

Abstract
Candida albicans is a dimorphic fungus that converts from a yeast form to a hyphae form during infection. This switch requires the formation of actin cable to coordinate polarized cell growth. It's known that nucleation of this cable requires a multiprotein complex localized at the tip called the polarisome, but the mechanisms underpinning this process were unclear. Here, we found that C. albicans Aip5, a homolog of polarisome component ScAip5 in Saccharomyces cerevisiae that nucleates actin polymerization and synergizes with the formin ScBni1, regulates actin assembly and hyphae growth synergistically with other polarisome proteins Bni1, Bud6, and Spa2. The C terminus of Aip5 binds directly to G-actin, Bni1, and the C-terminal of Bud6, which form the core of the nucleation complex to polymerize F-actin. Based on insights from structural biology and molecular dynamic simulations, we propose a possible complex conformation of the actin nucleation core, which provides cooperative positioning and supports the synergistic actin nucleation activity of a tri-protein complex Bni1-Bud6-Aip5. Together with known interactions of Bni1 with Bud6 and Aip5 in S. cerevisiae, our findings unravel molecular mechanisms of C. albicans by which the tri-protein complex coordinates the actin nucleation in actin cable assembly and hyphal growth, which is likely a conserved mechanism in different filamentous fungi and yeast.
Keywords: Candida albicans, polarisome complex, nucleation factor, nucleation promoting factor, actin cable establishment, filamentous growth, actin, cell polarity, computer modeling, crystal structure, protein complex
Polarized cell growth is essential for cell morphogenesis and development, which is tightly regulated by spatial-temporal recruitments of polarity proteins (1–3). Coordinated assembly of actin cables in filamentous fungi plays crucial roles in polarity establishment, cell protrusion, and continuous incorporation of biomolecules at the expanding edge, including lipids, proteins, and cell wall components. Budding yeast Saccharomyces cerevisiae polarisome complex proteins, including ScSpa2, ScBni1, ScBud6, and ScAip5, concentrate at the presumptive bud tip to mediate actin cable polymerization and polarized cell growth (4–11). Polarisome members ScBni1, ScBud6, and ScAip5 serve either as nucleation factor (NF) or nucleation promoting factor (NPF) to initiate actin cable assembly (4, 6, 10, 11). ScBni1 initiates barbed-end polymerization progressively from the polarized tip and also mediates subsequent fast-elongation through recruiting G-actins from profilin ScPfy1 (11–13). ScBud6 is an NPF by delivering G-actin to ScBni1 via ScBud6-C (residues 489–788 aa), which enhances ScBni1-mediated actin nucleation but not elongation (6, 14). By interacting with both ScBni1 and G-actin via its C terminus (residues 1110-1234 aa), ScAip5C functions as a modest NPF for ScBni1 as well as a weak actin nucleator on its own (4). ScAip5 N terminus is a mostly disordered region that was recruited into the macromolecular complex by polarisome scaffolder ScSpa2 via multivalent interactions (4). Interaction between the N terminus of ScBud6 and a ScAip5 region (1000–1131 aa), located at the N-terminal of the functional core (ScAip5-C, 1132–1234 aa), was also recently suggested (15). These cross-interactions between actin-polymerization factors through flexible intrinsically disordered regions (IDRs) have the advantage to provide dynamic assembly of actin NF and NPFs that might be able to tune the nucleation activities. Polarisome-mediated actin assembly is likely to be a conserved underlying mechanism in different fungal species because the folded domains of ScBni1, ScBud6, and ScAip5 showed high similarity in sequence among the fungi kingdom, including filamentous fungus, as well as the pattern of the ordered and disordered domains (4, 6).
As a polymorphic pathogenic fungus, Candida albicans switches morphology between yeast and hyphal forms of growth in response to environmental stimuli, such as a serum, host body temperature, peptidoglycan, and GlcNAc (16–18). For readability, all polarisome genes/proteins of C. albicans are shown without the prefix “Ca” hereafter. The underlying machinery for polarized cell growth in C. albicans resembles closely that of S. cerevisiae. Spa2, Bni1, and Bud6 are known to coordinate the functions of vesical-enriched Spitzenkörper and crescent surface-localized polarisome at the elongating hyphal tip (19–22). Disruptions of individual polarisome proteins, such as Spa2, Bni1, or Bud6, impair hyphal morphogenesis substantially. These genetic mutants form short and swollen filaments, which were similar to the morphological defects from the treatment with actin depolymerization drug cytochalasin A (20, 23–25), suggesting their collaborative role in actin assembly. The perturbation of actin cables in hyphal cells impairs the polarized assembly of vesicle transporting center Spitzenkörper, thereby leading to a swollen tip by depositing cell wall isotopically (20, 26).
In this report, we studied the orchestrated function of polarisome members in C. albicans, in actin polymerization and hyphal growth. We found that the C terminus-folded domain of Aip5 (Aip5C, residues 786-896 aa) directly interacted with both Bni1 (Bni1FH1C, residues 1043-1733 aa) and the C-terminal of Bud6 (Bud6C, residues 411-701 aa). We resolved the crystal structure of Aip5C at a 2.65 Å resolution, which shows a high similarity to ScAip5C. Whereas Aip5C promotes Bni1-mediated actin nucleation mildly, a strong synergy in elevating actin nucleation was observed when Aip5C applied to Bni1 in the presence of Bud6C. Double polarisome mutants of C. albicans in the absence of Aip5 showed additional defects in hyphal growth when compared with single mutants. To better understand the coordinated function among Aip5, Bni1, and Bud6, in polymerizing F-actin, we performed all-atom molecular dynamic (MD) simulation to investigate the complex assembly using protein domains that are physically associated with each other, including Aip5C, Bud6C, and C-terminal IDR of Bni1, Bni1C (residues 1545-1733 aa). We found that the most stable complex from MD simulations suggested a compatible positioning of each constituent of the tri-protein complex. The simulated complex conformation suggested that Bud6C and Aip5C occupy different binding regions of Bni1C, which allows both NPFs to stay close but not overlapping each other for G-actin delivery to the FH2 domain of Bni1. Together, our findings unravel molecular mechanisms of C. albicans polarisome protein in nucleation actin cable for hyphal growth, which is likely a common mechanism for other filamentous fungi.
Results
Aip5 regulates actin cable network with other polarisome components
The polarisome protein ScAip5 in budding yeast is highly conserved with Candida homologs (4). The C. albicans C6_00910C_A gene encodes a ScAip5 homolog, which we named AIP5, whose encoded protein Aip5 shares 90% similarity of the C terminus-folded domain with ScAip5. Similar to Aip5, other C. albicans polarisome proteins, Bni1, Bud6, and Spa2, are all highly conserved among the fungi kingdom (Fig. S1, A–C). To understand the physiological function of Aip5 in hyphal growth, we generated homozygous mutations of AIP5−/− in C. albicans, taking advantage of the CRISPR-Cas9 technology (27). Four DNA sites from the gene AIP5 following the PAM (NGG) sequence were selected for mutagenesis and also insertion of a stop codon (Fig. S1D). One of the four loci resulted in successful mutagenesis located at +76 bp from the start codon of AIP5 (Fig. 1A and Fig. S1D). Successful editing was evident by the BamHI digestion of a 520-bp fragment, which is PCR-amplified from the genomic DNA, into two pieces (76 and 444 bp) (Fig. 1B and Fig. S1E). Using the same editing strategy, AIP5 was knocked out in the background of four C. albicans homozygous strains, WT, bni1Δ/Δ, spa2Δ/Δ, and bud6Δ/Δ (Fig. 1B and Fig. S1E). All aip5Δ/Δ mutants were further validated by sequencing of the PCR products.
Figure 1.
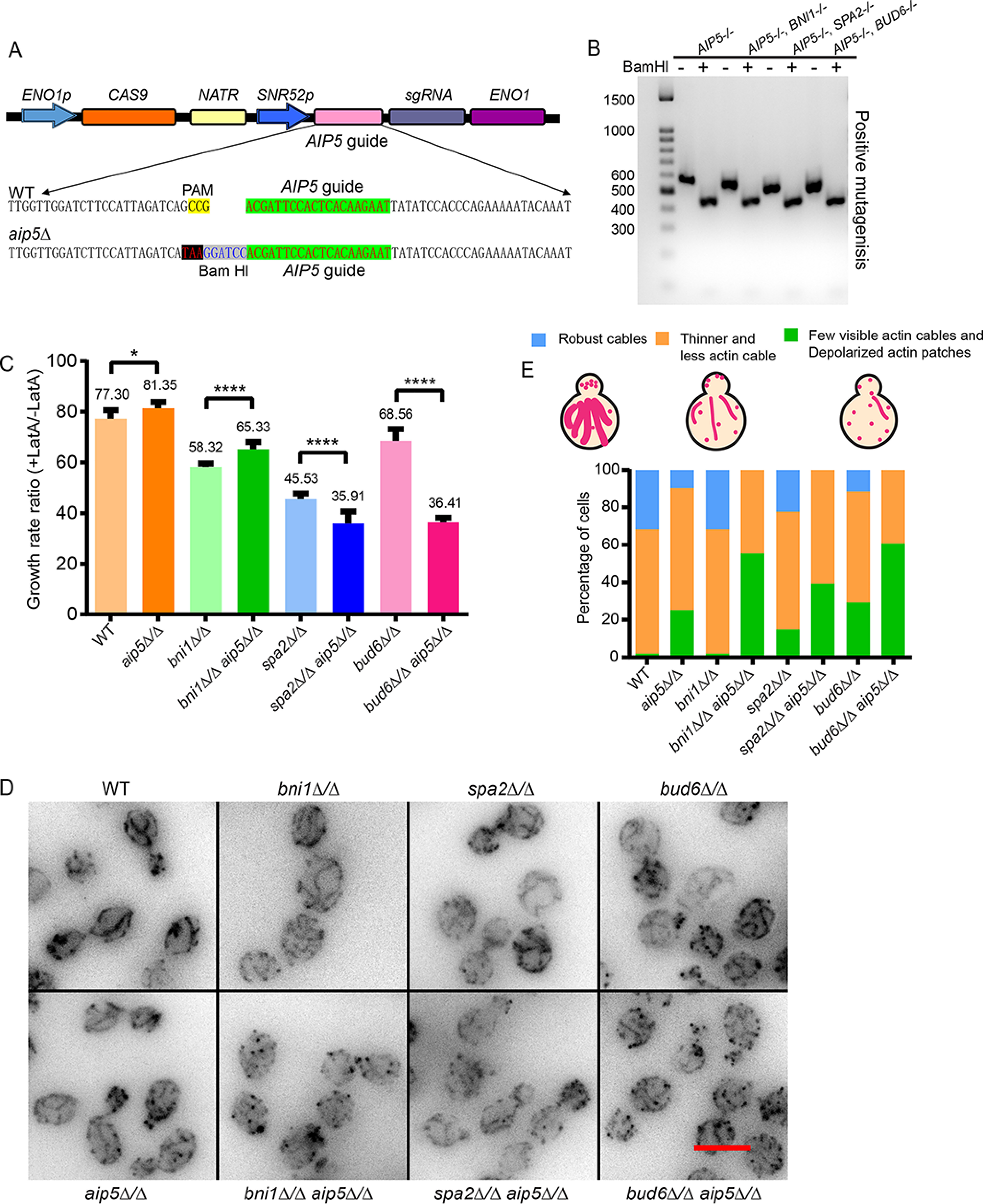
Polarisome mutants displayed thinner and less actin cable phenotype in C. albicans. A, design of the gRNA in pV1093 Solo system for editing AIP5. 20-bp gRNA sequences were selected, followed by the PAM sequence NGG. AIP5 guide sequence following a stop codon and the BamHI digestion site was inserted into the cas9 protein plasmid. B, verification of CRISPR/Cas9-edited AIP5 in various C. albicans WT and mutant strains by PCR amplification and enzyme digestion. The correct mutagenesis fragment can be amplified as a 520-bp fragment by the verification primers from the genomic DNA, and further digested by BamHI, which resulted in two fragments as 76 and 444 bp in length. C, the ratio of the quantified growth rate normalized by latrunculin A-treated conditions. The growth rate was calculated from the exponential phase (A600 = 0.5-1) of the indicated polarisome mutants and WT C. albicans with or without latrunculin A treatment (n = 4 for each condition; error bar, S.D.). D, the representative fluorescence microscopy images of actin networks stained by phalloidin green 488 from the indicated mutants in C. albicans. The scale bar represents 5 μm. E, characterization of the actin cable phenotype in various polarisome mutants as three categories, including robust actin cables, thinner and fewer actin cables, and few visible actin cables with depolarized patches. Cell number in each stain: WT, n = 224; aip5Δ/Δ, n = 198; bni1Δ/Δ, n = 145; bni1Δ/Δ aip5Δ/Δ, n = 175; spa2Δ/Δ, n = 153; spa2Δ/Δ aip5Δ/Δ, n = 160; bud6Δ/Δ, n = 184; bud6Δ/Δ aip5Δ/Δ, n = 219.
We first assessed the C. albicans growth sensitivity under mild actin depolymerization conditions using 1 μm latrunculin A (LatA) (28) (Fig. S1, F–I). The slopes of cell growth curves between A600 = 0.5-1.0 that indicate the exponential growth were used to calculate the growth rate. The ratio of growth rate normalized by LatA-treated conditions reflected how mutants growth were sensitized by actin disruption (Fig. 1C). aip5Δ/Δ showed a similar growth rate disruption (∼20%) compared with the WT strain upon LatA treatment, suggesting a modest sensitivity to actin depolymerization in aip5Δ/Δ (Fig. 1C). Both double mutants, spa2Δ/Δ aip5Δ/Δ, and bud6Δ/Δ aip5Δ/Δ, demonstrated high sensitivity to LatA, in which bud6Δ/Δ aip5Δ/Δ exhibited the strongest additive-sickness by showing a >30% more in growth reduction than bud6Δ/Δ single mutant (Fig. 1C), suggesting a potential synergy between Aip5 and Bud6 functions. We next investigated actin cable assembly in vivo by staining F-actin in C. albicans mutants using phalloidin green 488. Deletion of AIP5 increased the cell population with less-visible actin cables in the WT, bni1Δ/Δ, bud6Δ/Δ, and spa2Δ/Δ (Fig. 1, D and E). Consistent with the decrease in actin cables structures, there was an increase in the visibility of actin patches after the deletion of AIP5 (Fig. 1, D and E), which is due to the balance of actin patch and cable that share a same monomeric G-actin pool (28, 29). There was a ∼53% increase of highly disrupted actin cables (green population) with enhanced actin patches in bni1Δ/Δ aip5Δ/Δ compared with bni1Δ/Δ. Similarly, both double mutants of spa2Δ/Δ aip5Δ/Δ and bud6Δ/Δ aip5Δ/Δ displayed an increase of depolarized actin patches (green population) by ∼24 and ∼31%, respectively, compared with spa2Δ/Δ and bud6Δ/Δ single mutants (Fig. 1, D and E). Taken together, these results confirm the in vivo function of Aip5 in actin cable assembly, cooperatively, with other polarisome proteins.
Aip5 coordinates with polarisome components for hyphal establishment
The polarisome proteins Spa2, Bud6, and Bni1 are known to be involved in hyphal formation and growth (24, 30, 31). To examine the function of Aip5 in hyphal formation with other polarisome components, we used the Spider plates to induce the filamentous growth of polarisome mutants. Compared with the WT strain, aip5Δ/Δ showed apparent retardation in forming filamentous structures at the edge of C. albicans colonies (Fig. 2A and Fig. S2A). aip5Δ/Δ bni1Δ/Δ double mutant showed a complete abolishment of hyphal growth (Fig. 2A and Fig. S2A), compared with the single mutants. The additional effect of aip5Δ/Δ to spa2Δ/Δ and bud6Δ/Δ could not be determined, because neither spa2Δ/Δ nor bud6Δ/Δ displayed filamentous growth in the Spider plate assay (Fig. 2A and Fig. S2A). To better differentiate the polarisome members' functions, we investigated hyphal induction and morphogenesis at a single-cell level by switching the liquid culture added with 10% fetal bovine serum to a 37 °C condition (26). aip5Δ/Δ demonstrated a reduction in hyphal length compared with WT (Fig. 2, B and C). In the background of bni1Δ/Δ, spa2Δ/Δ and bud6Δ/Δ, additional AIP5 deletion further reduced the elongation of pseudohyphae significantly (Fig. 2, B and C), suggesting functional cooperation between Aip5 and the other polarisome components in hyphae elongation. In addition, we also evaluated C. albicans hyphae using the hydroxyurea (HU) induction system, which allows separation of filamentous hyphae to facilitate quantitative measurement of the polarized growth (32). Similarly, the removal of Aip5 lead to a higher rate of short hyphae in all tested genetic background (Fig. 2, B and C). The actin cables were also less visible in hyphae form once AIP5 was deleted (Fig. S2B), which is in agreement with the actin cable staining in their yeast forms (Fig. 1, D and E).
Figure 2.

Polarisome complex regulates hyphal formation in C. albicans. A, representative images of the indicated strain colonies with hyphae cells induced by solid agar spider plate at 37 °C. The images were shown with a magnification of ×40, the scale bar represents 0.1 cm. B, representative bright-field images of hyphal morphogenesis in polarisome mutants that were induced by 37 °C for 3 h in YPD medium. The scale bar represents 20 μm. C, quantification of hyphae length of indicated C. albicans strains, as shown in B, data are average from two independent experiments (WT: n = 76 cells; aip5Δ/Δ: n = 74 cells; bni1Δ/Δ: n = 81 cells; bni1Δ/Δ aip5Δ/Δ: n = 60 cells; spa2Δ/Δ: n = 72 cells; spa2Δ/Δ aip5Δ/Δ: n = 112 cells; bud6Δ/Δ: n = 63 cells; bud6Δ/Δ aip5Δ/Δ: n = 51 cells).
Aip5 directly interacts with Bni1 and Bud6 to stimulate actin assembly in vitro
To investigate the biochemical activities of C. albicans polarisome proteins, we first aligned the functional domains of ScBni1FH1C (residues 1227-1953 aa), ScBud6C (residues 498-789 aa), and ScAip5C (residues 1110-1234 aa) with the corresponding homologs of C. albicans (Fig. S3, A–C) (14, 33). All three polarisome proteins showed high sequence similarity within the folded active region between two fungal species. We first examined the physical interaction of Aip5C toward NP-actin, Bni1FH1C, and Bud6C using an anisotropy-binding assay. We purified recombinant Aip5C (residues 786-896 aa), Bni1FH1C (residues 1124-1727 aa), and Bud6C (residues 411-701 aa) from the bacteria system (Fig. 3A), and nonpolymerizable S. cerevisiae Act1 (NP-actin) from insect cell by introducing three mutations (D286A/V287A/D288A) (4, 34) (Fig. S4, A and B). Aip5C showed high affinity to NP-actin, Bni1FH1C, and Bud6C at KD of 205 ± 14 nm (n = 3), 126 ± 10 nm (n = 3), and 102 ± 13 nm (n = 3), respectively (Fig. 3, B–D). Although Bni1FH1C showed a dose-dependent function in actin polymerization (Fig. S4C), Bud6C on its own seemed to inhibit actin polymerization in a dose-dependent manner (Fig. S4D), which is consistent with the reported ScBud6C function (10). Surprisingly, Aip5C did not show obvious promotion in actin polymerization on its own (Fig. S4E), and displayed a marginal effect on either Bni1 or Bud6 for actin polymerization (Fig. S4, F and G). Bud6C enhanced Bni1FH1C-mediated actin polymerization (Fig. S4H), suggesting the possible NPF function like ScBud6 (6, 14, 35). Next, we combined all three actin-binding proteins of C. albicans for the actin polymerization assay. In the mixture of Bud6C and Bni1FH1C, a supplement of Aip5C exhibited synergy in nucleating the actin cable by increasing the initial assembly rate in a dose-dependent manner (Fig. 3, E and F). Such an overall increase in actin nucleation was unlikely due to actin severing, such as the activity of cofilin-enhanced actin polymerization by generating more free barbed-ends for actin polymerization (36). We did not observe obvious actin depolymerization from the tested protein combinations (Fig. S4I).
Figure 3.

Aip5C directly interacts with Bni1FH1C and Bud6C proteins. A, SDS-PAGE gel of purified proteins Aip5C, Bni1FH1C, and Bud6C. B–D, fluorescence anisotropy binding measurements of Alexa 488-labeled 30 nm Aip5C that was titrated by NP-actin, Bni1FH1C, and Bud6C. Data are represented by circles using the average value of three biological replicates and fitted by Hill equation to determine the KD. E, pyrene-actin polymerization by an increasing concentration of Aip5C in combination with Bni1FH1C and Bud6C. F, relative actin assembly rates through Aip5C titration on Bni1FH1C protein or in combination with Bud6C. At least three biological replicates were shown with an error bar of mean ± S.E.
We next sought to differentiate the actin nucleation from elongation using total internal reflection fluorescence (TIRF)-based actin assembly assay. The number of actin seeds was quantitatively measured during initial polymerization to determine the nucleation activities. Although 20 nm Bni1FH1C increased actin nucleation, neither Aip5C nor Bud6C enhanced initial actin nucleation noticeably on their own at a concentration of 10 nm (Fig. 4, A and B). However, both 50 nm Aip5C and 10 nm Bud6C could promote Bni1FH1C-mediated actin nucleation by ∼1.4-1.5–fold, respectively (Fig. 4, A and B), indicating their NPF activities for Bni1. Strikingly, once Bni1FH1C was incubated with both Bud6C and Aip5C, the Bni1-mediated actin nucleation was enhanced by ∼2.6-fold at the 5-min time point of polymerization, evidence of synergy in actin nucleation (Fig. S4, A and B). However, neither Aip5C nor Bud6C affected the barbed-end elongation in the presence of Bni1FH1C (Fig. S5, A and B). We next tested whether the Aip5 and Bni1 interaction could influence the association between Bni1FH1C and F-actin barbed-end. We performed an end-to-end F-actin annealing assay by mixing short F-actin filaments that were labeled by distinguishable fluorescent phalloidin. Although Bni1 impairs actin annealing, additional Aip5C significantly elevated such inhibitory effect by reducing the length of F-actin fragments from 4.3 ± 2.0 μm (Bni1FH1C) to 3.3 ± 1.2 μm (Bni1FH1C + Aip5C) (Fig. 4, C and D). However, Aip5C does not affect F-actin annealing directly (Fig. 4, C and D). Among different combinations, the protein mix of Aip5-Bud6-Bni1 yielded the shortest actin filament length of 2.2 ± 0.8 μm (Fig. 4, C and D). Because Bni1 was found to associate with barbed-end earlier through an immunoelectron microscopy experiment (33), the annealing assay results suggest a stronger barbed-end association with Bni1FH1C in the presence of Aip5, Bud6, or both.
Figure 4.

Polarisome proteins synergistically promote actin filament nucleation activity in vitro. A, the representative TIRF images of actin nucleation seeds formed at 5 and 10 min, respectively. The control actin filament was assembled from 0.5 μm actin (10% Oregon Green 488-labeled actin and 0.5% biotin-actin), and the used proteins were indicated as follows: 20 nm Bni1FH1C, 50 nm Aip5C, and 10 nm Bud6C. The scale bar represents 5 μm. B, quantification of actin nucleation seeds number at 5 min with the indicated protein combination and average seeds number (n = 45 for each sample from regions of interest = 64 µm2). C, representative merged fluorescence images from GFP and RFP excitation channels of annealed actin filaments. 250 nm sheared-actin filaments (rhodamine-phalloidin labeled) were mixed with 250 nm sheared-actin filaments (Alexa 488-phalloidin labeled) and annealed for 60 min at room temperature. 400 nm of each indicated protein was used. The scale bar represents 5 μm. D, quantification of annealed actin filament length at 60 min of the indicated protein combinations (n = 100 filaments for each sample). p values were determined by the one-way analysis of variance, ns, not significant; **, p < 0.01; ****, p < 0.0001. Error bar, S.D.
Aip5C shares a similar structural conformation as ScAip5C
To better understand the function of Aip5C, we were motivated to resolve the crystal structure of Aip5C. Using analytical gel filtration, we found that Aip5C has an equivalent elution volume (12 ml) to its homolog ScAip5C (4), implying dimer formation for both proteins (Fig. 5A). Indeed, the calculated molecular mass (25 kDa) of Aip5C suggests a dimeric state in solution when compared with its theoretical molecular mass (12.8 kDa) (Fig. 5A). Consistently, the resolved structure of Aip5C, determined through molecular replacement using the ScAip5C (PDB code 6ABR) structure as the search model (Table 1), also demonstrates that two molecules within the crystal lattice form a dimer, where two protomers (assigned A and B) showed high similarity (Fig. 5, B and C). The overall conformation of Aip5C and ScAip5C is highly similar, except for only a slight twist observed for Aip5C protomer B while aligning both protomers A (Fig. 5D). However, the alignment of individual protomers between Aip5C and ScAip5C highlighted three differences. First, the functional loop domains of Aip5C were present in both protomers, whereas the ScAip5C only showed one stable loop domain (Fig. 5, E and F), which might explain the difference in binding affinity to G-actin. On each of the protomers of the dimer, ScAip5C has one flexible loop and one stable loop, respectively, which directly interacts with G-actin and is essential for its activity in actin polymerization (4). The corresponding loop regions in both protomers of Aip5C showed clear electron density, suggesting the relatively stable conformation for both protomers (Fig. 5C). Perhaps such diverse structural features of ScAip5C and Aip5C could help us to rationalize our biochemical data, where Aip5C shows stronger binding to G-actin with KD ∼200 nm, whereas ScAip5C binds to G-actin with KD ∼1.5 μm (4). Second, the loop in Aip5C was shortened by a missing glycine (G), Gly-818 (Fig. S6A). Then we engineered Aip5C by inserting a glycine at the equivalent position of ScAip5C, namely Aip5C-G (Fig. S6B). However, the recombinant Aip5C-G protein was still unable to show detectable activity in actin polymerization on its own (Fig. S6, B and C), showing a negative correlation between Gly-818 of Aip5 and the nucleation activity. The third difference lies in the N terminus, which folded into an α-helix in Aip5C but remained as a loop in ScAip5C (Fig. 5E). However, such a difference of the N-terminal overhang is also less likely the reason for the missing activity of Aip5C in actin nucleation. Removal of the N-terminal sequence of ScAip5C did not abolish its nucleation activities, and the monomeric form of ScAip5-mC with a shorter N terminus was still able to promote actin nucleation on its own and ScBni1-mediated actin nucleation (4).
Figure 5.

Dimer formation of Aip5C and its structural comparison with the homolog ScAip5C. A, the elution profile of Aip5C and protein standards from calibrated Superdex 75 10/300 GL. The calibrated molecular masses of Aip5C are indicated as 25 kDa, suggesting that Aip5C forms a dimer in solution. B, crystal structure of Aip5C. Two molecules (protomers A & B) in an asymmetric unit were crystallized as a dimer. Protomer A and protomer B are highlighted in blue and yellow, respectively. C, alignment of protomer A with protomer B in Aip5C. The functional loop domains are present in both protomers. D, overall alignment of crystal structures of Aip5C with its budding yeast homolog ScAip5C. E, alignment of protomer A of Aip5C with that of ScAip5C, where a red box highlights the functional loop domain. The difference of the N-terminal region was highlighted by a black box, where Aip5C formed a α-helix, and ScAip5C was a loop structure. F, alignment of protomer B of Aip5C with that of ScAip5C, where a red box highlights the functional loop domain.
Table 1.
X-ray data and statistics
| Aip5C refinement statistics | |
|---|---|
| PDB code | 6M4C |
| Data collection | AS MX 2 |
| Space group | P6122 |
| Cell dimensions | |
| a (Å) | 95.116 |
| b (Å) | 95.116 |
| c (Å) | 74.119 |
| α (°) | 90.0 |
| β (°) | 90.0 |
| γ (°) | 120.0 |
| Resolution (Å) | 50.0-2.64 (2.80-2.64) |
| CC1/2 (%) | 100 (94.9) |
| <I/σ> | 37.83 (2.29) |
| Unique reflection | 6,164 (963) |
| Redundancy | 35.09 (34.42) |
| Completeness (%) | 100 (99.9) |
| Refinement | |
| Resolution (Å) | 41.19-2.65 (2.745-2.65) |
| No. reflections | 6,121 (586) |
| Rwork/Rfree (%) | 25.85/30.57 |
| R.m.s. deviation bonds/angle | 0.008/1.76 |
| Number of atoms | |
| Protein | 729 |
| Water | 15 |
| B factors | 30.50 |
| Ramachandran | |
| Favored (%) | 86.46% |
| Allowed (%) | 9.38% |
| Outlier (%) | 4.17% |
Molecular dynamics simulations reveal complex Bni1C-Bud6C-Aip5C organization
C-terminal IDR of ScBni1 (residues 1767-1953 aa) associates with ScAip5C directly, which is essential for NF-NPF pairing (4). Similarly, we found that Aip5C directly interacts with Bni1C (residues 1545-1733 aa) at a KD of 1.12 ± 0.34 μm in a fluorescence anisotropy assay (Fig. 6A and Fig. S7A). In addition, Bni1C also showed a high affinity to Bud6C with a KD ∼ 0.56 μm (Fig. 6B), similar to the intermolecular interaction of ScBud6C and ScBni1FH1C via the C-terminal tail region of ScBni1 (14). The flexible nature of intrinsically disordered Bni1C makes it challenging to study the assembly of the Bni1C-Bud6C-Aip5C tri-protein complex via crystallographic approaches. To understand the dynamic association of the members in a protein complex and their orchestrated functions, we next used all-atom MD simulations to identify the conformations with the lowest free energy of such an NF-NPF complex core. Bni1C was predicted to be intrinsically disordered (Fig. S7B) through the online software IUPRED2A (37), we assigned its initial structure as a random coil (Fig. S7C). Although there was no resolved crystal structure for Bud6C, we predicted it through I-TASSER (38) based on its homolog ScBud6core crystal structure (PDB code 3ONX) and the corresponding Bud6 protein sequence (Fig. S7D). The initial structure of Aip5C was resolved by crystallography (Fig. 5B). To avoid the bias from using a particular fixed initial structure for the protein complex in MD simulations, we randomly positioned the complex proteins in eight spatial combinations, where Bud6C and Aip5C were assigned at different positions relative to Bni1C, symmetrically or asymmetrically (simulations 1-8 in Fig. S7, E–L). After running the MD simulations for the eight initial structures for 200 ns, only six simulations were able to converge as shown by root mean square deviation (r.m.s. deviation) plots (Fig. S8A), suggesting a relatively stable complex conformation as a result of the simulation. We next determined the representative conformations of the tri-protein complex by choosing the lowest free energy conformation of each of the six simulation trajectories using principal component analysis (Fig. S8B). To further compare the relative stabilities of the six low free energy conformations, we calculated the scores of the conformations (Fig. S8C) by using the software Rosetta (39), where Rosetta uses an optimized energy function or score function to calculate the energy of all atomic interactions within the protein complex. Compared with other simulations with different initial structures, the low free energy conformation of simulation 1 showed the lowest Rosetta score, suggesting it is the most stable complex conformation calculated from the six selected conformations (Fig. 6C and Fig. S8D). Given the confirmed intramolecular interactions among all three constituents in the trip-protein complex Bni1-Bud6-Aip5, the next stable conformations are from simulations 4 and 5, in which relatively close contacts between Aip5 and Bud6 were retained but having much less favorable states than the simulation 1 conformation from Rosetta scoring (Fig. S8D). The simulation 1 conformation demonstrated that Bud6C preferentially associated with the N terminus (residues 1-35 aa) and middle region (residues 111-151 aa) of Bni1C, which interestingly avoided the favorable contacts between Aip5C and Bni1C at the C terminus (residues 150-188 aa) (Fig. 6D).
Figure 6.
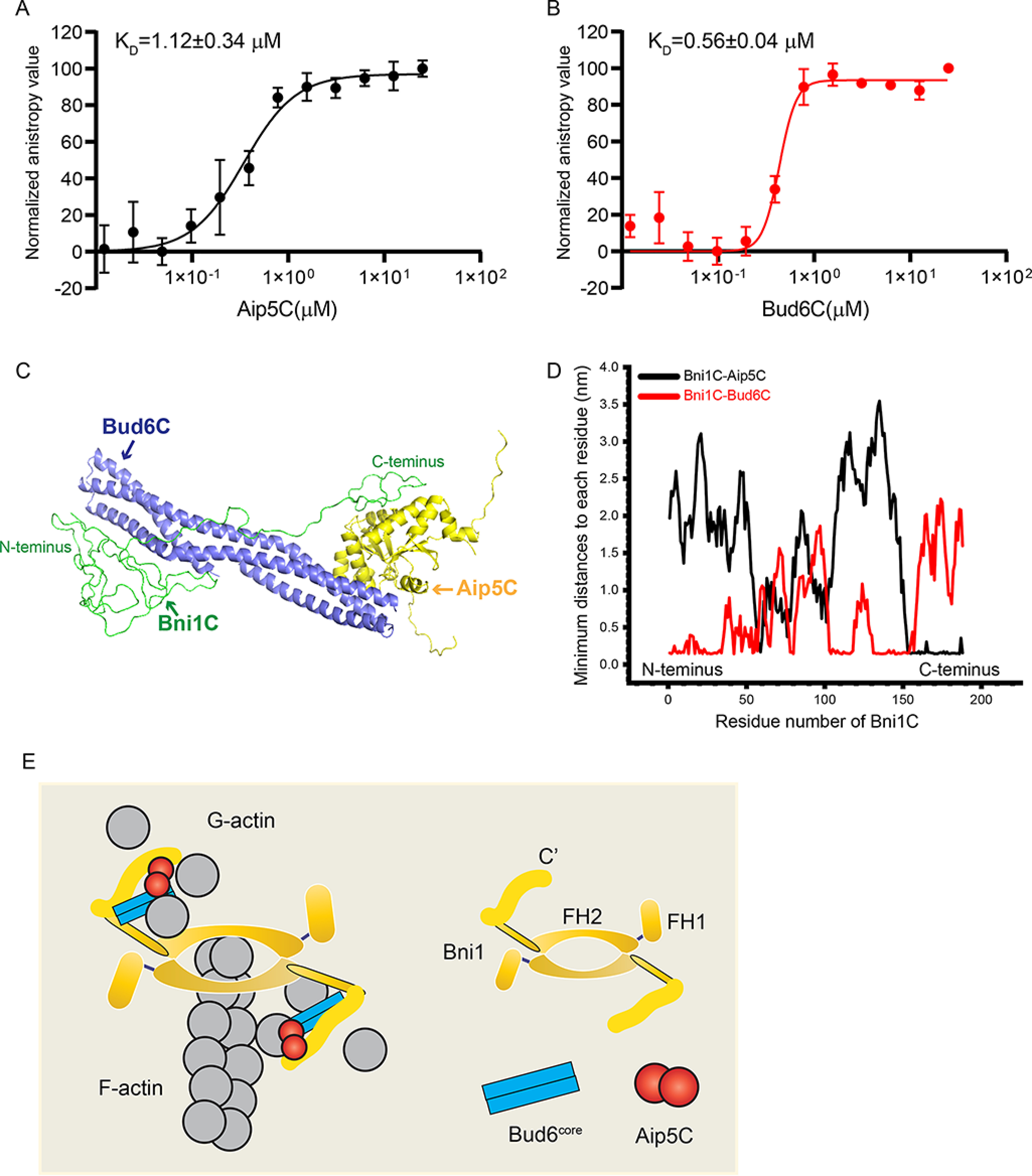
The complex organization of Bni1C-Bud6C-Aip5C. A, fluorescence anisotropy binding measurements of Alexa 488-labeled 30 nm Bni1C was titrated by a serial concentration of Aip5C. Data are represented by circles using the average value of three biological replicates and fitted by Hill equation to determine the KD with an error bar of mean ± S.E. B, fluorescence anisotropy binding measurements of Alexa 488-labelled 30 nm Bni1C was titrated by Bud6C. Data are represented by circles using the average value of three biological replicates and fitted by Hill equation to determine the KD with an error bar of mean ± S.E. C, the most stable complex conformation (simulation 1) among the eight MD simulations, where three proteins form stable protein-protein contact interfaces. Bni1C, Aip5C, and Bud6C are shown in green, yellow, and blue, respectively. D, the minimal distance plot of each residue between Bni1C and Aip5C (black)/Bud6C (red) in the final stable complex conformation, as shown in C. E, schematic model of actin nucleation from tri-protein complex Bni1-Aip5-Bud6.
Discussion
Numerous species of pathogenic fungi of mammals and plants undergo the dimorphic transition from yeast to hyphal growth during pathogenesis (26, 40–42). During infection, the hyphae of filamentous fungi locate predominately at the infection zone, whereas yeast stays more at the epithelial surface (43). Multiple signaling pathways were involved in morphological initiation of the hyphae, such as transcriptional rewiring, cell cycle signaling, vesicular transport, and cytoskeleton remodeling (17, 26, 32, 44). The polarized hyphal growth requires rapid membrane exchange and material deposition at the elongating zone, which is coordinated by the actin cables assembled by the polarisome complex at the budding tip. Appropriate assembly of the macromolecular complex of polarisome is critical to maintain the nucleation center at the elongating tip or adapt to environmental stresses by forming intracellular condensates. Orchestrated and reliable assembly of polarisome complex within the confined zone is critical for its activity in actin assembly under different cellular conditions.
The ribosome is an example of a flexible and orchestrated complex assembly in which ribosome proteins may have diverse participatory proteins and different manners in construction. However, the core ribosomal proteins are mainly the same (45, 46). Polarisome protein components bind with each other by inter- and intramolecular interactions via both folded domains and IDRs that drive molecular condensation of polarisome complex in vivo. The folded domains of each actin nucleation factors assemble into the functional core via high affinity. Flexible interactions result in tunable complex assembly and changes of surface physical chemistry by tuning the participation rates of the constituents into protein complex, the stoichiometry and affinity of subunits, the local protein concentration, diffusion rate, and possibly the biochemical activities, such as polarisome in actin nucleation (4). Spa2 is the master scaffolder of polarisome complex to recruit other actin-binding proteins members (4, 47, 48), Bni1, Bud6, and Aip5, in yeast and filamentous fungi. By condensing the polarisome proteins at the elongating tip with the crowding environment, the local concentration of actin monomers and actin-binding proteins are enriched near the filament ends in coordinating the efficient actin polymerization for hyphal growth. The condensation level of polarisome components changes dynamically from apical to isotropic growth throughout the cell cycle, as well as during re-localization to adapt to acute changes in cellular environments (4, 49, 50). During cell cycle progression from G1 to metaphase in budding yeast, polarisome complex switch from a condensed-assembly at the tip to a loose-packing as a surface crescent (4, 5, 51, 52). During hyphal growth, upon perceiving signals for dimorphic transition, the finetuning of condensation of the polarisome complex at the polarized tip is critical for the initiation and maintenance of fast hyphal elongation.
To better understand the dynamic activities in actin polymerization by polarisome, a critical step is to establish the working model of the functional core that drives actin nucleation. Here, we demonstrated a synergism of Bni1, Bud6, and Aip5 that form a tri-protein complex in generating actin NF-NPFs nucleation center for the polarized cell growth of C. albicans. Aip5C directly interacted with Bni1FH1C, Bud6C, and monomeric actin to synergize the Bni1-mediated actin nucleation. Consistently, in vivo multicomponent synergies were also collectively demonstrated, in both S. cerevisiae and C. albicans (4, 15). Here, the genetic studies showed that Aip5 contributed significantly to the hyphal growth of C. albicans by interacting with other polarisome proteins. The double mutants of aip5Δ/Δ showed higher sensitivity to actin perturbation by having a slower growth assay and a more disrupted actin cables in vivo. Using the C-terminal folded domain of Aip5, we have noticed a few points of divergence between the S. cerevisiae and C. albicans homologs. Aip5C showed much lower activity in promoting Bni1-mediated actin nucleation on its own and does not exhibit apparent actin-nucleation activity, which is different from ScAip5C. Instead, Aip5C demonstrated important roles for the teamwork in actin assembly by displaying pronounced promotion in actin nucleation for the Bni1-Bud6 complex. Such discrepancy in actin nucleation activity between Aip5C and ScAip5C is less likely due to the conformational difference because both of them showed high similarity with an r.m.s. deviation score of 2.676. Aip5C showed a higher affinity to G-actin than the ScAip5C, which might constrain the assembly of the actin filament as one plausible reason. The similar negative correlations between the efficacy in actin assembly and the affinity to G-actin were also reported before, in which a weak association with G-actin could be beneficial in maintaining dynamic F-actin treadmilling by having efficient disassociation. For example, G-actin–binding protein profilin requires rapid dissociation from the actin barbed-end and the polyproline region of formin FH1 domain (53–55). Nevertheless, genetic and biochemical assays both evident the physiological-relevant multicomponent synergy for Aip5, Bni1, and Bud6, in promoting actin assembly and polarized hyphal growth.
Here, our biochemistry experiments reconstituted the interactions and functions of the polarisome core in nucleating actin filaments. We also proposed a potential interactive mode between three components in the complex NF-NPFs core using all-atom molecular dynamics simulations (Fig. 6C). Our structural simulation and the evaluation of the energy landscape of the tri-protein complex revealed an assembly strategy of the nucleation core with compatible spatial positioning in polarisome based on the equilibrium thermal physics (Fig. 6E). Due to the nature of the tunable potential inter- or intra-molecular associations that are mediated by IDR of Bni1C, our simulation would not suggest the explicit conformation and interfaces of the packed complex, but rather an understanding of complex construction regarding the energy landscape. The biochemical assay using only the C terminus of Aip5 and Bud6 suggests that Aip5C may promote actin nucleation in a Bud6C-dependent manner, which is different from the activities of ScAip5. However, the double mutant bud6Δ/Δ aip5Δ/Δ demonstrated much more defects in cell growth and actin cable structures than the single mutants, suggesting either Aip5 has the additional function in actin assembly independent of Bud6, or Aip5 and Bud6 form multivalent interactions in vivo, which enhanced the protein connectivity and thereby activity drastically. It remains elusive how different regions of Aip5, Bud6, Bni1, and Spa2 create diverse assembly and packing modes of the biochemical core of the nucleation complex. To understand how IDRs coordinate the folded domains in the tri-protein complex in actin nucleation, in vitro reconstitution by having all full-length proteins would eventually facilitate our understanding of the dynamic complex function. Future co-crystallization or cryogenic EM (cryo-EM) of the core region of the tri-protein complex could also guide a refined simulation in viewing the dynamic complex formation with full-length proteins, which will be exceptionally technically challenging. Here, our work will guide future microscale simulation, via integrating coarse-grained modeling, toward the understanding of the macromolecular assembly of the polarisome IDR-containing members.
Materials and methods
Strains and media
C. albicans yeast cells were routinely cultured in YPD (10 g/liter of yeast extract, 20 g/liter of peptone, 20 g/liter of glucose) medium or 2% agar plates at 30 °C. Yeast strains used in this study are listed in Table S1. For expression and purification of recombinant proteins, E. coli BL21(DE3) Rosetta T1R bacteria was cultured with TB medium (24 g/liter of yeast extract, 20 g/liter of tryptone, 4 ml/liter of glycerol, phosphate buffer, pH 7.4).
Construction of aip5Δ/Δ mutant strains
The CRISPR/Cas9 system used to generate mutant strains was adapted from Gerald R. Fink laboratory Solo system (27). A pair of guide primers (SgAIP5-F, SgAIP5-R) were designed to modify pV1093, which can direct the guide RNA to the chosen PAM sites for mutagenesis. A pair of repair primers (RtAIP5-F, RtAIP5-R) were designed to disrupt the PAM sequence and introduced a stop codon as well as BamHI site for verification. The new construct pV1093-AIP5 was then linearized by digesting with KpnI and SacI, which were transformed into C. albicans yeast cells together with the 3 µg of repairing primers as previously described (56). The transformants were selected by Nourseothricin at a concentration of 200 µg/ml on YPD plates. Furthermore, a pair of verification primers (VerAIP5-F, VerAIP5-R) were designed to amplify the 520-bp fragment covering the mutagenesis sequence, where amplified DNA fragments from the positive mutagenesis colonies can be digested by BamHI enzyme into two fragments, corresponding to sizes 76 and 444 bp. All the primers used in this study are listed in Table S2.
Cell growth
Induction of filamentous growth in C. albicans
To induce hyphae in the liquid culture, the yeast strains were streaked out freshly on YPD agar plate, cultured overnight in YPD liquid medium at 30 °C until saturation. Usually, the A600 is able to reach 10-15 the next morning. The saturated cultures were diluted ×100 into fresh YPD medium (A600 around 0.1) containing 10% fetal bovine serum, and 800 μl of culture was applied to the imaging chambers with concanavalin A pre-treated coverslip to attach the yeast cells. Afterward, the imaging chambers were kept at 37 °C for 3 h to induce hyphal growth, and were subjected to the microscopic with ×63 objective lens for morphology analysis. To induce filamentous hyphal growth by HU, the saturated overnight culture of yeast cells were re-inoculated from A600 0.2 until log growth phase (A600 around 0.5-0.8) in YPD medium at 30 °C, a final concentration of 0.2 mm HU was added and grown for an additional 5 h in YPD medium at 30 °C. The filamentous cells were then fixed by 4% formaldehyde. To induce the hyphae growth on a spider plate (10 g/liter of yeast extract, 20 g/liter of peptone, 20 g/liter of glucose, 1% mannitol, 0.2% K2HPO4, and 1.5% agar), the saturated overnight culture of yeast cells were diluted to A600 = 1 and a 10-fold serial dilution was prepared with fresh YPD. Afterward, 4-μl aliquots of undiluted or diluted cells were spotted on the spider plate and kept at 37 °C for 72 h before scanning.
Cell growth liquid assay with LatA
The overnight culture of yeast cells was re-inoculated into fresh YPD medium starting from A600 = 0.2 and allowed to grow to A600 around 0.8 to 1. Subsequently, 130 µl of cultures were added into a transparent 96-well–plate with or without 1 μm latrunculin A. Each condition was replicated four times and monitored at A600 every 15 min at 25 °C. A constant shaking was maintained between each measurement to prevent cell precipitation. The A600 was recorded by the TECAN m200pro plate reader machine for 25 h. To quantify the growth rate of various conditions, the exported original data from Excel was used, and the growth curve was generated in Prism GraphPad 6. The cells in a A600 range of 0.5-1.0 were selected and applied with an exponential equation to analyze the growth rate (57).
Protein expression and purification
Aip5C, Bni1FH1C, Bud6C, Aip5C-G, and Bni1C were expressed in E. coli (BL21(DE3) Rosetta T1R). Cells were cultured in 5 ml of TB medium overnight and transferred to 1 liter of TB medium for 4-5 h at 37 °C. Afterward, 0.5 mm isopropyl thio-β-d-galactoside was added for protein induction at 18 °C for overnight culture. The cells were harvested the next morning and resuspended into 50 ml of lysis buffer (20 mm Hepes, pH 7.4, 500 mm NaCl, 10 mm imidazole) with 1 mm phenylmethylsulfonyl fluoride and 1 tablet of Pierce™ protease inhibitor (ThermoFisher). The cells were sonicated for 5 min (10 s on, 30 s off), and the resulting lysate further clarified by centrifugation at 40,000 × g, 1 h at 4 °C. The supernatant was filtered by 0.22-μm Minisart® Syringe Filter before loading into a 5-ml HisTrap HF column (GE Healthcare Life Sciences) connected to the FPLC system (GE ÄKTA FPLC™). The protein was eluted over a gradient injection of 500 mm imidazole. The collected protein peak fractions were examined by the SDS-PAGE and followed by GelCode™ Blue Stain (ThermoFisher Scientific). The pooled fractions were subjected to tobacco etch virus protease cleavage with buffer exchanging through dialysis against 20 mm Hepes (pH 7.4), 500 mm NaCl, 10 mm imidazole for 16 h at 4 °C. The dialyzed protein was injected into a 1-ml HisTrap HF column (GE Healthcare Life Sciences) to separate the untagged proteins from the free His tag. Afterward, the protein was further purified by size-exclusion chromatography using a HiLoad 16/600 Superdex200 column (GE Healthcare) in 20 mm Hepes (pH 7.4), 500 mm NaCl. The target protein was concentrated and aliquoted to 5-10 µl for each tube and frozen in liquid N2.
Rabbit muscle actin was purified as previously described (4). 2 g of rabbit muscle powder (PelFreez) was dissolved in 60 ml of G-buffer (5 mm Tris-HCl, pH 7.5, 0.5 mm DTT, 0.2 mm ATP, and 0.1 mm CaCl2) on ice for 30 min, and the supernatant was collected by filtering against cheesecloth. The crude actin extraction procedure was repeated three times. The obtained crude extracts were subjected to centrifugation at 18,000 × g at 4 °C for 30 min. The supernatant was transferred out for actin polymerization at 4 °C for 1 h with the supplementation of 50 mm KCl and 2 mm MgCl2. To further remove other F-actin–associated proteins, 0.8 m KCl powder was added to the solution and slowly stirred at 4 °C for 30 min. Afterward, the F-actin was pelleted by ultracentrifugation at 96,000 × g at 4 °C for 3 h. F-actin was solubilized by mild sonication (3 s on, 10 s off, repeated 4 times) and then homogenized using a Dounce homogenizer. To fully depolymerize the F-actin, the re-suspended actin was dialyzed 2-3 times against G-buffer. Furthermore, the G-actin was pre-washed by ultracentrifugation at 150,000 × g at 4 °C for 2.5 h. Two-thirds of the G-actin supernatant was collected and injected to gel filtration column Sephacryl S-300 HR for further purification. Peak actin-containing fractions were supplemented with 0.1% sodium azide and stored at 4 °C. To label the actin with either Biotin or Oregon Green dye, the F-actin pellet was sonicated and suspended using a Dounce homogenizer against G-buffer lacking DTT. The solubilized actin was dialyzed against DTT-lacking G-buffer overnight. Then the G-actin was diluted to 46 μm and polymerized by adding an equal volume of 2× labeling buffer (50 mm imidazole, pH 7.5, 200 mm KCl, 0.3 mm ATP, and 4 mm MgCl2) and gently mixed for 5 min. Subsequently, actin was mixed with a 10-fold molar excess of either NHS-dPEG®4-biotin (Sigma) or Oregon Green™ 488 iodoacetamide (ThermoFisher), which were re-suspended in anhydrous dimethylformamide, and incubated in the dark for 16 h at 4 °C. The labeled F-actin was pelleted as described before and solubilized by sonication and homogenized. The further depolymerization of F-actin was carried out as delineated above. In addition, the labeled G-actin was centrifuged at 150,000 × g at 4 °C for 2.5 h. The supernatant was purified by a Sephacryl S-300 HR gel filtration column. The actin containing fractions were dialyzed against G-buffer with 50% glycerol overnight to reduce the volume for storage in −80 °C.
Nonpolymerizable yeast actin expression and purification
Nonpolymerizable (NP)-actin protein was purified from insect cells as previously reported, in which three point-mutations at the barbed-end of Drosophila melanogaster 5C actin (D287A, V288A, D289A) were introduced (34). Using a similar strategy, we generated nonpolymerizable S. cerevisiae Act1 actin via mutagenesis of the homologous residues (D286A, V287A, D288A). Ligation-independent cloning was used to insert the PCR amplified gene containing S. cerevisiae NP-actin into the pFB-LIC-Bse cloning vector that includes an L21 enhancer sequence (58) driving expression of the gene. The cloned plasmid was then transformed into MAX EfficiencyTM DH10Bac E. coli cells (ThermoFisher), and successful transformants were selected using Blue-White screening. Bacmid extracted from DH10Bac was transfected into Sf9 cells using Cellfectin® II Reagent from ThermoFisher. Baculovirus generated from the transfection was further amplified for two passages, and the resulting P2 virus was used to infect Sf9 cells at 27 °C for 72 h. The Sf9 cells were resuspended in lysis buffer containing 20 mm Tris (pH 8.0), 50 mm KCl, 0.1 mm CaCl2, 0.2 mm ATP, and 2 mm β-mercaptoethanol and disrupted using SONICS Vibra-CellTM ultrasonic liquid processor. The lysate was clarified via centrifugation and filtration. Purification of NP-actin was performed at 4 °C by passing the crude lysate through nickel-nitrilotriacetic acid affinity column (GE Healthcare) and eluted with a gradient increase of imidazole from 20 to 500 mm. Additional purification was performed by gel filtration (HiLoad Superdex 200 16/60, GE Healthcare) using the same lysis buffer. The purified NP-actin was concentrated and aliquoted to 5-10 µl each tube and frozen in liquid N2.
TIRF microscope
To visualize the actin filament growth via TIRF microscope, the 24 × 60-mm coverslips were cleaned with 20% H2SO4 for overnight and rinsed thoroughly in sterile water before coating with 2 mg/ml of methoxy-polyethylene glycol-silane, and 2 µg/ml of biotin-polyethylene glycol-silane (Laysan Bio Inc.) in 80% ethanol (pH 2.0). The reaction was carried out at 70 °C overnight. Afterward, the coverslips were rinsed thoroughly by sterile water before drying by N2 gas. The functionalized coverslips were kept at −80 °C for long term storage. The flow cell chamber was assembled every time before use, where the coated coverslip can adhere to the sticky-Slide VI 0.4 (ibidi, Germany). Before each experiment, the flow cell was incubated with buffer HBSA (20 mm Hepes, pH 7.5, 1 mm EDTA, 50 mm KCl, and 1% BSA) for 30 s and washed by buffer HEKG10 (20 mm Hepes, pH 7.5, 1 mm EDTA, 50 mm KCl, and 10% glycerol) twice and followed by a 60-s incubation with 0.1 mg/ml of streptavidin in HEKG10 buffer. Then the flow cell was washed three times by 1× TIRF buffer (10 mm imidazole, 50 mm KCl, 1 mm MgCl2, 1 mm EGTA, 0.2 mm ATP, 10 mm DTT, 15 mm glucose, 20 µg/ml of glucose oxidase, and 0.5% methylcellulose (4000 cP), pH 7.4). Then 0.5 μm actin (10% Oregon Green labeled, 0.5% biotin labeled) with or without its binding partners were mixed with an equal volume of 2× TIRF buffer and applied to the flow cells for imaging. The still images were acquired at 4-5 min for quantifying actin nucleation seed numbers. 5-s interval movies were acquired for 10 min using Apochromat TIRF ×100 NA 1.49 (Nikon Instruments) on a Nikon ECLIPSE Ti-S inverted microscope with iLAS2 motorized TIRF illuminator (Roper Scientific, Evry Cedex, France). The illumination source, microscope stage, and a Prime 95B sCMOS camera (Photometrics) were all under the control of MetaMorph 7.8 software (Molecular Device, Sunnyvale, CA). The focus was maintained using the Perfect Focus System. To quantify the actin filament elongation rate, the individual filament in each sample was traced manually for at least 2 min each. The measured actin filament length was then divided by the corresponding time to determine the elongation rate. We used the conversion factor of 370 subunits per micrometer of F-actin to estimate the barbed-end elongation rate.
Bulk actin assembly assay
Kinetics of pyrene-actin assembly was measured as described (4). 10 μm G-actin (5% pyrene actin, Cytoskeleton Inc.) was first converted to Mg2+-ATP-actin for 5 min on ice and then mixed rapidly with various proteins in the G-buffer. To initiate the bulk actin polymerization, 10× KME buffer mix (10 mm MgCl2, 10 mm EGTA, and 500 mm KCl) was added to the 120-µl reaction and the pyrene-actin fluorescence signal was immediately recorded by the Cytation 5 plate reader (BioTek, USA) at excitation and emission wavelengths of 365 and 407 nm, respectively. To quantify the relative actin assembly rate as previously described (4), the raw data from 60 to 180 s were used to fit into a linear function where the R2 value was required to be greater than 0.95. The slope of the fitted curve represents the actin assembly rate of each condition; the relative actin assembly rate was derived by dividing each slope of the tested condition by the slope of the control actin curve. The values shown in the graph were averaged data from at least three independent experiments. The actin depolymerization assay was prepared by polymerizing 5 μm G-actin (30% pyrene actin) at room temperature for 2 h. To initiate the actin depolymerization, F-actin was diluted to 0.1 μm by F-buffer (5 mm Tris, pH 8.0, 0.2 mm ATP, 0.1 mm CaCl2, 0.5 mm DTT, 50 mm KCl, 1 mm MgCl2, and 1 mm EGTA) with its binding partners added. The decreased pyrene-actin fluorescence signal was immediately recorded by the Cytation 5 plate reader at excitation and emission wavelengths of 365 and 407 nm, respectively. The actin polymerization graphs were plotted using GraphPad Prism 6.
Actin filament annealing assay
The actin filament annealing assay was performed by polymerizing 4 μm actin in the presence of 6 μm of either acti-stainTM 488 phalloidin or rhodamine-phalloidin for 2 h. Equal volumes of phalloidin actin filaments (final concentration of 250 nm) were sheared and mixed through a gauge 27½ needle 20 times, followed by incubation with the desired proteins at room temperature for 1 h for annealing. The reactions were quenched by dilution of 250-fold with F-buffer and applied on the polylysine (0.01%)-coated cover glass for imaging using a ×100 oil objective lens under the fluorescence microscope. The annealed actin filaments were traced by line tool to quantify the length by ImageJ.
Fixed cell imaging
The yeast phase cells were grown to mid-log phase in YPD medium at 30 °C, and filamentous cells were induced by 0.2 mm HU in YPD medium at 30 °C for 5 h. 4% formaldehyde was added to the medium to fix cells. The cells were then collected by centrifugation and re-suspended in PBS medium with 4% formaldehyde to fix at room temperature for 1 h. After fixation, the cells were washed twice with PBS (pH 7.2) and re-suspended in 45 µl of PBS (pH 7.2) with 5 µl of actin-stainTM 488 phalloidin (Cytoskeleton, Inc.) and 0.1% Triton X-100 (Bio-Rad). The cells were stained in the dark for 1.5 h at room temperature and vortexed every 15 min to allow efficient staining. Afterward, the cells were washed twice by PBS (pH 7.2) before mounting to the glass slide for imaging acquisition. The whole-cell image was acquired through Z scanning with 250 nm stepwise by objective lens ×100 NA 1.4 on Leica DMi8 microscope equipped with a scientific CMOS camera ORCA-Flash4.0 LT (Hamamatsu, Japan). The fluorescence microscope was controlled by MetaMorph 7.8 software (Molecular Device, Sunnyvale, CA). Images were analyzed using ImageJ to generate the Z projection images, where the actin phenotypes were classified into three groups.
Crystallization
Crystallization trials of Aip5C were performed at 20 °C using crystal screening kits purchased from Hampton Research. Crystallization conditions that yielded the best diffracting Aip5C crystals were selected and optimized via a sitting drop vapor diffusion method with reservoir conditions containing 0.1 m Tris (pH 7.4), 0.2 m calcium acetate, 12.5% PEG 3000. Aip5C crystals appeared 24 h after optimization. All Aip5C crystals were soaked with cryoprotectant solution containing 0.1 m Tris (pH 7.4), 0.2 m calcium acetate, 12.5% PEG 3000, 20% glycerol and flash-frozen using liquid nitrogen before X-ray diffraction experiments. All diffraction data were collected at the Australian Synchrotron beamline MX2 and the data were processed using X-ray Detector Software (XDS) (59). Aip5C structure was determined by molecular replacement (Phenix) (60, 61) using the ScAip5C (PDB code 6ABR) (4) as a search model. The molecular structure of Aip5C was refined using the LORESTR pipeline (62) from the ccp4 program suite. Final refinement statistics are shown in Table 1. All figures of the molecular structures were prepared using PyMOL (RRID:SCR_00305, Schrödinger, L.L.C).
Characterization of Aip5C protein by size exclusion chromatography
To quantify the Aip5C solution molecular weight, 100 µl of 2 mg/ml of purified Aip5C was injected into a Superdex 75 10/300 GL (GE Healthcare) calibrated column in buffer containing 20 mm Hepes (pH 7.5), 150 mm NaCl. The Superdex column was first calibrated with five standard proteins (conalbumin, 76 kDa; ovalbumin, 43 kDa; carbonic anhydrase, 29 kDa; RNase A, 13.7 kDa and aprotinin, 6.5 kDa) and then Aip5C was injected into the column. The standard curve of protein size (yAxis: LogDa) was plotted against the standard protein elution volume (xAxis: ml). As a result, Aip5C protein size in the solution can be calculated based on its elution volume (around 12 ml). The UV280 elution profiles were plotted in GraphPad Prism 6.
MD simulations
The MD simulations were performed using the GROMACS 5.1.2 software (63). The Charmm36m force field (64) was used to describe proteins with the tip3p water model (65) for solvent molecules. The temperature of water and proteins was kept constant, coupling independently for each group of molecules at 300 K with a V-rescale thermostat (66). The pressure was coupled with a Parrinello-Rahman (67) barostat at 1 atm separately in each of the three dimensions. The temperature and pressure time constants of the coupling were 0.1 and 2 ps, respectively. The integration of the equations of motion was performed by using a leapfrog algorithm with a time step of 2 fs. Periodic boundary conditions were implemented in all systems. A cutoff of 1 nm was implemented for the Lennard–Jones interactions and for the direct space part of the Ewald sum for Coulombic interactions. The Fourier space part of the Ewald splitting was computed by using the particle-mesh-Ewald method (68), with a grid length of 0.16 nm and a cubic spline interpolation.
Protein sequence analysis
To identify Bni1, Bud6, and Spa2 homologs and preform conservative analysis, the full-length protein sequence was submitted as a query sequence in NCBI protein blast with default parameters. The top 250 hits of protein homologs were chosen from the species. Their correspondent sequence alignment was performed in the online server Clustal Omega (RRID:SCR_001591) and the phylogenetic tree was generated by the interactive tree of life (iTOL) (RRID:SCR_018174). Sequence alignment of Aip5C, Bni1FH1C, and Bud6C between S. cerevisiae and C. albicans was carried out through the online server Clustal Omega and subsequently generated by Jalview. The prediction of CaBni1C for the intrinsically disordered region was carried out in online software IUPRED2A (RRID:SCR_014632), and the intrinsically disordered tendency plot was generated in GraphPad Prism 6.
Statistical analysis
All statistical analyses were performed in GraphPad Prism 8. p Values were determined by the two-tailed Student's t test assuming equal variances and the one-way analysis of variance (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, and ns, not significant). Error bars indicate the standard deviation (S.D.).
Data availability
The coordinates and structure factors have been deposited in the Protein Data Bank with accession code 6M4C (Aip5C). The materials and datasets generated during the current study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We thank Gerald R. Fink (MIT, USA) for sharing the Candida CRISPR-Solo system. We thank the NTU Protein Production Platform for the protein expression test. We thank Min Wu (MBI, NUS) for help on the TIRF microscope system. We thank the National Supercomputing Center Singapore (https://www.nscc.sg/) for assisting the computational work.
This article contains supporting information.
Author contributions—Y. X., H. L., Y. W., L. L., Y. G.-G., and Y. M. conceptualization; Y. X., Z. Y. L., F. Z., J. S., Z. Q., and Y. M. data curation; Y. X., Z. Y. L., J. X., F. Z., J. S., Z. Q., S. J., and Y. M. formal analysis; Y. X., Z. Y. L., J. X., F. Z., J. S., and Y. M. investigation; .Y. X., Z. Y. L., J. X., F. Z., J. S., Z. Q., S. J., Y. D., Y. W., Y. G.-G., and Y. M. methodology; Y. X., Z. Y. L., F. Z., Y. W., L. L., Y. G.-G., and Y. M. writing-original draft; Y. X., Z. Y. L., and Y. M. writing-review and editing; Z. Y. L. and Z. Q. validation; F. Z., S. J., L. L., and Y. G.-G. software; Y. D., H. L., Y. W., L. L., Y. G.-G., and Y. M. resources; L. L., Y. G.-G., and Y. M. supervision; L. L., Y. G.-G., and Y. M. funding acquisition; Y. M. visualization; Y. M. project administration.
Funding and additional information—This study was supported by NTU startup Grant M4081533 (to Y. M.), Ministry of Education Tier 2 Grant MOE2016-T2-1-005S (to Y. M.), Skin Research Institute of Singapore (SRIS) Grant SIG18002 (to Y. M.), MOE Tier 2 Grant MOE2017-T2-1-106 (to Y. G.), and MOE Tier 1 Grant 2018-T1-001-096 (to L. L.).
Conflict of interest—The authors declare that they have no comflicts of interest with the contents of this article.
- NF
- nucleation factor
- NPF
- nucleation promoting factor
- IDR
- intrinsically disordered region
- aa
- amino acid(s)
- MD
- molecular dynamic
- LatA
- latrunculin A
- HU
- hydroxyurea
- TIRF
- total internal reflection fluorescence
- PAM
- protospacer adjacent motif
- NP-actin
- nonpolymerizable actin
- r.m.s. deviation
- root mean square deviation
- PDB
- Protein Data Bank
- gRNA
- guide RNA.
References
- 1. Irazoqui J. E., and Lew D. J. (2004) Polarity establishment in yeast. J. Cell Sci. 117, 2169–2171 10.1242/jcs.00953 [DOI] [PubMed] [Google Scholar]
- 2. Alberti S., and Hyman A. A. (2016) Are aberrant phase transitions a driver of cellular aging?. BioEssays 38, 959–968 10.1002/bies.201600042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Etienne-Manneville S. (2004) Cdc42-the centre of polarity. J. Cell Sci. 117, 1291–1300 10.1242/jcs.01115 [DOI] [PubMed] [Google Scholar]
- 4. Xie Y., Sun J., Han X., Turšić-Wunder A., Toh J. D., Hong W., Gao Y.-G., and Miao Y. (2019) Polarisome scaffolder Spa2-mediated macromolecular condensation of Aip5 for actin polymerization. Nat. Commun. 10, 1–18 10.1038/s41467-018-07882-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ozaki-Kuroda K., Yamamoto Y., Nohara H., Kinoshita M., Fujiwara T., Irie K., and Takai Y. (2001) Dynamic localization and function of Bni1p at the sites of directed growth in Saccharomyces cerevisiae. Mol. Cell Biol. 21, 827–839 10.1128/MCB.21.3.827-839.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Graziano B. R., DuPage A. G., Michelot A., Breitsprecher D., Moseley J. B., Sagot I., Blanchoin L., and Goode B. L. (2011) Mechanism and cellular function of Bud6 as an actin nucleation–promoting factor. Mol. Biol. Cell 22, 4016–4028 10.1091/mbc.E11-05-0404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kono K., Matsunaga R., Hirata A., Suzuki G., Abe M., and Ohya Y. (2005) Involvement of actin and polarisome in morphological change during spore germination of Saccharomyces cerevisiae. Yeast 22, 129–139 10.1002/yea.1205 [DOI] [PubMed] [Google Scholar]
- 8. Liu B., Larsson L., Caballero A., Hao X., Öling D., Grantham J., and Nyström T. (2010) The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140, 257–267 10.1016/j.cell.2009.12.031 [DOI] [PubMed] [Google Scholar]
- 9. Pruyne D. W., Schott D. H., and Bretscher A. (1998) Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143, 1931–1945 10.1083/jcb.143.7.1931 [DOI] [PubMed] [Google Scholar]
- 10. Moseley J. B., Sagot I., Manning A. L., Xu Y., Eck M. J., Pellman D., and Goode B. L. (2004) A conserved mechanism for Bni1-and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell 15, 896–907 10.1091/mbc.e03-08-0621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sagot I., Rodal A. A., Moseley J., Goode B. L., and Pellman D. (2002) An actin nucleation mechanism mediated by Bni1 and profilin. Nat. Cell Biol. 4, 626–631 10.1038/ncb834 [DOI] [PubMed] [Google Scholar]
- 12. Paul A. S., Paul A., Pollard T. D., and Pollard T. (2008) The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19 10.1016/j.cub.2007.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Otomo T., Tomchick D. R., Otomo C., Panchal S. C., Machius M., and Rosen M. K. (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433, 488–494 10.1038/nature03251 [DOI] [PubMed] [Google Scholar]
- 14. Tu D., Graziano B. R., Park E., Zheng W., Li Y., Goode B. L., and Eck M. J. (2012) Structure of the formin-interaction domain of the actin nucleation-promoting factor Bud6. Proc. Natl. Acad. Sci. U.S.A. 109, E3424–E3433 10.1073/pnas.1203035109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glomb O., Bareis L., and Johnsson N. (2019) YFR016c/Aip5 is part of an actin nucleation complex in yeast. Biol. Open 8, bio044024 10.1242/bio.044024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gow N. A., and Yadav B. (2017) Microbe profile: Candida albicans, a shape-changing, opportunistic pathogenic fungus of humans. Microbiology 163, 1145–1147 10.1099/mic.0.000499 [DOI] [PubMed] [Google Scholar]
- 17. Berman J. (2006) Morphogenesis and cell cycle progression in Candida albicans. Curr. Opin. Microbiol. 9, 595–601 10.1016/j.mib.2006.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim J., and Sudbery P. (2011) Candida albicans, a major human fungal pathogen. J. Microbiol. 49, 171–177 10.1007/s12275-011-1064-7 [DOI] [PubMed] [Google Scholar]
- 19. Jones L. A., and Sudbery P. E. (2010) Spitzenkörper, exocyst, and polarisome components in Candida albicans hyphae show different patterns of localization and have distinct dynamic properties. Eukaryot. Cell 9, 1455–1465 10.1128/EC.00109-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crampin H., Finley K., Gerami-Nejad M., Court H., Gale C., Berman J., and Sudbery P. (2005) Candida albicans hyphae have a Spitzenkörper that is distinct from the polarisome found in yeast and pseudohyphae. J. Cell Sci. 118, 2935–2947 10.1242/jcs.02414 [DOI] [PubMed] [Google Scholar]
- 21. Köhli M., Galati V., Boudier K., Roberson R. W., and Philippsen P. (2008) Growth-speed-correlated localization of exocyst and polarisome components in growth zones of Ashbya gossypii hyphal tips. J. Cell Sci. 121, 3878–3889 10.1242/jcs.033852 [DOI] [PubMed] [Google Scholar]
- 22. Zheng P., Nguyen T. A., Wong J. Y., Lee M., Nguyen T.-A., Fan J.-S., Yang D., and Jedd G. (2020) Spitzenkörper assembly mechanisms reveal conserved features of fungal and metazoan polarity scaffolds. Nat. Commun. 11, 1–13 10.1038/s41467-019-13993-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song Y., and Kim J.-Y. (2006) Role of CaBud6p in the polarized growth of Candida albicans. J. Microbiol. 44, 311–319 [PubMed] [Google Scholar]
- 24. Wang H., Huang Z. X., Au Yong J. Y., Zou H., Zeng G., Gao J., Wang Y., Wong A. H. H., and Wang Y. (2016) CDK phosphorylates the polarisome scaffold Spa2 to maintain its localization at the site of cell growth. Mol. Microbiol. 101, 250–264 10.1111/mmi.13386 [DOI] [PubMed] [Google Scholar]
- 25. Li C. R., Wang Y. M., De Zheng X., Liang H. Y., Tang J. C. W., and Wang Y. (2005) The formin family protein CaBni1p has a role in cell polarity control during both yeast and hyphal growth in Candida albicans. J. Cell Sci. 118, 2637–2648 10.1242/jcs.02393 [DOI] [PubMed] [Google Scholar]
- 26. Desai J. (2018) Candida albicans hyphae: from growth initiation to invasion. J. Fungi 4, 10 10.3390/jof4010010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vyas V. K., Barrasa M. I., and Fink G. R. (2015) A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci. Adv. 1, e1500248 10.1126/sciadv.1500248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miao Y., Han X., Zheng L., Xie Y., Mu Y., Yates J. R. III, and Drubin D. G. (2016) Fimbrin phosphorylation by metaphase Cdk1 regulates actin cable dynamics in budding yeast. Nat. Commun. 7, 11265 10.1038/ncomms11265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miao Y., Wong C. C., Mennella V., Michelot A., Agard D. A., Holt L. J., Yates J. R., and Drubin D. G. (2013) Cell-cycle regulation of formin-mediated actin cable assembly. Proc. Natl. Acad. Sci. U.S.A. 110, E4446–E4455 10.1073/pnas.1314000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin R., Walther A., and Wendland J. (2005) Ras1-induced hyphal development in Candida albicans requires the formin Bni1. Eukaryot. Cell 4, 1712–1724 10.1128/EC.4.10.1712-1724.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng X. D., Wang Y. M., and Wang Y. (2003) CaSPA2 is important for polarity establishment and maintenance in Candida albicans. Mol. Microbiol. 49, 1391–1405 10.1046/j.1365-2958.2003.03646.x [DOI] [PubMed] [Google Scholar]
- 32. Bachewich C., Nantel A., and Whiteway M. (2005) Cell cycle arrest during S or M phase generates polarized growth via distinct signals in Candida albicans. Mol. Microbiol. 57, 942–959 10.1111/j.1365-2958.2005.04727.x [DOI] [PubMed] [Google Scholar]
- 33. Pruyne D., Evangelista M., Yang C., Bi E., Zigmond S., Bretscher A., and Boone C. (2002) Role of formins in actin assembly: nucleation and barbed-end association. Science 297, 612–615 10.1126/science.1072309 [DOI] [PubMed] [Google Scholar]
- 34. Zahm J. A., Padrick S. B., Chen Z., Pak C. W., Yunus A. A., Henry L., Tomchick D. R., Chen Z., and Rosen M. K. (2013) The bacterial effector VopL organizes actin into filament-like structures. Cell 155, 423–434 10.1016/j.cell.2013.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moseley J. B., and Goode B. L. (2005) Differential activities and regulation of Saccharomyces cerevisiae formin proteins Bni1 and Bnr1 by Bud6. J. Biol. Chem. 280, 28023–28033 10.1074/jbc.M503094200 [DOI] [PubMed] [Google Scholar]
- 36. Breitsprecher D., Koestler S. A., Chizhov I., Nemethova M., Mueller J., Goode B. L., Small J. V., Rottner K., and Faix J. (2011) Cofilin cooperates with fascin to disassemble filopodial actin filaments. J. Cell Sci. 124, 3305–3318 10.1242/jcs.086934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mészáros B., Erdos G., and Dosztányi Z. (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 10.1093/nar/gky384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roy A., Kucukural A., and Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alford R. F., Leaver-Fay A., Jeliazkov J. R., O'Meara M. J., DiMaio F. P., Park H., Shapovalov M. V., Renfrew P. D., Mulligan V. K., Kappel K., Labonte J. W., Pacella M. S., Bonneau R., Bradley P., Dunbrack R. L., et al. (2017) The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comp. 13, 3031–3048 10.1021/acs.jctc.7b00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horwath M. C., Fecher R. A., and Deepe G. S. (2015) Histoplasma capsulatum, lung infection and immunity. Fut. Microbiol. 10, 967–975 10.2217/fmb.15.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang L., Zhai B., and Lin X. (2012) The link between morphotype transition and virulence in Cryptococcus neoformans. PLoS Pathog. 8, e1002765 10.1371/journal.ppat.1002765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee S. C., Li A., Calo S., and Heitman J. (2013) Calcineurin plays key roles in the dimorphic transition and virulence of the human pathogenic zygomycete Mucor circinelloides. PLoS Pathog. 9, e1003625 10.1371/journal.ppat.1003625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gow N. A., Van De Veerdonk F. L., Brown A. J., and Netea M. G. (2011) Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat. Rev. Microbiol. 10, 112–122 10.1038/nrmicro2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sudbery P. E. (2011) Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9, 737–748 10.1038/nrmicro2636 [DOI] [PubMed] [Google Scholar]
- 45. Zhou Y., Musalgaonkar S., Johnson A. W., and Taylor D. W. (2019) Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat. Commun. 10, 958 10.1038/s41467-019-08880-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gns H. S., Gr S., Murahari M., and Krishnamurthy M. (2019) An update on drug repurposing: re-written saga of the drug's fate. Biomed. Pharmacother. 110, 700–716 10.1016/j.biopha.2018.11.127 [DOI] [PubMed] [Google Scholar]
- 47. Fujiwara T., Tanaka K., Mino A., Kikyo M., Takahashi K., Shimizu K., and Takai Y. (1998) Rho1p-Bni1p-Spa2p interactions: implication in localization of Bni1p at the bud site and regulation of the actin cytoskeleton in Saccharomyces cerevisiae. Mol. Biol. Cell 9, 1221–1233 10.1091/mbc.9.5.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shih J. L., Reck-Peterson S. L., Newitt R., Mooseker M. S., Aebersold R., and Herskowitz I. (2005) Cell polarity protein Spa2P associates with proteins involved in actin function in Saccharomyces cerevisiae. Mol. Biol. Cell 16, 4595–4608 10.1091/mbc.e05-02-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheu Y.-J., Santos B., Fortin N., Costigan C., and Snyder M. (1998) Spa2p interacts with cell polarity proteins and signaling components involved in yeast cell morphogenesis. Mol. Cell. Biol. 18, 4053–4069 10.1128/mcb.18.7.4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buttery S. M., Yoshida S., and Pellman D. (2007) Yeast formins Bni1 and Bnr1 utilize different modes of cortical interaction during the assembly of actin cables. Mol. Biol. Cell 18, 1826–1838 10.1091/mbc.e06-09-0820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jerry H. Y., Crevenna A. H., Bettenbühl M., Freisinger T., and Wedlich-Söldner R. (2011) Cortical actin dynamics driven by formins and myosin V. J. Cell Sci. 124, 1533–1541 10.1242/jcs.079038 [DOI] [PubMed] [Google Scholar]
- 52. van Drogen F., and Peter M. (2002) Spa2p functions as a scaffold-like protein to recruit the Mpk1p MAP kinase module to sites of polarized growth. Curr. Biol. 12, 1698–1703 10.1016/S0960-9822(02)01186-7 [DOI] [PubMed] [Google Scholar]
- 53. Jégou A., Niedermayer T., Orban J., Didry D., Lipowsky R., Carlier M. F., and Romet-Lemonne G. (2011) Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLos Biol. 9, e1001161 10.1371/journal.pbio.1001161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun H., Qiao Z., Chua K. P., Tursic A., Liu X., Gao Y. G., Mu Y., Hou X., and Miao Y. (2018) Profilin negatively regulates formin-mediated actin assembly to modulate PAMP-triggered plant immunity. Curr. Biol. 28, 1882–1895e1887 10.1016/j.cub.2018.04.045 [DOI] [PubMed] [Google Scholar]
- 55. Courtemanche N., and Pollard T. D. (2013) Interaction of profilin with the barbed end of actin filaments. Biochemistry 52, 6456–6466 10.1021/bi400682n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zeng G., Wang Y.-M., Chan F. Y., and Wang Y. (2014) One-step targeted gene deletion in Candida albicans haploids. Nat. Protoc. 9, 464–473 10.1038/nprot.2014.029 [DOI] [PubMed] [Google Scholar]
- 57. Olivares-Marin I. K., González-Hernández J. C., Regalado-Gonzalez C., and Madrigal-Perez L. A. (2018) Saccharomyces cerevisiae exponential growth kinetics in batch culture to analyze respiratory and fermentative metabolism. J. Vis. Exp. 139, e58192 10.3791/58192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sano K.-I., Maeda K., Oki M., and Maéda Y. (2002) Enhancement of protein expression in insect cells by a lobster tropomyosin cDNA leader sequence. FEBS Lett. 532, 143–146 10.1016/S0014-5793(02)03659-1 [DOI] [PubMed] [Google Scholar]
- 59. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L.-W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., et al. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 10.1016/j.ymeth.2011.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 479–485 10.1107/S0907444909038360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kovalevskiy O., Nicholls R. A., and Murshudov G. N. (2016) Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallog. Sect. D Struct. Biol. 72, 1149–1161 10.1107/S2059798316014534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 64. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., and MacKerell A. D. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 10.1038/nmeth.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mackerell A. D. Jr, Feig M., and Brooks C. L. III (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 10.1002/jcc.20065 [DOI] [PubMed] [Google Scholar]
- 66. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 67. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
- 68. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 69. Wilson R. B., Davis D., and Mitchell A. P. (1999) Rapid hypothesis texting with Candida albicans through gene disruption and short homology regions. J. Bacteriol. 181, 1868–1874 10.1128/JB.181.6.1868-1874.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang H., Huang Z. X., Au Yong J. Y., Zou H., Zeng G., Gao J., Wang Y., Wong A. H. H., and Wang Y. (2016) CDK phosphorylates the polarisome scaffold Spa2 to maintain its localization at the site of cell growth. Mol. Microbiol. 101, 250–264 10.1111/mmi.13386 [DOI] [PubMed] [Google Scholar]
- 71. Li C. R., Wang Y. M., De Zheng X., Liang H. Y., Tang J. C. W., and Wang Y. (2005) The formin family protein CaBni1p has a role in cell polarity control during both yeast and hyphal growth in Candida albicans. J. Cell. Sci. 118, 2637–2648 10.1242/jcs.02393 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors have been deposited in the Protein Data Bank with accession code 6M4C (Aip5C). The materials and datasets generated during the current study are available from the corresponding author on reasonable request.