 4 h after reperfusion, but autophagosome formation is overtly reduced from four to 24 h post-reperfusion. Thereafter, the autophagosome fuses with lysosomes to form the autolysosome from one to 3 day post-reperfusion130. This conclusion is also highlighted in a number of high-profile thematic reviews131, 132, 133.
Actually, the early inactivation and late activation of mitophagy may be an adaptive and protective response. From a pathophysiological perspective, cardiomyocyte death mainly occurs within the early period of reperfusion due to ROS overproduction and calcium overload134,135. Under these conditions, cellular damage overwhelms the defense and/or repair systems of cardiomyocytes, including their anti-oxidative, anti-apoptotic and metabolism-remodeling capacities. Hence, either apoptosis or necroptosis is somewhat inevitable, so mitophagy is inhibited. Cardiomyocytes may thus avoid the possible activation of mitophagic cell death, which would otherwise accelerate or aggravate cardiomyocyte loss and myocardial dysfunction. However, in the late phase of reperfusion, the myocardium requires mitophagy to repair damaged mitochondria and restore cardiomyocyte viability, so the net result of mitophagy increases at this stage. Although mitophagy is inhibited or activated at different phases of reperfusion, we cannot conclude that various adaptors are inhibited or activated within a similar window. For example, in the early stage of reperfusion, PARKIN116 seems to be upregulated, whereas FUNDC1121 is rapidly inactivated. Thus, the ultimate effect of mitophagy results from the crosstalk among various mitophagy adaptors, although the time mapping of these adaptors has not yet been reported.
Although mitochondrial fission is reportedly to induce cardiomyocyte reperfusion damage through activating apoptosis, fission is supposed to occur prior to mitophagy at the stage of ischemia. A recent study114 reported that Unc-51 like autophagy activating kinase-1 (ULK1) phosphorylates RAB9 at Ser179, which promotes association between RAB9 and RIP1, followed by phosphorylation of DRP1 at Ser616 and its activation. Then, DRP1-induced fission sequesters damaged mitochondria and facilitates mitophagy to attenuate myocardial ischemic injury. Besides, DRP1 SUMOylation also contributes to mitophagy activation in hypoxia-treated cardiomyocyte, which is followed by sustained mitochondrial potential and decreased cardiomyocyte apoptosis136. These observations demonstrate that mitophagy is induced by moderate fission under ischemia/hypoxia conditions. In fact, an early study has proposed that PARKIN-independent mitophagy requires DRP1 to maintain the integrity of mammalian heart137. When DRP1 is absent, PARKIN becomes necessary to sustain mitochondrial function and structural integrity138. This notion is also supported by several following studies that FUNDC1 requires DRP1-dependent fission to control mitophagy139,140. Interestingly, at the stage of reperfusion, fission is significantly upregulated whereas protective mitophagy is largely inhibited. Re-introduction of mitophagy has been found to stop fatal fission79,141, resulting into mitochondrial potential stabilization and cardiomyocyte survival. These data suggest that mitophagy may in turn restrict abnormal mitochondrial fission. However, inhibition of DRP1-mediated mitochondrial fragmentation seems to impair autophagosome recognition and engulfing of damaged mitochondria142, reconfirming that fission is the prerequisite for mitophagy induction. Overall, although abnormal mitochondrial fission is followed by cardiomyocyte death in cardiac I/R injury, mitophagy requires moderate fission to sequester damaged mitochondria at the ischemic stage whereas reperfusion-induced excessive fission could be in turn corrected by mitophagy.
Notably, because mitophagy is regulated by various adaptors, when one adaptor is inhibited, another may be induced in compensation. For example, germline Parkin ablation in mice has proven not to be an ideal experimental model for mitophagy depletion, in part due to the compensation from mitochondrial E3 ubiquitin protein ligase 1 (MUL1)-induced mitophagy in the physiological state143. Further, although ATG32 is the primary mitophagy receptor in yeast, its mammalian homologue BCL2 like 13 (BCL2L13) compensates somewhat for basal mitophagy activity in Atg32-null yeast144. Moreover, under normal conditions, nonselective autophagy compensates for the lack of mitophagy in Mfn2-knockout mice145. Since compensatory mechanisms ensure that mitophagy occurs under various conditions, the next key question is how the various mitophagy adaptors interact with and compensate for one another in cardiac I/R injury.
5. Mitochondria-dependent cell death
Cardiac I/R injury involves the rapid loss of functional cardiomyocytes through programmed cell death, the final step of MQC. Mitochondria induce or inhibit cardiomyocyte death by two routes (Fig. 5). The first approach is the hyper-permeabilization of the OMM, followed by the leakage of Cyt-c from the mitochondria into the cytoplasm. There, Cyt-c activates caspase-9, which subsequently cleaves caspase-3146,147. This classical mitochondria-induced apoptotic pathway is also characterized by mitochondrial membrane potential reduction, ROS overload, BAX upregulation and BCL2 downregulation148,149. The second death pathway is induced by the protracted opening of the mPTP due to voltage-dependent anion-selective channel multimerization, CypD phosphorylation and adenine nucleotide translocator upregulation, although the primary constituents of the mPTP complex are being intensely debated150,151. The mPTP induces the opening of the IMM by forming a non-specific pore, leading to mitochondrial swelling, mitochondrial electron transport chain dysfunction and tricarboxylic acid cycle termination152,153. Subsequently, due to ATP exhaustion, the cell undergoes cytoplasmic swelling, membrane rupture and organelle breakdown, which lead to cell death through necroptosis154. In contrast to apoptosis, necroptotic cell death does not require energy, and exhibits features such as cell/organelle swelling, extensive mitochondrial disruption, blebbing and irreversible plasma membrane disintegration155,156.
Figure 5.

Mitochondrial death includes apoptosis and necroptosis. Apoptosis is regulated by outer mitochondrial membrane (OMM) permeabilization, mitochondrial membrane potential reduction, caspase-9 activation. Necroptosis is induced by the activation of RIPK3/MLKL pathway and the mPTP opening. Then, mitochondrial electron transport chain dysfunction and tricarboxylic acid cycle termination contribute to ATP exhaustion, cytoplasmic swelling, and membrane rupture.
Several regulators of mitochondrial apoptosis or necroptosis should be highlighted to illustrate the signal transduction pathways underlying mitochondria-induced cell death in cardiac I/R injury. First, with respect to apoptosis, BAX is an important inducer of OMM permeabilization, whereas BCL2 guards against BAX-induced OMM damage157,158. Under normal conditions, BCL2 heterodimerizes with BAX to inhibit its pro-apoptotic activity. However, certain stimuli upregulate the transcription of BAX, ultimately increasing the abundance of BAX proteins in the cytoplasm and enabling their homodimerization159. Subsequently, BAX homodimers migrate to and insert themselves into the OMM, thus permeabilizing it160,161. Accordingly, the levels of BCL2 and BAX, as well as the mitochondrial membrane potential, are usually used to monitor mitochondrial apoptosis162,163.
Regarding necroptosis, the initial signals include receptor interacting serine/threonine kinase 3 (RIPK3), phosphoglycerate mutase 5 (PGAM5) and mixed lineage kinase domain like protein (MLKL). At the stage of reperfusion, oxidative stress and calcium overload directly or indirectly activate RIPK3, which stimulates PGAM5 and MLKL phosphorylation and oligomerization on the membrane. MLKL then forms membrane pores to execute lytic cell death16,164. Of note, mPTP opening seems to result from RIPK3 activation in the setting of cardiac I/R injury, based on recent studies165, 166, 167. Ca2+–calmodulin-dependent protein kinase (CaMKII) is a substrate of RIPK3, and activated CaMKII promotes mPTP opening165,166. Additionally, RIPK3 upregulation augments the expression of PGAM5, which enhances CypD phosphorylation167,168, thus increasing the opening rate of the mPTP. Therefore, RIPK3 expression, MLKL phosphorylation and the mPTP opening rate are potential targets for the regulation of mitochondria-initiated necroptosis.
Apoptosis has traditionally been accepted as the main form of cardiomyocyte death responsible for myocyte loss during and after cardiac I/R injury. However, in recent studies120,165, only 30% of the cell death during I/R injury could be reversed by the pan-caspase inhibitor zVAD. In contrast, the depletion of necroptotic genes such as Ripk3 reduces cardiomyocyte death by 50% in I/R injury. These works demonstrate that the dominant form of programmed cell death in cardiac I/R injury is necroptosis, an unexpected result. Actually, the infarction site is known to have two different zones: the inner area of the infarcted myocardium (the umbra), and the surrounding ischemic penumbra. Necroptosis mainly presents in the core area of the infarcted zone, whereas apoptosis fills the ischemic region16,169,170.
Although the regulatory mechanisms of apoptosis and necroptosis are relatively clear, their interactive effects remain to be elucidated. RIPK3 is also an upstream activator of caspase 8-induced apoptosis in myocardial infarction171,172, but activated caspase-8 can degrade RIPK3 and thus inhibit necroptosis173,174. Mitochondrial apoptosis inhibitors such as c-IAP1 and c-IAP2 can induce RIPK3 ubiquitination and prevent necroptosis activation175,176. In a cellular reperfusion model177, RIPK3 is linked to DRP1 activation and mitochondrial membrane potential reduction, suggesting that RIPK3 may promote mitochondrial fission and subsequent mitochondrial apoptosis. Interestingly, the deletion of Ripk3 reverses FUNDC1-induced mitophagy and thus sends an anti-apoptotic signal to reperfused hearts120. On the other hand, the suppression of autophagy flux is found to trigger cardiomyocyte death via necroptosis178. These studies suggest that there is reciprocity between necroptosis and mitochondrial dynamics. Although necroptosis and apoptosis are regulated by completely different upstream signaling pathways, there is a striking pattern of overlap in their downstream events. Accordingly, when cardioprotective drugs are designed to reduce myocardial I/R injury, both anti-apoptotic and anti-necroptotic actions should be considered.
6. Conclusions
MQC is an adaptive response that adjusts the morphology and function of mitochondria during cardiac I/R injury (Fig. 6). After exposure to stress, cardiomyocytes employ anti-oxidative factors to neutralize mitochondrial ROS, reduce oxidative stress damage and ensure mitochondrial homeostasis. Concurrently, mitochondrial fission is activated so that damaged mitochondrial fractions can be removed from the mitochondrial network, with the cooperation of mitophagy. In contrast, healthy, long mitochondria can integrate with several small mitochondrial fragments to enhance the resistance of the entire mitochondrial population to stress. When these adaptive responses fail, programmed cell death by apoptosis or necroptosis is activated, and damaged mitochondria become the inducers of cell death, enabling the sequestration of incurable and dysfunctional cardiomyocytes. During this process, mitochondrial fission and mitophagy serve as a double-edged sword in the reperfused heart: on one hand, they exert pro-survival mechanisms by isolating damaged mitochondria, and on the other hand, if fission and mitophagy persist beyond a certain threshold, they may lead to cellular demise. Therefore, selective, effective, moderate and differential activation of mitophagy and mitigation of fission are essential for MQC, and could synergistically enhance cardiac function in I/R injury. Necroptosis and apoptosis, although activated by various stimulus, are functionally governed solely by mitochondria. As the final steps of MQC to maintain tissue homeostasis, necroptosis and apoptosis communicate with each other, and offer new targets for therapeutic approaches. The compounds or drugs targeting MQC are summarized in Table 1. More studies are required to further verify the therapeutic effects of these compounds/drugs in clinical practice.
Figure 6.

Involvement of mitochondrial quality control in cardiac I/R injury. Under physiological conditions, healthy mitochondria support the functions of cardiomyocytes by ensuring optimal catabolic and anabolic metabolism and regulating the intracellular trafficking of Ca2+. Additionally, an intact mitochondrial network maintains inflammatory homeostasis and tissue integrity by preventing the activation of signal transduction cascades that lead to pro-inflammatory factor secretion and regulated cell death. Mitochondrial dysfunction is accompanied by metabolic derangements and alterations in the intracellular Ca2+ flux, and also promotes an inflammatory milieu and regulated cell death, which culminates in tissue loss.
Table 1.
Compounds or drugs targeting mitochondrial quality control (MQC) in cardiac I/R injury.
| Name | Target | Ref. |
|---|---|---|
| Mdivi-1 | Mitochondrial fission (DRP1) | 179,180 |
| Propofol | Mitochondrial fission (DRP1) | 181 |
| Dapagliflozin | Mitochondrial fission (DRP1) | 182 |
| Dynasore | Mitochondrial fission (DRP1) | 183 |
| Isosteviol sodium | Mitochondrial fission (DRP1/FIS1) | 184 |
| Tetrahydrocurcumin | Mitochondrial fission and fusion (DRP1 and MFN2) | 185 |
| Pravastatin | Mitochondrial fission and fusion (DRP1 and MFN1) | 186 |
| Vildagliptin | Mitochondrial fusion (MFN2) | 64 |
| Mitochondrial fusion promoter M1 | Mitochondrial fusion (MFN1/2 and OPA1) | 187 |
| Melatonin | Mitochondrial fusion and mitophagy (OPA1 and FUNDC1) | 79 , 122 |
| Tongxinluo | Mitophagy (PARKIN) | 188 |
| Bicarbonate | Mitophagy (PARKIN) | 189 |
| Simvastatin | Mitophagy (PARKIN) | 190 |
| Ellagic acid | Mitophagy (BNIP3) | 117 |
| Hydrogen-rich saline | Mitophagy (PARKIN) | 191 |
| Metformin | Necroptosis (RIPK1 and RIPK3) | 192 |
| Dexmedetomidine | Necroptosis (RIPK3) | 193 |
| Necrostatin-1 | Necroptosis (RIPK1) | 194 |
| Baicalin | Necroptosis (RIPK3 and MLKL) | 195 |
| Ciclosporin A | Necroptosis (mPTP opening) | 167 |
| Tanshinone IIA | Apoptosis (BAX/BCL2) | 196 |
| Taxifolin | Apoptosis (BAX/BCL2) | 197 |
| Glutamine | Apoptosis (Cyt-c) | 198 |
| Febuxostat | Apoptosis (Cyt-c) | 199 |
| PD150606 | Apoptosis (Cyt-c) | 42 |
| Ru360 | Apoptosis (BAX) | 200 |
Ackownledgments
This work was partially supported by the China Postdoctoral Science Foundation (2019TQ0128) and the National Natural Science Foundation of China (NSFC; 81900252, 81900254 and 81870249).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Author contributions
Jin Wang and Hao Zhou were responsible for original draft and visualization. Hao Zhou was responsible for review and editing, supervision, and funding acquisition.
Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analysis or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- 1.Heusch G. 25 years of remote ischemic conditioning: from laboratory curiosity to clinical outcome. Basic Res Cardiol. 2018;113:15. doi: 10.1007/s00395-018-0673-2. [DOI] [PubMed] [Google Scholar]
- 2.Heusch G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol. 2019;114:45. doi: 10.1007/s00395-019-0756-8. [DOI] [PubMed] [Google Scholar]
- 3.Tai Y., Li L., Peng X., Zhu J., Mao X., Qin N. Mitochondrial uncoupler BAM15 inhibits artery constriction and potently activates AMPK in vascular smooth muscle cells. Acta Pharm Sin B. 2018;8:909–918. doi: 10.1016/j.apsb.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou H., Li N., Yuan Y., Jin Y.G., Guo H., Deng W. Activating transcription factor 3 in cardiovascular diseases: a potential therapeutic target. Basic Res Cardiol. 2018;113:37. doi: 10.1007/s00395-018-0698-6. [DOI] [PubMed] [Google Scholar]
- 6.Zhao J., Gao J.L., Zhu J.X., Zhu H.B., Peng X., Jiang M. The different response of cardiomyocytes and cardiac fibroblasts to mitochondria inhibition and the underlying role of STAT3. Basic Res Cardiol. 2019;114:12. doi: 10.1007/s00395-019-0721-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H., Ma Q., Zhu P., Ren J., Reiter R.J., Chen Y. Protective role of melatonin in cardiac ischemia–reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 2018;64:e12471. doi: 10.1111/jpi.12471. [DOI] [PubMed] [Google Scholar]
- 8.Ren J., Zhang Y. Editorial: new therapetic approaches in the management of ischemia reperfusion injury and cardiometabolic diseases: opportunities and challenges. Curr Drug Targets. 2017;18:1687–1688. doi: 10.2174/138945011815171019092703. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H., Wang S., Hu S., Chen Y., Ren J. ER–mitochondria microdomains in cardiac ischemia–reperfusion injury: a fresh perspective. Front Physiol. 2018;9:755. doi: 10.3389/fphys.2018.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lochner A., Marais E., Huisamen B. Melatonin and cardioprotection against ischaemia/reperfusion injury: what's new? J Pineal Res. 2018;65:e12490. doi: 10.1111/jpi.12490. [DOI] [PubMed] [Google Scholar]
- 11.Wang M., Smith K., Yu Q., Miller C., Singh K., Sen C.K. Mitochondrial connexin 43 in sex-dependent myocardial responses and estrogen-mediated cardiac protection following acute ischemia/reperfusion injury. Basic Res Cardiol. 2019;115:1. doi: 10.1007/s00395-019-0759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H.F., Wang Y.L., Tan Y.Z., Wang H.J., Tao P., Zhou P. Enhancement of cardiac lymphangiogenesis by transplantation of CD34+VEGFR-3+ endothelial progenitor cells and sustained release of VEGF-C. Basic Res Cardiol. 2019;114:43. doi: 10.1007/s00395-019-0752-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuznetsov A.V., Javadov S., Margreiter R., Grimm M., Hagenbuchner J., Ausserlechner M.J. The role of mitochondria in the mechanisms of cardiac ischemia–reperfusion injury. Antioxidants (Basel) 2019;8:e454. doi: 10.3390/antiox8100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scarabelli T.M., Gottlieb R.A. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: old and new concepts after 10 years of contributions. Cell Death Differ. 2004;11:S144–S152. doi: 10.1038/sj.cdd.4401544. [DOI] [PubMed] [Google Scholar]
- 15.Vela D. Keeping heart homeostasis in check through the balance of iron metabolism. Acta Physiol (Oxf) 2019;228:e13324. doi: 10.1111/apha.13324. [DOI] [PubMed] [Google Scholar]
- 16.Del Re D.P., Amgalan D., Linkermann A., Liu Q., Kitsis R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–1817. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maneechote C., Palee S., Chattipakorn S.C., Chattipakorn N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J Cell Mol Med. 2017;21:2643–2653. doi: 10.1111/jcmm.13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyman L., Karbowski M., Lederer W.J. Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol Med. 2020;26:21–39. doi: 10.1016/j.molmed.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprenger H.G., Langer T. The good and the bad of mitochondrial breakups. Trends Cell Biol. 2019;29:888–900. doi: 10.1016/j.tcb.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Forini F., Nicolini G., Kusmic C., Iervasi G. Protective effects of euthyroidism restoration on mitochondria function and quality control in cardiac pathophysiology. Int J Mol Sci. 2019;20:e3377. doi: 10.3390/ijms20143377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yellon D.M., He Z., Khambata R., Ahluwalia A., Davidson S.M. The GTN patch: a simple and effective new approach to cardioprotection? Basic Res Cardiol. 2018;113:20. doi: 10.1007/s00395-018-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverblatt J.A., Ziff O.J., Dancy L., Daniel A., Carter B., Scott P. Therapies to limit myocardial injury in animal models of myocarditis: a systematic review and meta-analysis. Basic Res Cardiol. 2019;114:48. doi: 10.1007/s00395-019-0754-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seidel T., Fiegle D.J., Baur T.J., Ritzer A., Nay S., Heim C. Glucocorticoids preserve the t-tubular system in ventricular cardiomyocytes by upregulation of autophagic flux. Basic Res Cardiol. 2019;114:47. doi: 10.1007/s00395-019-0758-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mughal W., Martens M., Field J., Chapman D., Huang J., Rattan S. Myocardin regulates mitochondrial calcium homeostasis and prevents permeability transition. Cell Death Differ. 2018;25:1732–1748. doi: 10.1038/s41418-018-0073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wider J., Undyala V.V.R., Whittaker P., Woods J., Chen X., Przyklenk K. Remote ischemic preconditioning fails to reduce infarct size in the Zucker fatty rat model of type-2 diabetes: role of defective humoral communication. Basic Res Cardiol. 2018;113:16. doi: 10.1007/s00395-018-0674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H.H., Wu Y.J., Tseng Y.M., Su C.H., Hsieh C.L., Yeh H.I. Mitochondrial fission protein 1 up-regulation ameliorates senescence-related endothelial dysfunction of human endothelial progenitor cells. Angiogenesis. 2019;22:569–582. doi: 10.1007/s10456-019-09680-2. [DOI] [PubMed] [Google Scholar]
- 27.Scarpelli P.H., Tessarin-Almeida G., Vicoso K.L., Lima W.R., Borges-Pereira L., Meissner K.A. Melatonin activates FIS1, DYN1, and DYN2 Plasmodium falciparum related-genes for mitochondria fission: mitoemerald-GFP as a tool to visualize mitochondria structure. J Pineal Res. 2019;66:e12484. doi: 10.1111/jpi.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosdah A.A., Holien J.K., Delbridge L.M., Dusting G.J., Lim S.Y. Mitochondrial fission—a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect. 2016;4:e00235. doi: 10.1002/prp2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz R., Agg B., Ferdinandy P. Survival pathways in cardiac conditioning: individual data vs. meta-analyses. What do we learn? Basic Res Cardiol. 2018;113:4. doi: 10.1007/s00395-017-0661-y. [DOI] [PubMed] [Google Scholar]
- 30.Dorn G., II Mitochondrial fission/fusion and cardiomyopathy. Curr Opin Genet Dev. 2016;38:38–44. doi: 10.1016/j.gde.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jhun B.S., Ou J., Adaniya S.M., Cypress M.W., Yoon Y. Adrenergic regulation of Drp1-driven mitochondrial fission in cardiac physio-pathology. Antioxidants. 2018;7:e195. doi: 10.3390/antiox7120195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu S., Wang P., Zhang H., Gong G., Gutierrez Cortes N., Zhu W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun. 2016;7:13189. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharp W.W., Fang Y.H., Han M., Zhang H.J., Hong Z., Banathy A. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia–reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. Faseb J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trindade F., Vitorino R., Leite-Moreira A., Falcao-Pires I. Pericardial fluid: an underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res Cardiol. 2019;114:10. doi: 10.1007/s00395-019-0716-3. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H., Wang J., Zhu P., Zhu H., Toan S., Hu S. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol. 2018;113:23. doi: 10.1007/s00395-018-0682-1. [DOI] [PubMed] [Google Scholar]
- 37.Wells R.C., Picton L.K., Williams S.C., Tan F.J., Hill R.B. Direct binding of the dynamin-like GTPase, Dnm1, to mitochondrial dynamics protein Fis1 is negatively regulated by the Fis1 N-terminal arm. J Biol Chem. 2007;282:33769–33775. doi: 10.1074/jbc.M700807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atkins K., Dasgupta A., Chen K.H., Mewburn J., Archer S.L. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease. Clin Sci (Lond) 2016;130:1861–1874. doi: 10.1042/CS20160030. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z., Liu L., Wu S., Xing D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation-induced apoptosis. FASEB J. 2016;30:466–476. doi: 10.1096/fj.15-274258. [DOI] [PubMed] [Google Scholar]
- 40.Zaja I., Bai X., Liu Y., Kikuchi C., Dosenovic S., Yan Y. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem Biophys Res Commun. 2014;453:710–721. doi: 10.1016/j.bbrc.2014.09.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou H., Hu S., Jin Q., Shi C., Zhang Y., Zhu P. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. 2017;6:e005328. doi: 10.1161/JAHA.116.005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo T., Yue R., Hu H., Zhou Z., Yiu K.H., Zhang S. PD150606 protects against ischemia/reperfusion injury by preventing mu-calpain-induced mitochondrial apoptosis. Arch Biochem Biophys. 2015;586:1–9. doi: 10.1016/j.abb.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H., Shi C., Hu S., Zhu H., Ren J., Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018;21:599–615. doi: 10.1007/s10456-018-9611-z. [DOI] [PubMed] [Google Scholar]
- 44.Ding M., Ning J., Feng N., Li Z., Liu Z., Wang Y. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J Pineal Res. 2018;64:e12447. doi: 10.1111/jpi.12447. [DOI] [PubMed] [Google Scholar]
- 45.Ter Horst E.N., Krijnen P.A.J., Hakimzadeh N., Robbers L., Hirsch A., Nijveldt R. Elevated monocyte-specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res Cardiol. 2018;114:1. doi: 10.1007/s00395-018-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu P., Zhang J., Yu S., Luo Z., Hua F., Yuan L. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 2015;10:e0134666. doi: 10.1371/journal.pone.0134666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su H.H., Liao J.M., Wang Y.H., Chen K.M., Lin C.W., Lee I.H. Exogenous GDF11 attenuates non-canonical TGF-beta signaling to protect the heart from acute myocardial ischemia–reperfusion injury. Basic Res Cardiol. 2019;114:20. doi: 10.1007/s00395-019-0728-z. [DOI] [PubMed] [Google Scholar]
- 48.Ding M., Dong Q., Liu Z., Liu Z., Qu Y., Li X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc Diabetol. 2017;16:19. doi: 10.1186/s12933-017-0501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu J., Maimaitili Y., Xie P., Wu J.J., Wang J., Yang Y.N. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol (Oxf) 2017;220:83–98. doi: 10.1111/apha.12812. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber T., Salhofer L., Quinting T., Fandrey J. Things get broken: the hypoxia-inducible factor prolyl hydroxylases in ischemic heart disease. Basic Res Cardiol. 2019;114:16. doi: 10.1007/s00395-019-0725-2. [DOI] [PubMed] [Google Scholar]
- 51.Gharanei M., Hussain A., Janneh O., Maddock H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: a mitochondrial division/mitophagy inhibitor. PLoS One. 2013;8:e77713. doi: 10.1371/journal.pone.0077713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue R.Q., Sun L., Yu X.J., Li D.L., Zang W.J. Vagal nerve stimulation improves mitochondrial dynamics via an M3 receptor/CaMKKbeta/AMPK pathway in isoproterenol-induced myocardial ischaemia. J Cell Mol Med. 2017;21:58–71. doi: 10.1111/jcmm.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Totzeck M., Hendgen-Cotta U.B., Rassaf T. Nitrite-nitric oxide signaling and cardioprotection. Adv Exp Med Biol. 2017;982:335–346. doi: 10.1007/978-3-319-55330-6_18. [DOI] [PubMed] [Google Scholar]
- 54.Maneechote C., Palee S., Kerdphoo S., Jaiwongkam T., Chattipakorn S.C., Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond) 2018;132:1669–1683. doi: 10.1042/CS20180510. [DOI] [PubMed] [Google Scholar]
- 55.Dong Y., Undyala V.V.R., Przyklenk K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: critical importance of the timing of treatment. Basic Res Cardiol. 2016;111:59. doi: 10.1007/s00395-016-0578-x. [DOI] [PubMed] [Google Scholar]
- 56.Ikeda Y., Shirakabe A., Maejima Y., Zhai P., Sciarretta S., Toli J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 57.Rossello X., Yellon D.M. The RISK pathway and beyond. Basic Res Cardiol. 2018;113:2. doi: 10.1007/s00395-017-0662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer J.N., Leuthner T.C., Luz A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 2017;391:42–53. doi: 10.1016/j.tox.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park M., Sandner P., Krieg T. cGMP at the centre of attention: emerging strategies for activating the cardioprotective PKG pathway. Basic Res Cardiol. 2018;113:24. doi: 10.1007/s00395-018-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen M.M., Tareste D. Recent insights into the structure and function of Mitofusins in mitochondrial fusion. F1000Res. 2018;7:e1983. doi: 10.12688/f1000research.16629.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang M., Linn B.S., Zhang Y., Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia–reperfusion injury. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2293–2302. doi: 10.1016/j.bbadis.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 62.MacVicar T., Langer T. OPA1 processing in cell death and disease—the long and short of it. J Cell Sci. 2016;129:2297–2306. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 63.Guan L., Che Z., Meng X., Yu Y., Li M., Yu Z. MCU up-regulation contributes to myocardial ischemia–reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy inhibition. J Cell Mol Med. 2019;23:7830–7843. doi: 10.1111/jcmm.14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pirzeh L., Babapour V., Badalzadeh R., Panahi N. Pretreatment with vildagliptin boosts ischemic–postconditioning effects on cardioprotection and expression profile of genes regulating autophagy and mitochondrial fission/fusion in diabetic heart with reperfusion injury. Naunyn-Schmiedeberg’s Arch Pharmacol. 2019;392:1371–1382. doi: 10.1007/s00210-019-01660-z. [DOI] [PubMed] [Google Scholar]
- 65.Wang Q., Xu J., Li X., Liu Z., Han Y., Xu X. Sirt3 modulate renal ischemia–reperfusion injury through enhancing mitochondrial fusion and activating the ERK–OPA1 signaling pathway. J Cell Physiol. 2019;234:23495–23506. doi: 10.1002/jcp.28918. [DOI] [PubMed] [Google Scholar]
- 66.Chen H., Ren S., Clish C., Jain M., Mootha V., McCaffery J.M. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol. 2015;211:795–805. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Y., Csordas G., Jowdy C., Schneider T.G., Csordas N., Wang W. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y., Sparks M., Bhandari P., Matkovich S.J., Dorn G.W., 2nd Mitochondrial genome linearization is a causative factor for cardiomyopathy in mice and Drosophila. Antioxidants Redox Signal. 2014;21:1949–1959. doi: 10.1089/ars.2013.5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Papanicolaou K.N., Ngoh G.A., Dabkowski E.R., O'Connell K.A., Ribeiro R.F., Jr., Stanley W.C. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papanicolaou K.N., Khairallah R.J., Ngoh G.A., Chikando A., Luptak I., O'Shea K.M. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ndongson-Dongmo B., Lang G.P., Mece O., Hechaichi N., Lajqi T., Hoyer D. Reduced ambient temperature exacerbates SIRS-induced cardiac autonomic dysregulation and myocardial dysfunction in mice. Basic Res Cardiol. 2019;114:26. doi: 10.1007/s00395-019-0734-1. [DOI] [PubMed] [Google Scholar]
- 72.Shen T., Zheng M., Cao C., Chen C., Tang J., Zhang W. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- 73.Song M., Mihara K., Chen Y., Scorrano L., Dorn G.W., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metabol. 2015;21:273–286. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mouton A.J., DeLeon-Pennell K.Y., Rivera Gonzalez O.J., Flynn E.R., Freeman T.C., Saucerman J.J. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. 2018;113:26. doi: 10.1007/s00395-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Detmer S.A., Chan D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405–414. doi: 10.1083/jcb.200611080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morell M., Burgos J.I., Gonano L.A., Vila Petroff M. AMPK-dependent nitric oxide release provides contractile support during hyperosmotic stress. Basic Res Cardiol. 2018;113:7. doi: 10.1007/s00395-017-0665-7. [DOI] [PubMed] [Google Scholar]
- 78.Dorn G.W., 2nd, Clark C.F., Eschenbacher W.H., Kang M.Y., Engelhard J.T., Warner S.J. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y., Wang Y., Xu J., Tian F., Hu S., Chen Y. Melatonin attenuates myocardial ischemia–reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK–OPA1 signaling pathways. J Pineal Res. 2019;66:e12542. doi: 10.1111/jpi.12542. [DOI] [PubMed] [Google Scholar]
- 80.Nan J., Nan C., Ye J., Qian L., Geng Y., Xing D. EGCG protects cardiomyocytes against hypoxia–reperfusion injury through inhibition of OMA1 activation. J Cell Sci. 2019;132:e220871. doi: 10.1242/jcs.220871. [DOI] [PubMed] [Google Scholar]
- 81.Ma S., Dong Z. Melatonin attenuates cardiac reperfusion stress by improving OPA1-related mitochondrial fusion in a Yap–Hippo pathway-dependent manner. J Cardiovasc Pharmacol. 2019;73:27–39. doi: 10.1097/FJC.0000000000000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo Y., Wang Z., Qin X., Xu J., Hou Z., Yang H. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart. Cardiovasc Res. 2018;114:979–991. doi: 10.1093/cvr/cvy052. [DOI] [PubMed] [Google Scholar]
- 83.Moore J.B., Tang X.L., Zhao J., Fischer A.G., Wu W.J., Uchida S. Epigenetically modified cardiac mesenchymal stromal cells limit myocardial fibrosis and promote functional recovery in a model of chronic ischemic cardiomyopathy. Basic Res Cardiol. 2018;114:3. doi: 10.1007/s00395-018-0710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ferreira J.C.B., Campos J.C., Qvit N., Qi X., Bozi L.H.M., Bechara L.R.G. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat Commun. 2019;10:329. doi: 10.1038/s41467-018-08276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pyakurel A., Savoia C., Hess D., Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. 2015;58:244–254. doi: 10.1016/j.molcel.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y., Dorn G.W., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leboucher G.P., Tsai Y.C., Yang M., Shaw K.C., Zhou M., Veenstra T.D. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Y., Su W., Zhang Q., Xu J., Liu H., Luo J. Glycine protects H9C2 cardiomyocytes from high glucose- and hypoxia/reoxygenation-induced injury via inhibiting PKCbeta2 activation and improving mitochondrial quality. J Diabetes Res. 2018;2018:9502895. doi: 10.1155/2018/9502895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsushima K., Bugger H., Wende A.R., Soto J., Jenson G.A., Tor A.R. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res. 2018;122:58–73. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nan J., Hu H., Sun Y., Zhu L., Wang Y., Zhong Z. TNFR2 stimulation promotes mitochondrial fusion via Stat3- and NF-κB-dependent activation of OPA1 expression. Circ Res. 2017;121:392–410. doi: 10.1161/CIRCRESAHA.117.311143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen P.J., Shang A.Q., Yang J.P., Wang W.W. microRNA-874 inhibition targeting STAT3 protects the heart from ischemia–reperfusion injury by attenuating cardiomyocyte apoptosis in a mouse model. J Cell Physiol. 2019;234:6182–6193. doi: 10.1002/jcp.27398. [DOI] [PubMed] [Google Scholar]
- 92.Zuurbier C.J., Jong W.M., Eerbeek O., Koeman A., Pulskens W.P., Butter L.M. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. PLoS One. 2012;7:e40643. doi: 10.1371/journal.pone.0040643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meyer I.S., Leuschner F. The role of Wnt signaling in the healing myocardium: a focus on cell specificity. Basic Res Cardiol. 2018;113:44. doi: 10.1007/s00395-018-0705-y. [DOI] [PubMed] [Google Scholar]
- 94.Samant S.A., Zhang H.J., Hong Z., Pillai V.B., Sundaresan N.R., Wolfgeher D. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807–819. doi: 10.1128/MCB.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Makino A., Suarez J., Gawlowski T., Han W., Wang H., Scott B.T. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1296–R1302. doi: 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mehra P., Guo Y., Nong Y., Lorkiewicz P., Nasr M., Li Q. Cardiac mesenchymal cells from diabetic mice are ineffective for cell therapy-mediated myocardial repair. Basic Res Cardiol. 2018;113:46. doi: 10.1007/s00395-018-0703-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu L., Jin X., Hu C.F., Zhang Y.P., Zhou Z., Li R. Amphiregulin enhances cardiac fibrosis and aggravates cardiac dysfunction in mice with experimental myocardial infarction partly through activating EGFR-dependent pathway. Basic Res Cardiol. 2018;113:12. doi: 10.1007/s00395-018-0669-y. [DOI] [PubMed] [Google Scholar]
- 98.Liu D., Zeng X., Li X., Mehta J.L., Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113:5. doi: 10.1007/s00395-017-0663-9. [DOI] [PubMed] [Google Scholar]
- 99.Yu J., Wu J., Xie P., Maimaitili Y., Wang J., Xia Z. Sevoflurane postconditioning attenuates cardiomyocyte hypoxia/reoxygenation injury via restoring mitochondrial morphology. PeerJ. 2016;4:e2659. doi: 10.7717/peerj.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Merz J., Albrecht P., von Garlen S., Ahmed I., Dimanski D., Wolf D. Purinergic receptor Y2 (P2Y2)-dependent VCAM-1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res Cardiol. 2018;113:45. doi: 10.1007/s00395-018-0702-1. [DOI] [PubMed] [Google Scholar]
- 101.Kim Y.R., Baek J.I., Kim S.H., Kim M.A., Lee B., Ryu N. Therapeutic potential of the mitochondria-targeted antioxidant MitoQ in mitochondrial-ROS induced sensorineural hearing loss caused by Idh2 deficiency. Redox Biol. 2019;20:544–555. doi: 10.1016/j.redox.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Morales P.E., Arias-Duran C., Avalos-Guajardo Y., Aedo G., Verdejo H.E., Parra V. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspect Med. 2019;3:e100822. doi: 10.1016/j.mam.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 103.Pietzsch S., Ricke-Hoch M., Stapel B., Hilfiker-Kleiner D. Modulation of cardiac AKT and STAT3 signalling in preclinical cancer models and their impact on the heart. Biochim Biophys Acta Mol Cell Res. 2019;1867:118519. doi: 10.1016/j.bbamcr.2019.07.014. [DOI] [PubMed] [Google Scholar]
- 104.Kowaltowski A.J. Strategies to detect mitochondrial oxidants. Redox Biol. 2019;21:101065. doi: 10.1016/j.redox.2018.101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cadete V.J.J., Vasam G., Menzies K.J., Burelle Y. Mitochondrial quality control in the cardiac system: an integrative view. Biochim Biophys Acta Mol Basis Dis. 2019;1865:782–796. doi: 10.1016/j.bbadis.2018.11.018. [DOI] [PubMed] [Google Scholar]
- 106.Li J., Cai S.X., He Q., Zhang H., Friedberg D., Wang F. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. 2018;113:36. doi: 10.1007/s00395-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 107.Strappazzon F., Di Rita A., Peschiaroli A., Leoncini P.P., Locatelli F., Melino G. HUWE1 controls MCL1 stability to unleash AMBRA1-induced mitophagy. Cell Death Differ. 2020;27:1155–1168. doi: 10.1038/s41418-019-0404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Landry N.M., Cohen S., Dixon I.M.C. Periostin in cardiovascular disease and development: a tale of two distinct roles. Basic Res Cardiol. 2018;113:1. doi: 10.1007/s00395-017-0659-5. [DOI] [PubMed] [Google Scholar]
- 109.Shimizu S., Yoshida T., Tsujioka M., Arakawa S. Autophagic cell death and cancer. Int J Mol Sci. 2014;15:3145–3153. doi: 10.3390/ijms15023145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mekala N.K., Kurdys J., Depuydt M.M., Vazquez E.J., Rosca M.G. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. The protective role of the redox compound methylene blue. Redox Biol. 2019;20:107–117. doi: 10.1016/j.redox.2018.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Button R.W., Luo S., Rubinsztein D.C. Autophagic activity in neuronal cell death. Neurosci Bull. 2015;31:382–394. doi: 10.1007/s12264-015-1528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu Y., Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367–376. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jin Q., Li R., Hu N., Xin T., Zhu P., Hu S. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saito T., Nah J., Oka S.I., Mukai R., Monden Y., Maejima Y. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129:802–819. doi: 10.1172/JCI122035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun T., Ding W., Xu T., Ao X., Yu T., Li M. Parkin regulates programmed necrosis and myocardial ischemia/reperfusion injury by targeting cyclophilin-D. Antioxidants Redox Signal. 2019;31:1177–1193. doi: 10.1089/ars.2019.7734. [DOI] [PubMed] [Google Scholar]
- 116.Zhou H., Zhang Y., Hu S., Shi C., Zhu P., Ma Q. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017;63:e12413. doi: 10.1111/jpi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dhingra A., Jayas R., Afshar P., Guberman M., Maddaford G., Gerstein J. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med. 2017;112:411–422. doi: 10.1016/j.freeradbiomed.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 118.Paradies G., Paradies V., Ruggiero F.M., Petrosillo G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia–reperfusion injury: implications for pharmacological cardioprotection. Am J Physiol Heart Circ Physiol. 2018;315:H1341–H1352. doi: 10.1152/ajpheart.00028.2018. [DOI] [PubMed] [Google Scholar]
- 119.Morton A.B., Smuder A.J., Wiggs M.P., Hall S.E., Ahn B., Hinkley J.M. Increased SOD2 in the diaphragm contributes to exercise-induced protection against ventilator-induced diaphragm dysfunction. Redox Biol. 2019;20:402–413. doi: 10.1016/j.redox.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou H., Zhu P., Guo J., Hu N., Wang S., Li D. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou H., Zhu P., Wang J., Zhu H., Ren J., Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25:1080–1093. doi: 10.1038/s41418-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou H., Li D., Zhu P., Hu S., Hu N., Ma S. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res. 2017;63:e12438. doi: 10.1111/jpi.12438. [DOI] [PubMed] [Google Scholar]
- 123.Yang K.C., Ma X., Liu H., Murphy J., Barger P.M., Mann D.L. Tumor necrosis factor receptor-associated factor 2 mediates mitochondrial autophagy. Circ Heart Fail. 2015;8:175–187. doi: 10.1161/CIRCHEARTFAILURE.114.001635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Florido A., Saraiva N., Cerqueira S., Almeida N., Parsons M., Batinic-Haberle I. The manganese(III) porphyrin MnTnHex-2-PyP5+ modulates intracellular ROS and breast cancer cell migration: impact on doxorubicin-treated cells. Redox Biol. 2019;20:367–378. doi: 10.1016/j.redox.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang J., Nadtochiy S.M., Urciuoli W.R., Brookes P.S. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am J Physiol Heart Circ Physiol. 2016;310:H29–H38. doi: 10.1152/ajpheart.00926.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Esposti D.D., Domart M.C., Sebagh M., Harper F., Pierron G., Brenner C. Autophagy is induced by ischemic preconditioning in human livers formerly treated by chemotherapy to limit necrosis. Autophagy. 2010;6:172–174. doi: 10.4161/auto.6.1.10699. [DOI] [PubMed] [Google Scholar]
- 127.Otani H. Ischemic preconditioning: from molecular mechanisms to therapeutic opportunities. Antioxidants Redox Signal. 2008;10:207–247. doi: 10.1089/ars.2007.1679. [DOI] [PubMed] [Google Scholar]
- 128.Ji J., Zhou X., Xu P., Li Y., Shi H., Chen D. Deficiency of apoptosis-stimulating protein two of p53 ameliorates acute kidney injury induced by ischemia reperfusion in mice through upregulation of autophagy. J Cell Mol Med. 2019;23:2457–2467. doi: 10.1111/jcmm.14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Feng J., Li H., Zhang Y., Wang Q., Zhao S., Meng P. Mammalian STE20-like kinase 1 deletion alleviates renal ischaemia–reperfusion injury via modulating mitophagy and the AMPK–YAP signalling pathway. Cell Physiol Biochem. 2018;51:2359–2376. doi: 10.1159/000495896. [DOI] [PubMed] [Google Scholar]
- 130.Li L., Wang Z.V., Hill J.A., Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25:305–335. doi: 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin F. Autophagy in renal tubular injury and repair. Acta Physiol (Oxf) 2017;220:229–237. doi: 10.1111/apha.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaushal G.P., Shah S.V. Autophagy in acute kidney injury. Kidney Int. 2016;89:779–791. doi: 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Havasi A., Dong Z. Autophagy and tubular cell death in the didney. Semin Nephrol. 2016;36:174–188. doi: 10.1016/j.semnephrol.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nybo T., Dieterich S., Gamon L.F., Chuang C.Y., Hammer A., Hoefler G. Chlorination and oxidation of the extracellular matrix protein laminin and basement membrane extracts by hypochlorous acid and myeloperoxidase. Redox Biol. 2019;20:496–513. doi: 10.1016/j.redox.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Narzt M.S., Nagelreiter I.M., Oskolkova O., Bochkov V.N., Latreille J., Fedorova M. A novel role for NUPR1 in the keratinocyte stress response to UV oxidized phospholipids. Redox Biol. 2019;20:467–482. doi: 10.1016/j.redox.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bian X., Xu J., Zhao H., Zheng Q., Xiao X., Ma X. Zinc-induced SUMOylation of dynamin-related protein 1 protects the heart against ischemia–reperfusion injury. Oxid Med Cell Longev. 2019;2019:1232146. doi: 10.1155/2019/1232146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kageyama Y., Hoshijima M., Seo K., Bedja D., Sysa-Shah P., Andrabi S.A. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Roy M., Kageyama Y., Iijima M., Sesaki H. PARK2/Parkin becomes critical when DNM1L/Drp1 is absent. Autophagy. 2015;11:573–574. doi: 10.1080/15548627.2015.1017193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen M., Chen Z., Wang Y., Tan Z., Zhu C., Li Y. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wu W., Lin C., Wu K., Jiang L., Wang X., Li W. FUNDC1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–1384. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yu J., Li Y., Liu X., Ma Z., Michael S., Orgah J.O. Mitochondrial dynamics modulation as a critical contribution for Shenmai injection in attenuating hypoxia/reoxygenation injury. J Ethnopharmacol. 2019;237:9–19. doi: 10.1016/j.jep.2019.03.033. [DOI] [PubMed] [Google Scholar]
- 142.Huang C.Y., Lai C.H., Kuo C.H., Chiang S.F., Pai P.Y., Lin J.Y. Inhibition of ERK–Drp1 signaling and mitochondria fragmentation alleviates IGF-IIR-induced mitochondria dysfunction during heart failure. J Mol Cell Cardiol. 2018;122:58–68. doi: 10.1016/j.yjmcc.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 143.Yun J., Puri R., Yang H., Lizzio M.A., Wu C., Sheng Z.H. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3:e01958. doi: 10.7554/eLife.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mao K., Wang K., Zhao M., Xu T., Klionsky D.J. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193:755–767. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Song M., Chen Y., Gong G., Murphy E., Rabinovitch P.S., Dorn G.W., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Amanakis G., Kleinbongard P., Heusch G., Skyschally A. Attenuation of ST-segment elevation after ischemic conditioning maneuvers reflects cardioprotection online. Basic Res Cardiol. 2019;114:22. doi: 10.1007/s00395-019-0732-3. [DOI] [PubMed] [Google Scholar]
- 147.Honda T., He Q., Wang F., Redington A.N. Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res Cardiol. 2019;114:15. doi: 10.1007/s00395-019-0724-3. [DOI] [PubMed] [Google Scholar]
- 148.Opferman J.T., Kothari A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018;25:37–45. doi: 10.1038/cdd.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pohl S.O., Pervaiz S., Dharmarajan A., Agostino M. Gene expression analysis of heat-shock proteins and redox regulators reveals combinatorial prognostic markers in carcinomas of the gastrointestinal tract. Redox Biol. 2019;25:101060. doi: 10.1016/j.redox.2018.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Someda M., Kuroki S., Miyachi H., Tachibana M., Yonehara S. Caspase-8, receptor-interacting protein kinase 1 (RIPK1), and RIPK3 regulate retinoic acid-induced cell differentiation and necroptosis. Cell Death Differ. 2020;27:1539–1553. doi: 10.1038/s41418-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xiong Y., Li L., Zhang L., Cui Y., Wu C., Li H. The bromodomain protein BRD4 positively regulates necroptosis via modulating MLKL expression. Cell Death Differ. 2019;26:1929–1941. doi: 10.1038/s41418-018-0262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morciano G., Bonora M., Campo G., Aquila G., Rizzo P., Giorgi C. Mechanistic role of mPTP in ischemia–reperfusion injury. Adv Exp Med Biol. 2017;982:169–189. doi: 10.1007/978-3-319-55330-6_9. [DOI] [PubMed] [Google Scholar]
- 153.Pozzer D., Varone E., Chernorudskiy A., Schiarea S., Missiroli S., Giorgi C. A maladaptive ER stress response triggers dysfunction in highly active muscles of mice with SELENON loss. Redox Biol. 2019;20:354–366. doi: 10.1016/j.redox.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Frank T., Tuppi M., Hugle M., Dotsch V., van Wijk S.J.L., Fulda S. Cell cycle arrest in mitosis promotes interferon-induced necroptosis. Cell Death Differ. 2019;26:2046–2060. doi: 10.1038/s41418-019-0298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Robinson N., Ganesan R., Hegedus C., Kovacs K., Kufer T.A., Virag L. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239. doi: 10.1016/j.redox.2019.101239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhou X., Wu Y., Ye L., Wang Y., Zhang K., Wang L. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm Sin B. 2019;9:711–723. doi: 10.1016/j.apsb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Singh P.K., Roukounakis A., Weber A., Das K.K., Sohm B., Villunger A. Dynein light chain binding determines complex formation and posttranslational stability of the Bcl-2 family members Bmf and Bim. Cell Death Differ. 2020;27:434–450. doi: 10.1038/s41418-019-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hadebe N., Cour M., Lecour S. The SAFE pathway for cardioprotection: is this a promising target? Basic Res Cardiol. 2018;113:9. doi: 10.1007/s00395-018-0670-5. [DOI] [PubMed] [Google Scholar]
- 159.Lomonosova E., Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008;27 Suppl 1:S2–S19. doi: 10.1038/onc.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar D., Jugdutt B.I. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003;142:288–297. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 161.Kleinbongard P., Skyschally A., Gent S., Pesch M., Heusch G. STAT3 as a common signal of ischemic conditioning: a lesson on “rigor and reproducibility” in preclinical studies on cardioprotection. Basic Res Cardiol. 2018;113:3. doi: 10.1007/s00395-017-0660-z. [DOI] [PubMed] [Google Scholar]
- 162.Qi S., Guo L., Yan S., Lee R.J., Yu S., Chen S. Hypocrellin A-based photodynamic action induces apoptosis in A549 cells through ROS-mediated mitochondrial signaling pathway. Acta Pharm Sin B. 2019;9:279–293. doi: 10.1016/j.apsb.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu C., Zhang X., Wei W., Zhang N., Wu H., Ma Z. Matrine attenuates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via maintaining AMPKalpha/UCP2 pathway. Acta Pharm Sin B. 2019;9:690–701. doi: 10.1016/j.apsb.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Quispe R.L., Jaramillo M.L., Galant L.S., Engel D., Dafre A.L., Teixeira da Rocha J.B. Diphenyl diselenide protects neuronal cells against oxidative stress and mitochondrial dysfunction: involvement of the glutathione-dependent antioxidant system. Redox Biol. 2019;20:118–129. doi: 10.1016/j.redox.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 166.Zhu P., Hu S., Jin Q., Li D., Tian F., Toan S. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhou H., Li D., Zhu P., Ma Q., Toan S., Wang J. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia–reperfusion injury. J Pineal Res. 2018;65:e12503. doi: 10.1111/jpi.12503. [DOI] [PubMed] [Google Scholar]
- 168.Fuhrmann D.C., Wittig I., Brune B. TMEM126B deficiency reduces mitochondrial SDH oxidation by LPS, attenuating HIF-1alpha stabilization and IL-1beta expression. Redox Biol. 2019;20:204–216. doi: 10.1016/j.redox.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Oerlemans M.I., Koudstaal S., Chamuleau S.A., de Kleijn D.P., Doevendans P.A., Sluijter J.P. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int J Cardiol. 2013;165:410–422. doi: 10.1016/j.ijcard.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 170.Schmidt H.M., Kelley E.E., Straub A.C. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019;21:101072. doi: 10.1016/j.redox.2018.101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Luedde M., Lutz M., Carter N., Sosna J., Jacoby C., Vucur M. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103:206–216. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 172.Lalaoui N., Lindqvist L.M., Sandow J.J., Ekert P.G. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol. 2015;39:63–69. doi: 10.1016/j.semcdb.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 173.Zhang J., Yang Y., He W., Sun L. Necrosome core machinery: MLKL. Cell Mol Life Sci. 2016;73:2153–2163. doi: 10.1007/s00018-016-2190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shih Y.M., Cooke M.S., Pan C.H., Chao M.R., Hu C.W. Clinical relevance of guanine-derived urinary biomarkers of oxidative stress, determined by LC–MS/MS. Redox Biol. 2019;20:556–565. doi: 10.1016/j.redox.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bertrand M.J., Milutinovic S., Dickson K.M., Ho W.C., Boudreault A., Durkin J. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 176.Graczyk-Jarzynka A., Goral A., Muchowicz A., Zagozdzon R., Winiarska M., Bajor M. Inhibition of thioredoxin-dependent H2O2 removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019;21:101062. doi: 10.1016/j.redox.2018.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hou H., Wang Y., Li Q., Li Z., Teng Y., Li J. The role of RIP3 in cardiomyocyte necrosis induced by mitochondrial damage of myocardial ischemia–reperfusion. Acta Biochim Biophys Sin (Shanghai) 2018;50:1131–1140. doi: 10.1093/abbs/gmy108. [DOI] [PubMed] [Google Scholar]
- 178.Ogasawara M., Yano T., Tanno M., Abe K., Ishikawa S., Miki T. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J Mol Cell Cardiol. 2017;108:203–213. doi: 10.1016/j.yjmcc.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 179.So E.C., Hsing C.H., Liang C.H., Wu S.N. The actions of mdivi-1, an inhibitor of mitochondrial fission, on rapidly activating delayed-rectifier K+ current and membrane potential in HL-1 murine atrial cardiomyocytes. Eur J Pharmacol. 2012;683:1–9. doi: 10.1016/j.ejphar.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 180.Ong S.B., Kwek X.Y., Katwadi K., Hernandez-Resendiz S., Crespo-Avilan G.E., Ismail N.I. Targeting mitochondrial fission using Mdivi-1 in a clinically relevant large animal model of acute myocardial infarction: a pilot study. Int J Mol Sci. 2019;20:e3972. doi: 10.3390/ijms20163972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhao L., Zhuang J., Wang Y., Zhou D., Zhao D., Zhu S. Propofol ameliorates H9c2 cells apoptosis induced by oxygen glucose deprivation and reperfusion injury via inhibiting high levels of mitochondrial fusion and fission. Front Pharmacol. 2019;10:61. doi: 10.3389/fphar.2019.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tanajak P., Sa-Nguanmoo P., Sivasinprasasn S., Thummasorn S., Siri-Angkul N., Chattipakorn S.C. Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia–reperfusion injury. J Endocrinol. 2018;236:69–84. doi: 10.1530/JOE-17-0457. [DOI] [PubMed] [Google Scholar]
- 183.Gao D., Zhang L., Dhillon R., Hong T.T., Shaw R.M., Zhu J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One. 2013;8:e60967. doi: 10.1371/journal.pone.0060967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sun X., Yang Y., Xie Y., Shi X., Huang L., Tan W. Protective role of STVNa in myocardial ischemia reperfusion injury by inhibiting mitochondrial fission. Oncotarget. 2018;9:1898–1905. doi: 10.18632/oncotarget.22969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mondal N.K., Behera J., Kelly K.E., George A.K., Tyagi P.K., Tyagi N. Tetrahydrocurcumin epigenetically mitigates mitochondrial dysfunction in brain vasculature during ischemic stroke. Neurochem Int. 2019;122:120–138. doi: 10.1016/j.neuint.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Oi M., Donner D., Peart J., Beck B., Wendt L., Headrick J.P. Pravastatin improves risk factors but not ischaemic tolerance in obese rats. Eur J Pharmacol. 2018;826:148–157. doi: 10.1016/j.ejphar.2018.02.050. [DOI] [PubMed] [Google Scholar]
- 187.Maneechote C., Palee S., Kerdphoo S., Jaiwongkam T., Chattipakorn S.C., Chattipakorn N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin Sci (Lond) 2019;133:497–513. doi: 10.1042/CS20190014. [DOI] [PubMed] [Google Scholar]
- 188.Yang H.X., Wang P., Wang N.N., Li S.D., Yang M.H. Tongxinluo ameliorates myocardial ischemia–reperfusion injury mainly via activating parkin-mediated mitophagy and downregulating ubiquitin–proteasome system. Chin J Integr Med. 2019 doi: 10.1007/s11655-019-3166-8. Available from: [DOI] [PubMed] [Google Scholar]
- 189.Queliconi B.B., Kowaltowski A.J., Gottlieb R.A. Bicarbonate increases ischemia–reperfusion damage by inhibiting mitophagy. PLoS One. 2016;11:e0167678. doi: 10.1371/journal.pone.0167678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Andres A.M., Hernandez G., Lee P., Huang C., Ratliff E.P., Sin J. Mitophagy is required for acute cardioprotection by simvastatin. Antioxidants Redox Signal. 2014;21:1960–1973. doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yao L., Chen H., Wu Q., Xie K. Hydrogen-rich saline alleviates inflammation and apoptosis in myocardial I/R injury via PINK-mediated autophagy. Int J Mol Med. 2019;44:1048–1062. doi: 10.3892/ijmm.2019.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li C., Mu N., Gu C., Liu M., Yang Z., Yin Y. Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell. 2020;19:e13096. doi: 10.1111/acel.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen J., Jiang Z., Zhou X., Sun X., Cao J., Liu Y. Dexmedetomidine preconditioning protects cardiomyocytes against hypoxia/reoxygenation-induced necroptosis by inhibiting HMGB1-mediated inflammation. Cardiovasc Drugs Ther. 2019;33:45–54. doi: 10.1007/s10557-019-06857-1. [DOI] [PubMed] [Google Scholar]
- 194.Liu H., Zhang M., Dong X., Liu Y., Hao Y., Wang Y. Necrostatin-1 protects against ischemia/reperfusion injury by inhibiting receptor-interacting protein 1 in a rat flap model. J Plast Reconstr Aesthetic Surg. 2019;72:194–202. doi: 10.1016/j.bjps.2018.10.019. [DOI] [PubMed] [Google Scholar]
- 195.Bai J., Wang Q., Qi J., Yu H., Wang C., Wang X. Promoting effect of baicalin on nitric oxide production in CMECs via activating the PI3K-AKT-eNOS pathway attenuates myocardial ischemia–reperfusion injury. Phytomedicine. 2019;63:153035. doi: 10.1016/j.phymed.2019.153035. [DOI] [PubMed] [Google Scholar]
- 196.Jin H.J., Xie X.L., Ye J.M., Li C.G. TanshinoneIIA and cryptotanshinone protect against hypoxia-induced mitochondrial apoptosis in H9c2 cells. PLoS One. 2013;8:e51720. doi: 10.1371/journal.pone.0051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tang Z., Yang C., Zuo B., Zhang Y., Wu G., Wang Y. Taxifolin protects rat against myocardial ischemia/reperfusion injury by modulating the mitochondrial apoptosis pathway. PeerJ. 2019;7:e6383. doi: 10.7717/peerj.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li K., Cui Y.C., Zhang H., Liu X.P., Zhang D., Wu A.L. Glutamine reduces the apoptosis of H9C2 cells treated with high-glucose and reperfusion through an oxidation-related mechanism. PLoS One. 2015;10:e0132402. doi: 10.1371/journal.pone.0132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang S., Li Y., Song X., Wang X., Zhao C., Chen A. Febuxostat pretreatment attenuates myocardial ischemia/reperfusion injury via mitochondrial apoptosis. J Transl Med. 2015;13:209. doi: 10.1186/s12967-015-0578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Correa F., Soto V., Zazueta C. Mitochondrial permeability transition relevance for apoptotic triggering in the post-ischemic heart. Int J Biochem Cell Biol. 2007;39:787–798. doi: 10.1016/j.biocel.2007.01.013. [DOI] [PubMed] [Google Scholar]