

1. Introduction
Peptide therapeutics are acclaimed as a promising addition to the pharmaceutical arena and they continue to attract interest due to their high potency, specificity, and fewer concerns with toxicology, drug to drug crossreactions and tissue accumulation.1,2 Although poor drug delivery and low in vivo stability are still issues to be addressed, peptides look poised to play an important role in the treatment of diseases ranging from Alzheimer’s disease to cancer.2,3 The huge biodiversity offered by venom peptides, especially conotoxins isolated from the venom of predatory marine snails, holds an enormous promise for the development of future drugs.
2. Cone Snails
Around 700 species of marine snails of the genus Conus are distributed throughout tropical and sub-tropical waters.4 As different species preferentially hunt fish, worms or molluscs they are categorized as piscivorous, vermivorous or mollusciverous, respectively, although some cone snail species can feed on more than one prey type. These slow moving creatures evolved into predators through the incorporation of a specialized envenomation apparatus that enables them to quickly subdue their fast moving prey. Their envenomation apparatus comprises a hollow radular tooth, similar to a harpoon or disposable needle, connected to the venom bulb via a tubular venom duct.5,6 Cone snail venom first attracted research interest in the 1960s in an attempt to understand the pharmacological basis for human fatalities resulting from cone snail stings. These early studies showed Conus venom to be a complex mixture of biologically active components, including a large collection of neuroactive peptides termed conotoxins.7–9 This cocktail of neurotoxins produced in the venom duct is injected into the prey through the radular tooth leading to almost instantaneous paralysis. For a long time it was thought that each cone snail species expresses a distinct set of 50-200 peptides that is influenced by environmental and dietary factors.5,10 From these numbers, which were originally derived from HPLC fractionation and mass spectrometry (MS) analysis, a repertoire of over 70 000 mostly neuroactive peptides was estimated to be produced by the Conus species.4,11 With the advance of more sensitive technology and alternative methods to measure this diversity, it is now clear that this estimate is at the lower end of the scale, with newer estimates going into the hundreds of thousands of distinct neuroactive conopeptides produced by cone snails (see also section 4.1.2 and. 4.1.3).
Conopeptides can be broadly classified into two categories. The first group comprises disulfide-poor peptides, which include contulakins,12 conantokins,13 conorfamides,14 conolysins,15 conophans,16 conomarphins,17 contryphans,18 conomap,19 and conopressins.20 The second group, termed conotoxins, represents the majority of the venom peptide repertoire and contains multiple disulfide bonds. Conotoxins target a wide range of receptors and ion channels with unparalleled potency and selectivity. They have consequently become the subject of intense research in light of their immense diagnostic and therapeutic potential, and are the focus of this review.
3. Conotoxin Classification and Nomenclature
Around 1700 mature conotoxin sequences have been identified to date and this number is rapidly increasing as next generation gene sequencing and proteomic costs continue to fall. This diverse group of peptides was originally organized into various superfamilies based on two sequence elements namely, the conserved signal sequence and the characteristic cysteine framework (i.e. cysteine residue arrangement). Historically, they were further categorized into families based on their receptor target.21 As the number of sequences expands, it seems that a wide array of conotoxin cysteine frameworks are shared between sequences with highly similar signal sequences.22 For example, the M-superfamily includes 9 cysteine frameworks (I,II,III,IV,VI/VII,IX,X,XIV,XVI) and nearly all of them have been found in other superfamilies. Thus, the classification system is undergoing continuous modifications in light of new information becoming available. Figure 1 shows the conotoxin superfamilies, disulfide frameworks and families identified to date. Among these, the α-conotoxins, μ-conotoxins and ω-conotoxins are among the most characterized families so far.
Figure 1. Classification of conotoxins into various families and superfamilies.

Conotoxins are classified into various superfamilies based on their conserved signal sequence homology. Further classification into families is based on their disulfide bond framework and their target receptor. The target receptors for the conotoxin families, which do not have a specified receptor shown in this figure, are yet to be identified. NE-Norepinephrine; nAChR-nicotinic acetylcholine receptor.
The convention in use for naming novel conotoxins is based on the NC-IUPHAR (the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification) system.23 As illustrated in Figure 2, the first (Greek) letter indicates the conotoxin’s pharmacological target. The next one or two letters (Roman, first letter uppercase) derive from the species name from which the conotoxin was isolated. This letter (or letters in the case of ambiguity) is followed by a Roman numeral, which provides information on the disulfide framework. Finally, an uppercase letter denotes the order of discovery of the conotoxin within that category (species + cysteine framework). If the target receptor of the peptide is yet to be determined, then the Greek letter is omitted, lowercase letters are used to indicate the species, Arabic numerals to indicate the framework and the order of discovery is indicated by a lowercase letter.24,25
Figure 2. Conotoxin nomenclature.

Conotoxin naming convention is based on the NC-IUPHAR system. The first letter in Greek indicates the conotoxin’s pharmacological target i.e. α-conotoxin targeting the nicotinic acetylcholine receptors. The next one or two uppercase letters represent the species from which it was isolated in this case, Conus Pennaceus. This is followed by a Roman numeral, I, providing information on the disulfide framework (e.g.: CC-C-C). Finally, an uppercase letter denotes the order of discovery of the conotoxin within that category. If the mechanism of action of the conotoxin is yet to be determined, the Greek letter is omitted, the species name is in lower case letters, an Arabic numeral is used to designate the disulfide bonding pattern, and a small letter is used to specify the peptide variant.
4. Conotoxin Diversity
In the last few years new discoveries on the biology of cone snails and on conotoxin evolution have led to dramatically growing estimates of conotoxin diversity.11,26,27 The ~1700 toxins for which we have information are now considered to be only a small subset of the available diversity. In this section, conotoxin diversity is described at the sequence and structural level, providing critical information for chemists wishing to engineer conotoxins. At the sequence level, our current understanding of the natural mechanisms driving conotoxin chemical diversity is described along with artificial chemical modifications that have been introduced to engineer the properties of conotoxins. At the structural level, conotoxin folds and sequence/structure relationships are discussed. ConoServer - a database that curates the sequences and the three-dimensional structures of conotoxins - was the main source of information in this section.28,29
4.1. Sequence Diversity
4.1.1. Techniques used to study conotoxin sequence diversity
Conotoxins were initially discovered at the peptide level using a combination of fractionation and liquid chromatography,30,31 but the advent of molecular biology techniques substantially accelerated the discovery process by accessing information at the nucleic acid level.25,32–36 More recently, massive amounts of sequence information have been generated using second generation sequencing technology applied to the transcriptomes or genomes of cone snails.37–41 Two factors render the identification of conotoxins from nucleotide sequences incomplete: firstly, conotoxins are produced as precursors and the identification of the mature sequence in the precursor is often ambiguous; secondly, conotoxins often display many types of post-translational modifications,42 most of which cannot be predicted from precursor sequences. In drug discovery and development programs these post-translational modifications (except for the disulfide bonds and C-terminal amidation) are often ignored, since it is cheaper and easier to synthesize the unmodified synthetic analogues for initial lead identification. Nevertheless, isolation and characterization of conotoxins at the peptide level is time consuming, and most known conotoxins have been inferred from transcript sequences.29 In early studies, conotoxins were sequenced using Edman degradation,31,43 but mass spectrometry is now the method of choice for the sequencing of mature conotoxins.44–47 Mass spectrometry is significantly less expensive and allows direct identification of post-translational modifications. Combined proteomic and transcriptomic approaches recently have been employed to explore the venom content of individual Conus species with second generation sequencing providing an unbiased list of precursor sequences whose mature peptide and post-translational modifications could be identified using modern proteomic techniques.41,48
One post-translational modification, the formation of disulfide bond cross-links between cysteine residues, has a major impact on protein structure, and the determination of disulfide connectivities is therefore an important characterization step for conotoxins.1 Various techniques have been used to determine conotoxin disulfide connectivities, including the proteolytic cleavage and analysis of conotoxin fragments using mass spectrometry,49,50 the chromatographic co-elution of native and synthetic peptides displaying selectively formed disulfide bonds,51,52 or the direct structural determination by X-ray crystallography53 or by NMR NOESY54 / ROESY experiments.55
4.1.2. Conotoxin genetic diversity
More than 2000 conotoxin nucleotide sequences are cataloged in ConoServer, of which only 15% are genomic sequences. Conotoxin precursor transcript sequences comprise three regions: an endoplasmic reticulum (ER) signal peptide, a mature peptide region, and pre- and/or post-propeptide regions.32 The sequence of the mature peptide regions is highly variable, and only a handful of conotoxins have been found to be expressed by more than one Conus species.42 In contrast, the ER signal peptide is highly conserved among particular subgroups of conotoxins and for this reason it is used to cluster conotoxins into gene superfamilies.
The currently known gene superfamilies are summarized in Table 1, which shows the ER signal peptide consensus sequence and the corresponding number of identified conotoxins for each gene superfamily. The classification into gene superfamilies using the degree of sequence identity between ER signal peptides has been validated using a clustering analysis, available on the ConoServer website. A recent phylogenetic analysis also supports the proposed divisions in gene superfamilies.56 Whereas, originally, only the conotoxins (disulfide-rich conopeptides) were classified into gene superfamilies, this classification has now been extended to some of the disulfide-poor conopeptide families.56 Several new gene superfamilies have been identified in an early divergent cone snail clade, and it was suggested that these gene superfamilies might be specific to this clade.36
Table 1. Conopeptide gene superfamilies.
| Gene superfamily | # Protein precursorsb | # Nucleic sequencesb | Cysteine frameworks occurring in superfamily | Consensus of signal peptide sequencec |
|---|---|---|---|---|
| A | 202 | 234 | I, II, IV, XIV | MGMRMMFTVFLLVVLATTVVSXTS |
| B1 | 14 | 5 | (disulfide poor conantokins) | MXLYTYLYLLVPLVTFHLILGXGT |
| B2 | 1 | VIII | MLRLITAAVLVSACLA | |
| B3 | 1 | 1 | XXIV | METLTLLWRASSSCLLVVLSHSLLRLLG |
| C | 4 | 7 | (disulfide poor contulakins) | MXXAYWVMVMMMVXIXAPLSEG |
| D | 28 | 14 | XV, XX | MPKLEMMLLVLLILPLXYFDAAGG |
| E | 1 | XXII | MMTRVFFAMFFLMALTEG | |
| F | 1 | MQRGAVLLGVVALLVLWPQAGA | ||
| G | 1 | 1 | XIII | MSGMGVLLLVLLLVMPLAA |
| H | 7 | VI/VII | MNTAGRLLLLCLALGVLVFESLG | |
| I1 | 17 | 10 | VI/VII, XI | MKLXXTFLLXLXILPXXXG |
| I2 | 57 | 38 | XI, XII | MMFRXTSVXCFLLVIXXLNL |
| I3 | 7 | 8 | VI/VII, XI | MKLVLAIVXILMLLSLSTGA |
| J | 12 | 12 | XIV | MPSVRSVTCCCLLWXMLSXXLVTPGSP |
| K | 4 | 1 | XXIII | MIMRMTLTLFVLVVMTAASASG |
| L | 13 | 8 | XIV | MXXXVMFXVXLXLTMPLTX |
| M | 361 | 333 | I, II, III, IV, VI/VII, IX, X, XIV, XVI | MMXKXGVXMLXIXLXLFPLXXXQLDA |
| N | 3 | XV | MSTLKMMLLILLLLLPXATFDSDG | |
| O1 | 460 | 559 | I, VI/VII, XII, XIV | MMKLTCVXIVAVLFLTAXXLXTAXXSA |
| O2 | 84 | 73 | VI/VII, XIV, XV | MEKLTILLLVAAVLMSTQALXQS |
| O3 | 28 | 22 | VI/VII | MSGLGIMVLTLLLLVFMXTSHQ |
| P | 9 | 5 | IX | MHXXLXXSAVLXLXLLXAXXNFXXVQ |
| S | 13 | 8 | VIII | MMXKMGAMFVLLLLFXLXSSQQ |
| T | 157 | 143 | I, V, X, XVI | MRCLPVFXILLLLIXSAPSVDA |
| V | 2 | 2 | XV | MMPVILLLLLSLAIRXXDG |
| Y | 1 | 1 | XVII | MQKATVLLLALLLLLPLSTA |
Conopeptides gene superfamilies are defined by the similarity of the signal peptide regions in conopeptide precursors. The founding reference for each superfamily can be found on the ConoServer website (http://www.conoserver.org), which also provides regular updates to this table and additional statistics. Definitions of the cysteine frameworks are in Table 2.
Number of protein and nucleic precursors catalogued in ConoServer.
The consensus signal sequence was obtained by establishing determining amino acids with at least 60% of conservation at each position of the signal sequence in all protein precursors in ConoServer.
As it is apparent from Table 1, the gene superfamilies, A, M, O1 and T have been extensively sampled, whereas the other gene superfamilies describe fewer than 20 conotoxins in each. This unbalanced number of conotoxins in the gene superfamilies might not completely reflect natural distributions but rather derive from biased sampling methods based on known pharmacology that targets specific gene superfamilies. Recent transcriptomic studies have provided a less biased insight into the relative size of each gene superfamily, confirming that superfamilies O1, M, A and T are indeed the largest superfamilies found in studies to date.37–39,41,48 The number of different transcripts recorded in a single cone snail specimen is 50 to 100 for the most complete studies,39–41,48 but estimating the number of different conotoxin genes in a single species is problematic due to the large intra-species variability,11,57,58 and differential expression in particular regions of the venom duct.59 Furthermore, a reliable estimate of the number of cone snail species still needs to be established,60,61 and only a lower estimate to the total number of conotoxin genes, around 50 000 genes, can therefore be suggested.
Several mechanisms have been proposed to explain this huge genetic diversity. The conotoxin gene family is thought to be one of the fastest evolving gene family of the animal kingdom62 due to extensive and continuous gene duplications and positive selection.62–65 In addition, it has been proposed that allelic variations57,66 and genetic recombination58 might also play a significant role in creating conotoxin genetic diversity. Most of our knowledge on conotoxin genetics was gained at the transcript level, but a recent study focusing on genomic sequences from the A gene superfamily revealed the existence of a large number of conotoxin pseudogenes, indicating that conotoxin genes have highly accelerated turnover.65 On the basis of geographical gene variability for the same species, ecological adaptation to different prey has been suggested as the main driving force of fast conotoxin evolution.57,58,67 Comparative studies between the conotoxin genes, whose main purpose is to allow the organism to attack prey, and rapidly evolving gene families related to host defense, including immunoglobulins, T-cell receptors, major histocompatibility complex proteins and defensins, will certainly be one of the most fascinating future investigations in this field.
4.1.3. Natural conotoxin chemical diversity
Conotoxin precursors are translated in the ER and subsequently undergo several maturation steps, including a range of post-translational modifications that dramatically increase their chemical diversity.68 Our knowledge of the mature toxins is currently limited to the ~250 conotoxins that have been directly isolated from venoms. The most frequent post-translational modification is the formation of disulfide bonds,69 but 12 other modifications have been identified in wild-type conotoxins. The most common modifications besides disulfide bond formation are C-terminal amidation, proline hydroxylation and glutamate γ-carboxylation. A list of posttranslational modifications and associated conotoxins is regularly updated on ConoServer.
Size is one measure of diversity. Figure 3 shows the distribution of conotoxin sequence lengths for all the ~1700 currently known peptides either isolated from the venom or predicted from a precursor sequence. Most are small, with a median size of 26 (with the majority ranging from 10 to 45 amino acids), although recently much larger proteins were identified.70 The pattern of cysteine residues is another measure of diversity, and disulfide-rich conotoxins are categorized into cysteine ‘frameworks’ according to the arrangement of cysteines in the mature peptide region of the precursor. Table 2 summarizes the currently recognized frameworks.
Figure 3. Distribution of mature conopeptide sequence lengths in the ConoServer dataset.

Table 2. Conotoxin cysteine frameworks.
| Cysteine frameworka | Cysteine patternb | # Cysteines | # Mature proteins | Gene superfamilies |
|---|---|---|---|---|
| I | CC-C-C | 4 | 293 | A, M, O1, T |
| II | CCC-C-C-C | 6 | 3 | A, M |
| III | CC-C-C-CC | 6 | 299 | M |
| IV | CC-C-C-C-C | 6 | 51 | A, M |
| V | CC-CC | 4 | 128 | T |
| VI/VII | C-C-CC-C-C | 6 | 517 | H, I1, I3, M, O1, O2, O3 |
| VIII | C-C-C-C-C-C-C-C-C-C | 10 | 11 | B2, S |
| IX | C-C-C-C-C-C | 6 | 29 | M, P |
| X | CC-C[PO]C | 4 | 11 | M, T |
| XI | C-C-CC-CC-C-C | 8 | 89 | I1, I2, I3 |
| XII | C-C-C-C-CC-C-C | 8 | 49 | I2, O1 |
| XIII | C-C-C-CC-C-C-C | 8 | 2 | G |
| XIV | C-C-C-C | 4 | 56 | A, I2, J, L, M, O1, O2 |
| XV | C-C-CC-C-C-C-C | 8 | 23 | D, N, O2, V |
| XVI | C-C-CC | 4 | 7 | M, T |
| XVII | C-C-CC-C-CC-C | 8 | 1 | Y |
| XVIII | C-C-CC-CC | 6 | 2 | |
| XIX | C-C-C-CCC-C-C-C-C | 10 | 2 | |
| XX | C-CC-C-CC-C-C-C-C | 10 | 21 | D |
| XXI | CC-C-C-C-CC-C-C-C | 10 | 1 | |
| XXII | C-C-C-C-C-C-C-C | 8 | 8 | E |
| XXIII | C-C-C-CC-C | 6 | 6 | K |
| XXIV | C-CC-C | 4 | 1 | B3 |
| XXV | C-C-C-C-CC | 6 | 1 | |
| XXVI | C-C-C-C-CC-CC | 8 | 1 |
The conotoxin cysteine frameworks are defined according to a specific pattern of cysteines in the mature peptide region of conopeptide precursors. The founding reference for each cysteine framework can be found on the ConoServer website (http://www.conoserver.org), which provides updates to this table and additional statistics.
Hyphens separating the cysteines (C) stand for a protein segments of one or more amino acids. “[PO]” indicate a position that could be occupied by a proline (P) or hydroxy-proline (O). “.” represents a single position that could be occupied by any amino acid.
Associated with the classic categorization of conotoxins into cysteine frameworks, it has become common practice to refer to the backbone residues between cysteines as ‘loops’. The simplest and most common use of this terminology occurs in Framework I, with the Cys spacing CC-Xm-C-Xn-C defining two loops of size m and n residues respectively. Table 3 summaries the various loop length categories for Framework I conotoxins, from which it is apparent that the most common loop subfamilies are the 4/3 and 4/7 subfamilies.
Table 4-3. Cysteine Framework I conotoxins.
| Loop length class | # of conopeptides | Representative conopeptide |
|---|---|---|
| 3/5 | 52 | α-GI |
| 3/6 | 4 | α-Mn1.1 |
| 4/2 | 1 | α-Cl1.1 |
| 4/3 | 63 | α-ImI |
| 4/4 | 14 | α-BuIA |
| 4/5 | 5 | α-Ca1.1 |
| 4/6 | 15 | α-AuIB |
| 4/7 | 237 | α-Vc1.1 |
| 4/8 | 2 | α-Vt1.24 |
| 5/2 | 3 | α-Cal1a |
| 5/8 | 1 | α-Cl1.2 |
The most optimistic estimates of conotoxin diversity at the peptide level are an order of magnitude higher than at the transcript level, with 1000 to 9000 peptides per species.11,48 Considering that only a small number of conotoxins are shared between cone snail venoms from different species,11,26 it can be estimated that only 0.1% of the total pool of conotoxins has been investigated. The most recent proteomic studies suggest that venoms in different species have vastly different complexity: 419 different peptide masses were found in C. consors, 455 in C. novaehollandiae,71 650 in C. victoria,71 845 in C. imperialis,11 1147 in C. marmoreus 11 and 2428 in C. textile.11 These numbers are lower estimates because a large intra-species variability,11,71,72 and even intra-specimen variability,11,73,74 was observed, indicating that further investigations of already studied species are likely to unravel additional conotoxins.
Several mechanisms have been shown to contribute to the increased diversity at the protein level compared to the already large genetic diversity of conotoxins. These include the differential incorporation of post-translational modifications,48,72,74,75 the conservation of bioactive propeptide regions in the venom.41,48 and the non-specific truncations of conotoxin termini,48 in a process defined as variable peptide processing.48
4.1.4. Synthetic conotoxin diversity
Adding to the natural sequence diversity of conotoxins, synthetic conotoxins have been engineered to better understand their sequence/structure/function relationship or for pharmaceutical applications.76 A total of 26 different types of non-natural amino acids have been used in chemically synthesized conotoxins in addition to the modified amino acids that occur naturally in conotoxins. The sequence/function relationships of ω-MVIIA, the only conotoxin approved for the clinic so far,77 have been probed particularly thoroughly. For example, in one early study, iodotyrosine, norleucine or oxomethionine were introduced to create subtle chemical variations from the wild-type tyrosine, leucine and methionine residues.78 These modifications helped to identify side chains important for ω-MVIIA function. The N-terminus of ω-MVIIA was also N-acetylated to probe the effects of N-terminal charge.78 A similar strategy was used with conotoxins α-AuIB79 and α-ImI,80 and with κ-RIIIK.81 An interesting strategy reported by Kasheverov et al. was to substitute some residues of α-GI with benzoylphenylalanines, which create cross links with the receptor after photoactivation, unraveling the conotoxin binding site.82 Non-natural amino acids have also been used to improve the selectivity and affinity for molecular targets83–85 or to render conotoxins more suitable as drugs by simplifying their structure.86
Another important area in conotoxin drug development is the improvement of conotoxin bioavailability, stability and absorption via chemical re-engineering studies.87 For example, introduction of lipo-amino acids was employed to improve the oral bioavailability of conotoxin α-MII,88 whereas approaches to enhance stability have so far focused on peptide backbone cyclization,79,89–91 or on substitutions of cysteine residues by carba bridges,92 by non-peptidic backbone spacers,93 or by diselenide bridges.94,95
4.2. Structural Diversity
4.2.1. Techniques used to explore conotoxin structural diversity
Table 4 summarizes the currently available structural information on conotoxins. The majority of structures determined so far have utilized NMR spectroscopy rather than X-ray crystallography.96 There are two main reasons for this; the first is that conotoxins are difficult to crystallize and the second is that, being small, they are an ideal size for NMR structure determination and eminently suitable for homonuclear NMR methods, where there is no need for labeling.97 Thus it is easy to determine structures for either native peptides or for samples produced by solid phase chemistry, and thus the limitation of having to recombinantly express labeled conotoxins is not a consideration.
Table 4. Classification of all known three-dimensional structures of wild-type and synthetic conopeptides into folds and sub-folds.
| Namea | Speciesb | Cys frame workc | Size (aa) | # Cysd | Loop sizee | Gene super family | Methodf | BMRB IDg | PDB IDg | Cono Server IDg |
|---|---|---|---|---|---|---|---|---|---|---|
| Fold A: four cysteines, globular [connectivity 1-3, 2-4] | ||||||||||
| Sub-fold A1 (one turn of helix in first loop) | ||||||||||
| α-ImI | C. imperialis | I | 12 | 4 | 4/3 | A | NMR | 1G2G, 1IMI, 1CNL, 1IM1 | 5, 24, 25, 27 | |
| X-ray | 2BYP, 2C9T | 34, 35 | ||||||||
| α-ImI [D5N] | I | 12 | 4 | 4/3 | NMR | 4847 | 1E76 | 10 | ||
| α-ImI [R7L] | I | 12 | 4 | 4/3 | NMR | 4846 | 1E75 | 9 | ||
| α-ImI [A9L,W10Y,R11ABA] | I | 12 | 4 | 4/3 | NMR | 20107 | 131 | |||
| α-ImI [R11E] | I | 12 | 4 | 4/3 | NMR | 4845 | 1E74 | 8 | ||
| α-ImI [C2Agl,C8Agl] | I | 12 | 2 | 4/3 | NMR | 20033 | 128 | |||
| α-ImI [C2U,C8U] | I | 12 | 4 | 4/3 | NMR | 6897 | 2BC7 | 97 | ||
| α-ImI [C2U,C3U,C8U,C12U] | I | 12 | 4 | 4/3 | NMR | 6896 | 2BC8 | 98 | ||
| α-RgIA | C. regius | I | 12 | 4 | 4/3 | A | NMR | 20002, 15435 | 2JUT | 118, 123 |
| α-RgIA [D5E] | I | 12 | 4 | 4/3 | NMR | 15367 | 2JUR | 119 | ||
| α-RgIA [P6V] | I | 12 | 4 | 4/3 | NMR | 15436 | 2JUQ | 121 | ||
| α-BuIA | C. bullatus | I | 13 | 4 | 4/4 | A | NMR | 15031 | 2I28 | 7 |
| α-AuIB | C. aulicus | I | 15 | 4 | 4/6 | A | NMR | 1MXN, 1DG2 | 31, 13 | |
| cyclic-AuIB-4 (GGAA) | I | 19 | 4 | 4/6 | NMR | 142 | ||||
| cyclic-AuIB-5 (AGAGA) | I | 20 | 4 | 4/6 | NMR | 143 | ||||
| cyclic-AuIB-6 (GGAAGG) | I | 21 | 4 | 4/6 | NMR | 144 | ||||
| α-EI | C. ermineus | I | 18 | 4 | 4/7 | A | NMR | 1K64 | 18 | |
| α-Epi [sTy15>Y] | I | 16 | 4 | 4/7 | X-ray | 1A0M | 20 | |||
| α-GIC | C. geographus | I | 16 | 4 | 4/7 | A | NMR | 5985 | 1UL2 | 26 |
| α-GID | C. geographus | I | 18 | 4 | 4/7 | A | NMR | 5585 | 1MTQ | 15 |
| α-MII | C. magus | I | 16 | 4 | 4/7 | A | NMR | 1M2C, 1MII | 21, 29 | |
| α-MII [E11A] | I | 16 | 4 | 4/7 | NMR | 145 | ||||
| cyclic-MII-6 | I | 22 | 4 | 4/7 | NMR | 6818 | 2AJW | 32 | ||
| cyclic-MII-7 | I | 23 | 4 | 4/7 | NMR | 6817 | 2AK0 | 33 | ||
| α-OmIA | C. omaria | I | 17 | 4 | 4/7 | A | NMR | 6237 | 2GCZ | 5 |
| α-PeIA | C. pergrandis | I | 16 | 4 | 4/7 | A | NMR | 139 | ||
| α-PIA | C. purpurascens | I | 18 | 4 | 4/7 | A | NMR | 6720 | 1ZLC | 36 |
| α-Pni1 | I | 16 | 4 | 4/7 | X-ray | 1PEN | 12 | |||
| α-PnIA [A10L,D14K,sTy15Y] | I | 16 | 4 | 4/7 | X-ray | 2BR8 | 30 | |||
| α-PnIB | C. pennaceus | I | 16 | 4 | 4/7 | A | X-ray | 1AKG | 16 | |
| ρ-TIA | C. tulipa | I | 19 | 4 | 4/7 | A | NMR | 1IEN | 65 | |
| α-TxIA | C. textile | I | 16 | 4 | 4/7 | A | X-ray | 2UZ6 | 110 | |
| α-Vc1.1 | C. victoriae | I | 16 | 4 | 4/7 | NMR | 7177 | 2H8S | 4 | |
| cyclic-Vc1.1 | I | 22 | 4 | 4/7 | NMR | 149 | ||||
| α-Vc1.2 | C. victoriae | I | 16 | 4 | 4/7 | A | NMR | 20126 | 141 | |
| Sub-fold A2 (no turn of helix in first loop) | ||||||||||
| α-CnIA | C. consors | I | 14 | 4 | 3/5 | NMR | 1B45 | 53 | ||
| α-GI | C. geographus | I | 13 | 4 | 3/5 | A | NMR | 1XGA | 22 | |
| X-ray | 1NOT | 11 | ||||||||
| α-GI [N4Benzoy1-phenylalanine] | I | 13 | 4 | 3/5 | NMR | 2FRB | 3 | |||
| α-GI [S12Benzoy1-phenylalanine] | I | 13 | 4 | 3/5 | NMR | 2FR9 | 2 | |||
| α-SI | C. striatus | I | 13 | 4 | 3/5 | A | NMR | 4503 | 1QMW | 1 |
| X-ray | 1HJE | 17 | ||||||||
| α-LtXIVA | C. litteratus | XIV | 13 | 4 | 3/3/2 | L | NMR | 21014 | 148 | |
| Sub-fold A3 (no turn of helix in first loop, second loop similar to sub-fold A1) | ||||||||||
| χ-CMrVIA [K6P] | X | 11 | 4 | 4/2 | NMR | 2IH6 | 111 | |||
| χ-CMrVIA [K6P] amidated | X | 11 | 4 | 4/2 | NMR | 2IH7 | 112 | |||
| Fold B: six cysteines, three disulfide bonds not in a knotted arrangement [connectivity 1-4, 2-5, 3-6] | ||||||||||
| Sub-fold B1 (one turn of helix in second loop, two turns of helix overall) | ||||||||||
| μ-CnIIIC | C. consors | III | 22 | 6 | 5/4/5 | NMR | 2YEN | 150 | ||
| μ-GIIIA | C. geographus | III | 22 | 6 | 5/4/4 | M | NMR | 1664, 1665 | 1TCG, 1TCJ | 82, 84, 134, 135 |
| μ-GIIIA [R13A] | III | 22 | 6 | 5/4/4 | NMR | 1TCH,1TCK | 83, 85 | |||
| μ-GIIIB | C. geographus | III | 22 | 6 | 5/4/4 | M | NMR | 1GIB | 64 | |
| μ-KIIIA | C. kinoshitai | III | 16 | 6 | 5/4/4 | M | NMR | 20048 | 129 | |
| μ-PIIIA | C. purpurascens | III | 22 | 6 | 5/4/4 | M | NMR | 6027 | 1R9I | 79 |
| μ-RIIIK [T24A] | III | 24 | 6 | 6/4/4 | NMR | 146 | ||||
| μ-SIIIA | C. striatus | III | 20 | 6 | 1/4/5 | M | NMR | 20025 | 125 | |
| μ-SmIIIA | C. stercusmuscarum | III | 22 | 6 | 5/4/5 | M | NMR | 5881, | 1Q2J | 77 |
| μ-TIIIA | C. tulipa | III | 22 | 6 | 5/4/4 | M | NMR | 20024 | 126 | |
| Sub-fold B2 (no turn of helix in second loop, one turn of helix overall) | ||||||||||
| α-PIIIE | C. purpurascens | III | 24 | 6 | 4/5/4 | M | NMR | 5113 | 1AS5, 1JLO | 51, 68 |
| α-PIIIF | C. purpurascens | III | 24 | 6 | 4/5/4 | M | NMR | 5112 | 1JLP | 69 |
| Fold C: six cysteines, three disulfide bonds forming a cystine knot [connectivity 1-4, 2-5, 3-6] | ||||||||||
| Sub-fold C1 (six residues in first loop) | ||||||||||
| δ-Am2766 | C. amadis | VI/VII | 26 | 6 | 6/6/3/3 | O1 | NMR | 1YZ2 | 94 | |
| ω-CVID | C. catus | VI/VII | 27 | 6 | 6/6/3/6 | O1 | NMR | 138 | ||
| δ-EVIA | C. ermineus | VI/VII | 32 | 6 | 6/9/3/3 | O1 | NMR | 1G1P, 1G1Z | 62, 63 | |
| ω-FVIA | C. fulmen | VI/VII | 25 | 6 | 6/6/3/4 | NMR | 2KM9 | 137 | ||
| μ-conotoxin-GS | C. geographus | VI/VII | 27 | 6 | 6/3/4/7 | O1 | NMR | 1AG7 | 50 | |
| ω-GVIA | C. geographus | VI/VII | 27 | 6 | 6/6/2/6 | O1 | NMR | 2CCO, 1TTL, 1OMC | 72, 89, 100 | |
| ω-GVIA [O10>K] | VI/VII | 27 | 6 | 6/6/2/6 | NMR | 1TR6 | 86 | |||
| μ-MrVIB | C. marmoreus | VI/VII | 31 | 6 | 6/9/4/4 | O1 | NMR | 6135 | 1RMK | 80 |
| ω-MVIIA | C. magus | VI/VII | 25 | 6 | 6/6/3/4 | O1 | NMR | 1DW4, 1DW5, 1MVI, 1OMG, 1TTK | 55, 56, 70, 73, 88 | |
| ω-MVIIA with C-terminal Gly | VI/VII | 26 | 6 | 6/6/3/4 | NMR | 1FEO | 59 | |||
| ω-MVIIA [R10>K] | VI/VII | 25 | 6 | 6/6/3/4 | NMR | 1TT3 | 87 | |||
| ω-MVIIC | C. magus | VI/VII | 26 | 6 | 6/6/3/5 | NMR | 4500 | 1CNN, 1OMN | 74 | |
| ω-MVIIC [S17K,S19R,K25R] | VI/VII | 26 | 6 | 6/6/3/5 | NMR | 1V4Q | 90 | |||
| κ-PVIIA | C. purpurascens | VI/VII | 27 | 6 | 6/6/3/5 | O1 | NMR | 1AV3, 1KCP | 46 | |
| ω-SO3 | C. striatus | VI/VII | 25 | 6 | 6/6/3/4 | O1 | NMR | 1FYG | 61 | |
| ω-SVIB | C. striatus | VI/VII | 26 | 6 | 6/6/3/5 | O1 | NMR | 1MVJ | 71 | |
| t7a | C. tulipa | VI/VII | 30 | 6 | 6/3/4/4 | O1 | NMR | 1EYO | 57 | |
| ω-TxVII | C. textile | VI/VII | 26 | 6 | 6/6/3/3 | O1 | NMR | 1F3K | 58 | |
| δ-TxVIA | C. textile | VI/VII | 27 | 6 | 6/6/3/4 | O1 | NMR | 1FU3 | 60 | |
| ι-RXIA | C. radiatus | XI | 46 | 8 | 6/5/2/4 | I1 | NMR | 15175 | 2P4L, 2JTU | 104, 130 |
| ι-RXIA [BTr33>W] | XI | 46 | 8 | 6/5/2/4 | NMR | 15174 | 2JRY | 105 | ||
| Sub-fold C2 (three residues in first loop) | ||||||||||
| gm9a | C. gloriamaris | IX | 27 | 6 | 3/5/3/1/4 | P | NMR | 1IXT | 67 | |
| Fold D: four cysteines, disulfide bonds with ribbon connectivity [connectivity 1-4, 2-3] | ||||||||||
| Sub-fold D1 (disulfide 2-3 in a staple conformation) | ||||||||||
| χ-MrIA | C. marmoreus | X | 13 | 4 | 4/2 | T | NMR | 6891 | 2EW4 | 102 |
| cyclic-MrIA | X | 15 | 4 | 4/2 | NMR | 2J15 | 49 | |||
| χ-MrIB amidated | C. marmoreus | X | 13 | 4 | 4/2 | NMR | 1IEO | 66 | ||
| *α-GI ribbon isoform | I | 13 | 4 | 3/5 | NMR | 1XGB | 23 | |||
| Sub-fold D2 (disulfide 2-3 in a hook conformation) | ||||||||||
| *α-AuIB ribbon isoform | I | 15 | 4 | 4/6 | NMR | 1MXP | 14 | |||
| *α-BuIA ribbon isoform | I | 4 | 4/4 | NMR | 2NS3 | 114 | ||||
| *α-ImI deamidated ribbon isoform | I | 12 | 4 | 4/3 | NMR | 2IGU | 106 | |||
| *α-ImI [P6A] ribbon isoform | I | 12 | 4 | 4/3 | NMR | 2IFI | 108 | |||
| *α-ImI [P6K] ribbon isoform | I | 12 | 4 | 4/3 | NMR | 2IFZ | 107 | |||
| *α-ImI [P6K] ribbon deamidated isoform | I | 12 | 4 | 4/3 | NMR | 2IFJ | 109 | |||
| *χ-CMrVIA ribbon isoform | 11 | 4 | 4/2 | 2B5P | 95 | |||||
| *χ-CMrVIA amidated ribbon isoform | X | 11 | 4 | 4/2 | NMR | 2IHA | 113 | |||
| Fold E: four cysteines, mirror of fold A [connectivity 1-3, 2-4] | ||||||||||
| χ-CMrVIA | C. marmoreus | X | 11 | 4 | 4/2 | NMR | 2B5Q | 96 | ||
| Fold F: four cysteines, disulfide bonds collinear [connectivity 1-3, 2-4] | ||||||||||
| α-Pu14a | C. pulicarius | XIV | 23 | 4 | 10/1/3 | A | NMR | 21015 | 147 | |
| Fold G: four cysteines, parallel disulfide bonds [connectivity 1-3, 2-4] | ||||||||||
| κ-PlXIVA | C. litteratus | XIV | 25 | 4 | 3/10/1 | J | NMR | 6951 | 2FQC | 103 |
| Kunitz fold: large protein with two disulfide bonds [connectivity 1-4,2-3] | ||||||||||
| Conkunitzin-S1 | C. striatus | XIV | 60 | 4 | 24/20/3 | X-ray | 1Y62 | 48 | ||
| Conkunitzin-S2 | C. striatus | XIV | 65 | 4 | 24/20/3 | NMR | 2j6d | 117 | ||
| Fold H: six cysteines [connectivity 1-5, 2-4, 3-6] | ||||||||||
| mr3e | C. marmoreus | III | 16 | 6 | 4/3/1 | M | NMR | 15195 | 2EFZ | 101 |
| Fold I: six cysteines [connectivity 1-5, 2-3, 4-6] | ||||||||||
| α-PIVA [Hyp7P,Hyp13P] | IV | 25 | 6 | 7/2/1/6 | NMR | 1P1P | 75 | |||
| α-EIVA | C. ermineus | IV | 30 | 6 | 7/2/1/7 | NMR | 5869 | 1PQR | 76 | |
| Fold J: two cysteines, cystine stabilized turn | ||||||||||
| contryphan-R | C. radiatus | 8 | 2 | 5 | NMR | 1QFB | 47 | |||
| contryphan-R [Δ1] | 7 | 2 | 5 | NMR | 1DG0 | 45 | ||||
| contryphan-Sm | C. stercusmuscarum | 8 | 2 | 5 | NMR | 1DFY, 1DFZ | 38, 39 | |||
| contryphan-Vn | C. ventricosus | 9 | 2 | 5 | NMR | 1NXN | 43 | |||
| cyclic-contryphan | 8 | 2 | 5 | NMR | 1D7T | 37 | ||||
| conopressin-T | C. tulipa | 9 | 2 | 4 | NMR | 20007 | 124 | |||
| Fold K: no cysteine, fully helical | ||||||||||
| conantokin-G | C. geographus | 17 | 0 | B | NMR | 1AD7, 1AWY, 1ONU | 40, 41, 44 | |||
| conantokin-T | C. tulipa | 21 | 0 | NMR | 1ONT | 42 | ||||
| Fold L: no cysteine, 3/10 helix and coil | ||||||||||
| conomarphin | C. marmoreus | 15 | 0 | M | NMR | 7397 | 2YYF | 115 | ||
| conomarphin [d13>D] | 15 | 0 | NMR | 2JQC | 116 | |||||
A brief description of the folds and sub-folds is provided in Figures 4a or 4b. The names of non-natural synthetic variants are indented in the case where the fold is the same as the wild-type conopeptide, whereas the name of the variant is preceded by an asterisk in the case where it adopts a different fold from the wild-type.
Only wild-type conopeptides are provided with a Conus (C.) species.
Cysteine frameworks are defined in Table 2.
The number of cysteine residues (# cysteines) is counted in the sequence of the mature peptide region in the precursor, before modification to cystines.
The “loop size” designates the length of the inter-cysteine segments defined in the cysteine frameworks, whose description is in Table 2.
“Method” refers to the experimental method used to determine the three-dimensional structures. If two different experimental methods were used for the same conopeptide, identifiers are provided on two separate lines.
The database identifiers in the Biological Magnetic Resonance dataBank (BMRB), Protein Data Bank (PDB) and ConoServer database are provided. Distinct structural studies are catalogued as different entries in ConoServer, and therefore each entry in ConoServer can be associated with a BMRB and/or a PDB entry. Some conopeptide three-dimensional structures are only found in ConoServer as they were not deposited by their authors in the PDB or BMRB.
Table 4 divides conotoxin structures into fold and sub-fold families, illustrated in Figures 4a and 4b. So far there has been no uniform nomenclature to describe the overall folds of conotoxins and hence here we propose grouping them into ‘fold classes’ sharing similar shapes and spatial position of disulfide cross-links. The discussion below defines these fold classes (A-L).
Figure 4.

a: Most commonly discovered or studied conopeptide folds. All available three-dimensional structures in ConoServer corresponding to the four folds A to D were overlaid. The peptide backbone of each conopeptide is shown using a ribbon representation. The alpha carbon of cystine residues or equivalent (i.e. selenocysteines or half-carba-bridge) are represented as spheres, and the cross-links are shown using orange sticks. The most structurally conserved regions are highlighted in red or in orange. Some structures presenting interesting differences to the fold and discussed in the text are colored in green or blue. The half-cystines have been numbered according to their sequential position in the primary sequence, allowing to clearly distinguish the cross-link connectivities. A description of all the structures is provided in Table 4. This figure was partly drawn using PyMol.1
b: Conopeptide folds with only a few representatives. All available three-dimensional structures in ConoServer corresponding to the four folds E to L and Kunitz are overlaid. The peptide backbone structure of each conopeptide is shown using a ribbon representation, and also using a cartoon representation for fold G, K and Kunitz. The alpha carbons of the cystine residues are represented as spheres, and the cross-links are shown using orange sticks. The most structurally conserved regions are highlighted in red for the Kunitz fold. The half-cystines have been numbered according to their sequential position in the primary sequence to clearly distinguish the cross-link connectivities. A description of all the structures is provided in Table 4. The figure was partly drawn using PyMol.1
4.2.2. Folds with four cysteine residues
The most studied class of conotoxins from a structural perspective is the α-conotoxins, which display cysteine Framework I (CC-C-C).98,99 Most of these peptides adopt fold A (with the globular disulfide connectivity [1-3, 2-4]). Framework I peptides that have four residues in their first loop form a small helical region (sub-fold A1), whereas this helical region disappears when the loop is shortened to three residues (sub-fold A2). Framework X differs from Framework I only by the requirement to have two residues in loop 2, with the second position being occupied by a proline or hydroxy-proline.100 The three currently known wild-type structures of Framework X peptides, i.e., conotoxins χ-MrIA, χ-MrIB, and μ-MrVIA, display two different disulfide connectivities: χ-MrIA and χ-MrIB form a ribbon structure (fold D)90,101 whereas the structure of χ-CMrVIA55 is a mirror of fold A (denoted as fold E in Figure 4b). Interestingly, a synthetic variant of χ-CMrVIA, in which the central position of the first loop is mutated to proline, adopts a type A fold (sub-fold A3),102 suggesting that the simultaneous presence of a proline in both loop 1 and 2 might “compensate” their effects on the fold.
Ribbon-type structures with four cysteines (fold D, connectivity [1-4, 2-3]) can be divided in two groups depending on the disulfide bond 2-3 adopting a staple (sub-fold D1), or a hook conformation (sub-fold D2). Most synthetic Framework I conotoxins synthesized with a ribbon disulfide connectivity adopt a sub-fold D2, whereas wild-type χ-MrIA displays a sub-fold D1. Interestingly, Framework I conotoxin α-AuIB has been shown to be also active in its ribbon isoform,103,104 which was considered as non-natural for Framework I peptides until the recent discovery of minute amounts of a Framework I conotoxin with a ribbon disulfide connectivity in the venom of C. imperialis.105 Framework XIV is the latest framework with four cysteines to have been structurally characterized.106 Perhaps due to the looser definition of this framework, the corresponding conotoxins show very different shapes, including folds A, F, G, and a Kunitz-type fold.
4.2.3. Folds with six cysteine residues
Fold B is characteristic of conotoxins with cysteine Framework III and its characteristic shape is strikingly similar to that of fold A but the spatial arrangement of the disulfide bonds is different. A slight decrease in size of the second loop from 4 to 5 amino acids corresponds to the loss of an helical segment (sub-folds B1 and B2, respectively), which seems to correlate with a change of pharmacological target, from voltage-gated sodium channels (μ-conotoxins) to nicotinic acetylcholine receptors (nAChRs, α-conotoxins), respectively. Framework III conotoxin mr3e has a different connectivity, i.e., connectivity [1-5, 2-4, 3-6], to the other Framework III conotoxins (connectivities [1-6, 2-4, 3-5] and [1-4, 2-5, 3-6]), and its structure determined by NMR reveals a new fold (fold H).107 Interestingly, a recent re-evaluation of another Framework III conotoxin, μ-KIIIA, indicates that it might use the same connectivity as mr3e and therefore might also adopt fold H.108
Fold C has the same disulfide bond connectivity as fold B (connectivity [1-4, 2-5, 3-6]) but here the disulfide bonds are in a knotted arrangement, forming a so-called cystine knot, a stable structural scaffold that has been found in all kingdoms of life.109 Conotoxins displaying the cystine knot motif target various voltagegated ion channels corresponding to the ω, κ, μ, and δ pharmacological families.110 It has been hypothesized that the degree of flexibility of certain loops, as monitored by NMR experiments, could play an important role in target specificity.96,111 The NMR structure of ι-conotoxin ι-RXIA, which has cysteine Framework XI (eight cysteines, C-C-CC-CC-C-C),112 forms a cystine knot fold that superimposes very well with sub-fold C1 structures. The structure of Framework IX gm9a also displays a cystine knot,113 but is classified as a separate sub-fold C2 because only two of the disulfide bridges could be overlaid with corresponding disulfide bonds of sub-fold C1 conotoxins. The sub-folds C1 and C2 correspond to the classes “conotoxin-1” and “conotoxin-2” in the Knottin database, respectively.109 Cysteine Framework IV conotoxins so far studied display different disulfide bond connectivities (connectivity [1-5, 2-3, 4-6]) to the other six cysteine conotoxins. Despite, their radically different structures (fold I), these peptides act on nAChRs,21 similarly to fold A conotoxins.
4.2.4. Disulfide poor conopeptides
Some disulfide-poor conopeptides, including contulakin-G,114 have been shown to be intrinsically disordered, whereas others have well-defined structures. For instance, fold J of contryphans and conopressins includes a turn whose extremities are stabilized by a disulfide bond, creating stable cyclic structures.115,116 Folds K and L that describe conomarphin and conantokins respectively, are not stabilized by disulfide bonds but are either partially (fold K) or totally (fold L) helical.17,117
5. Conotoxin Synthesis
The sparse availability of Conus venom from natural sources presents a major limitation towards utilization of conotoxins for research and clinical applications. Two approaches are currently employed to produce significant amounts of pure material required to carry out structure activity relationship (SAR) studies. The first is recombinant expression of conotoxins in heterologous expression systems such as E. coli and yeast, an approach that is generally applied for protein production.118,119 This method is used to produce cDNA libraries for the screening of a wide range of conotoxins, and while one of the advantages is to have rapid access to longer peptides as well as to a diverse range of superfamilies, it also holds several limitations.120 Incorporation of the vast array of PTMs, characteristically observed in conotoxins, and unnatural amino acids is challenging, and difficulties in isolating the desired products in high purity remain. Many laboratories therefore opt for the viable alternative of chemical synthesis, particularly feasible due to the rather small size of the conotoxins. This approach eliminates undesired (host) protein contamination often faced in recombinant protein production and gives researchers more control and freedom for modifications. Chemical synthesis is rapid, highly automated and scalable, providing significant quantities of native and modified peptides in high purity. It allows the incorporation of unnatural amino acids, a wide array of PTMs, imaging tags and structural modifications such as backbone cyclization or disulfide bond replacements that improve stability against enzymes. The first chemical syntheses of conotoxins were accomplished in the early to mid 1980s on α-conotoxins α-GI, α-MI,121–123 and ω-conotoxin ω-GVIA.124,125 Since then, thousands of conotoxin analogues have been synthesized, typically providing multi-milligram amounts for structural or biological studies.
5.1. Solid phase peptide synthesis – major chemical approaches
Solution chemical peptide synthesis has been used for more than 100 years, until Bruce Merrifield pioneered a major methodological advance in 1963 with the introduction of the concept of solid phase peptide synthesis (SPPS).126 In this method peptide assembly is carried out on a cross-linked solvent accessible polymer. The Nα amino residues and side chain groups are protected in order to avoid side reactions during chain assembly. Solid phase chemistry generally excels over solution phase synthesis as it employs an excess of reagents at high concentrations to drive amine acylation to completion, synthetic steps can be performed in the same vessel, and side products and excess reagents are easily washed from the growing, tethered peptide chain. The two protection strategies most commonly in use are the acid-labile tert-butyloxycarbonyl (Boc) method and the base-labile fluorenylmethyloxycarbonyl (Fmoc) method.127,128 Regardless of the protecting strategy, the key steps of assembly are the same and are depicted in Figure 5. Briefly, the C-terminal amino acid of the peptide is covalently linked to an insoluble polymeric support. The temporary α-amino protecting group of each newly amino acid is removed before the next chemically activated amino acid is added. Stepwise assembly from the C- to the N-terminus is achieved through peptide bond formation between successively added amino acids according to the target sequence. Excess reagents throughout the process of assembly are removed by simple filtration thereby eliminating the requirement of intermediate purification steps. The deprotection and coupling steps can be fully automated and peptide synthesizers produce peptides routinely up to 50 amino acids long, ideal for conotoxin synthesis. Upon completion of chain assembly, the peptide is cleaved from the solid support (with concomitant removal of labile protecting groups) using either hydrogen fluoride (HF) or trifluoroacetic acid (TFA) for Boc and Fmoc methods respectively. The crude peptide is then purified by RP-HPLC and analyzed by high-resolution mass spectrometry. Numerous resins types, linkers, activating agents and protecting groups available confer enormous versatility for SPPS chemistry.127,129
Figure 5. Key steps involved in the assembly of peptides by solid phase peptide synthesis.

Peptides are assembled via successive rounds of Nα deprotection and addition of activated amino acid. In the final step the peptide is cleaved from the solid support with simultaneous removal of the side chain protecting groups.
Certain regions in peptides can be difficult to couple during chain assembly or have incomplete Nα-deprotection during assembly due to aggregation or secondary structure formation. The optimized Boc in situ neutralization protocol coupled with SPPS significantly improves the synthesis of peptides with such “difficult” sequences.130 Additional optimization and introduction of rapid synthetic procedures that improve speed and efficiency of SPPS result in powerful syntheses as illustrated with the Boc-SPPS of α-conotoxin α-[A10L]-PnIA, which was fully assembled, cleaved, and oxidized within a single working day.131 Native chemical ligation (NCL) became another important addition to the repertoire of SPPS, which allows the synthesis of peptides up to 200 amino acids long.132,133 In this approach, two unprotected peptide segments, one containing a C-terminal thioester and the other a cysteine residue at its N-terminus react chemoselectively to give the native amide (peptide) bond.132 NCL is generally not used for standard synthesis since conotoxins fall in the category of small peptides, but NCL plays an important role in combinatorial SAR studies, N-C-terminal backbone cyclization, or in the avoidance of difficult sequences. It is anticipated that the emergence of improved coupling reagents, protecting groups, resins and purification methods will further facilitate the synthesis of longer and more complex peptides, and it is expected that the synthesis of high purity conotoxin libraries will soon be fully automated.
High-throughput synthesis and generation of combinatorial conotoxin libraries are receiving more and more attention due to the importance of efficient SARs in the quest for more potent, selective and stable conotoxin analogues for drug development. Combinatorial techniques are able to rapidly generate numerous libraries within a fraction of the time it would otherwise take for individual peptide synthesis.134,135 The applicability of such library design was demonstrated in a recent study where a mixture-based positional scan of α-ImI was achieved.84 Analogue mixtures with mutations positions 9, 10 and 11 were screened for pharmacological activity at α7 nAChRs, providing candidates for second and third generation analogue syntheses. Through this high-throughput procedure 96 analogues of α-ImI were generated. Whereas the disulfide-rich character and uncontrolled folding are still a major issue in these high-throughput or combinatorial approaches, implementation of novel techniques such as the use of selenocysteine136 or other folding enhancers69 will help to create libraries of more complex conotoxin analogues with directed folding in the future.
5.2. Oxidative folding strategies employed and challenges
The highly conserved cysteine frameworks in the conotoxin superfamilies have one major function: to form and stabilize the rigid 3D structures that comprise a vast array of secondary structures, including α-helices, β-sheets and turns crucial for receptor recognition, potency and selectivity. In contrast to non-disulfide peptides, which follow Anfinsen’s rules for folding,137 correct folding of disulfide-rich peptides involves a complex process involving covalent reactions such as oxidation (S-S formation), reduction (S-S breakage) and isomerization or scrambling (S-S rearrangement).138 Depending on the number of disulfide bonds present, this folding process increases rapidly in complexity due to the increasing number of possible isomers (2n)!/(2nn!), with n being the number of disulfide bonds formed. Thus, where two, three or four disulfides are present, 3, 15 or 105 isomers are theoretically possible.139 So far, a maximum of five disulfide bonds have been observed within the conotoxin family. Hence it is no surprise that correct folding is a significant requirement for the chemical synthesis of conotoxins, particularly as generally only one of the isomers corresponds to the bioactive conformation (Figure 6).140–143
Figure 6. Non-selective isomer formation in a two-disulfide bond containing peptide.

Cone snails deal with this folding challenge through a combination of posttranslational processing, N- and C-terminal propeptides that can act as intramolecular chaperones, folding catalysts and the intrinsic folding properties within the amino acid sequence itself.138,144–146 By contrast, in vitro folding can be quite inefficient: the conceptually simplest approach involves random oxidation of the fully deprotected peptide to the native isomer. The clear advantage for this approach is that it requires only a single cysteine protecting group during SPPS and a single purification step. However, the inherent problem is that the formation of disulfide bonds is a random process and the final yield highly depends on the encoded structural information within the amino acid sequence and the thermodynamic stability of the native conformation versus non-native conformations, which is often only marginally different.69,147 Common folding conditions include slightly alkaline (pH 7.5-8.5) aqueous or aqueous/organic buffers in high dilutions (200-500 μM) to prevent dimer formation. Thiol/disulfide exchange reactions are often induced by addition of GSH/GSSG/peptide ratios (eg. 100:10:1) to mimic physiological conditions that reshuffle misformed disulfide bonds directing the fold into the thermodynamically favored and assumed native conformation. Denaturants such as 0.5-3.0 M urea or 0.1-1.5 M guanidine hydrochloride are occasionally added to prevent aggregation. Table 5 lists examples of folding conditions that have been used successfully to fold various conotoxin analogues. Once a major isomer is obtained, validation of the native/desired fold is necessary either by reductive alkylation, tryptic digest followed by MS/MS, by NMR or by biological activity determination. Even though this approach has its disadvantages, it is still the most employed for native and unmodified conotoxins based on the belief that the native and bioactive isomer is the thermodynamic most stable fold.
Table 5. Common buffer conditions for oxidative folding of conotoxins.
| Standard conotoxin folding conditions |
| 0.1M NH4HCO3; pH 7.5 - 8.5; r.t. 0.1M NH4HCO3 / 30% i-PrOH (or ACN); pH 7.5-8.5; r.t. 0.33M NH4OAc / 0.5M GnHCl, GSH:GSSG:peptide (100:10:1); pH 7.8; 4°C 30% DMSO / 5% acetic acid / 65% water; r.t. 30% DMSO / 0.1 M KH2PO4; pH 6, r.t. |
| Additional conditions |
| 0.33M NH4OAc / 0.5M GnHCl; pH 7.8; 4°C 2M (NH4)2SO4 / 0.1M NH4OAc; pH 7.7; 4°C 2M GnHCl / 50mM NH4OAc; pH 7.7; 4°C 0.1M NH4HCO3; pH 8; 4°C 0.05M - 0.1M NH4OAc; pH 7.8-8.4; 4°C or r.t. 0.1M NH4HCO3 / 30% i-PrOH; pH 8; r.t. 0.1M NH4HCO3 / 15% DMSO / 30% TFE; pH 8; r.t. 0.1M Tris / 6M GnHCl; pH 8.5, 4°C or r.t. 0.1M NH4HCO3 / 50% TFE; pH 8; r.t. |
Recommended peptide concentration = 200-500 μM
GSH / GSSG … reduced / oxidized glutathione
GnHCl … guanidine hydrochloride
DMSO … dimethyl sulfoxide
TFE … 2,2,2-trifluoroethanol
ACN … acetonitrile
i-PrOH … isopropanol
DMSO-promoted oxidation has also found frequent application since it can be applied over an extended pH range from 1 to 8. Generally faster oxidation rates are obtained with DMSO in acidic media and DMSO is known to disrupt aggregates and to assist in dissolving hydrophobic peptides. Oxidation with 10% DMSO or even better 50% DMSO in 1M HCl leads to disulfide bond formation in high yields, yet problems may arise in removing DMSO from the reaction mixture. In general, the more reactive the oxidizing agent, the more side reactions are to be expected, particularly at sensitive amino acid residues such as methionine, tryptophan and tyrosine.
Correct conotoxin folding becomes more difficult as soon as non-native modifications are embedded in the synthetic design. These modifications can disrupt encoded folding information, switching the thermodynamic equilibrium to favor non-native (and therefore often inactive) disulfide bond isomers (having the same mass and similar retention times). Given that the main reason to select synthetic chemistry approaches over peptide expression systems is to introduce non-native modifications for such purposes as SAR studies, ligation chemistry, cyclization, fluorescent tagging, PEGylation etc., it is imperative to have efficient regioselective control over disulfide bond formation even though this may mean lower yields due to additional purification steps. Currently, this is mainly achieved via orthogonal thiol-protecting groups.148–150
5.3. Chemically directed regioselective disulfide bond formation
5.3.1. Chemical strategies
The regioselective approach enables directed formation of individual disulfide bonds, thereby ensuring the desired isomer. The general strategy is based on selective deprotection and oxidation of pair-wise cysteine residues. Since multiple disulfide bonds are formed step-by-step, reaction conditions are required that prevent breaking or scrambling of the disulfide bond already formed. Therefore exposure to alkaline conditions, thiols or other nucleophiles and lengthy reaction times that could allow competitive disulfide bond disproportionation must be avoided. Cysteine protecting groups fall within four general categories consisting of base-labile, acid-labile, metallic ion-labile and mixed alkyl/aryl disulfide (Table 6). The versatility of the S-Acm group in combination with acid labile protecting groups makes it one of the most utilized protecting groups in Fmoc- and Boc-SPPS.151 S-Acm can either be cleaved with mercury salts to obtain free thiols or, as it is mainly the case during conotoxin synthesis, it can be oxidatively cleaved by iodine or thallium trifluoroacetate to form a disulfide bond with another S-Acm group.151,152 Reaction conditions for cleavage153 and deprotection have to be chosen carefully to avoid side reactions such as oxidation of methionine, histidine, tryptophan or tyrosine residues,151 as well as an S to O Acm shift in peptides serine- and threonine-rich peptides.154 For a more detailed review on existing cysteine protecting groups the reader is referred to the following reviews.155,156 Here we only listed sulfur/selenium protecting groups that have been successfully used in conotoxin synthesis.
Table 6. Overview of commonly used cysteine and selenocysteine protecting groups, their stability and standard cleavage conditions.
The most utilized protecting groups are marked in grey. A more complete overview of cysteine and selenocysteine protecting groups and cleavage conditions can be found in these reviews.155,156
| Chemistry | Protecting Group | Structure | Stability | Removal conditions |
|---|---|---|---|---|
| General Synthesis | Sulfur | |||
| Boc (Fmoc) |
4-Methylbenzyl (S-Meb) |
|
Base TFA |
HF 5% DMSO/TFA 60ºC |
| Boc (Fmoc) |
4-Methoxybenzyl (S-Mob) |
|
Base TFA |
HF TFMSA |
| Fmoc | Triphenylmethyl (S-Trt) |
|
Base | 1% TFA Tl(III) I2 |
| Fmoc | 2,4,6-Trimethoxybenzyl (S-Tmob) |
|
Base | 7% TFA/scavengers |
| Orthogonal Synthesis | ||||
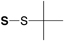
| Boc | 9-Fluorenylmethyl (S-Fm) |
|
TFA HF |
Base |
| Boc | 3-Nitro-2-pyridylsulfenyl (S-Npys) |
|
TFA HF |
Reducing agents Thiols |
| Boc Fmoc |
Acetomidomethyl (S-Acm) |
|
Base TFA HF |
I2 Hg(II); Ag(II); Tl(II); |
| Boc Fmoc |
tert-Butylsulfenyl (S-S-tBu) |
|
TFA HF (partial) |
Reducing agents Thiols |
| Fmoc (Boc) |
tert-Butylmercapto (S-tBu) |
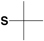
|
TFA Base |
5% DMSO/TFA 25ºC HF (20ºC) |
| Fmoc | 9H-Xanthen-9-yl (S-Xan) |
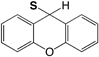
|
Base | 1% TFA/scavengers I2 Tl(III) |
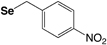
| Selenium | ||||
| Boc | Benzyl (Se-Bzl) |
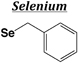
|
Base | Na/NH3 |
| Boc | 4-Methylbenzyl (Se-Meb) |
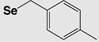
|
Base TFA |
HF |
| Boc Fmoc |
4-Nitro-benzyl (Se-pNB) |
|
Base TFA HF |
Zn, then I2
SnCl2, then I2 |
| Fmoc (Boc) |
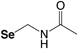
Acetomidomethyl (Se-Acm) |
|
Base TFA HF (partial) |
I2 |
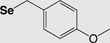
| Fmoc | 4-Methoxybenzyl (Se-Mob) |
|
Base | HF TFMSA or TMSBr TFA/DMSO/scavenger DTNP/TFA |
5.4. Off-resin approaches
5.4.1. Syntheses of conotoxins with 1, 2, 3 and 4 disulfide bonds
Regioselective formation of disulfide bonds can either be carried out while the peptide is still attached to the solid support or upon cleavage in solution. A very common regioselective off-resin approach utilizes the thiol protecting group acetamidomethyl (S-Acm). Typically, the peptide is cleaved from the solid support together with all protecting groups with the exception of S-Acm. The free cysteine residues are oxidized by mild oxidants to form the first disulfide bridge. The second disulfide bond is generally formed directly by treatment with iodine. This approach was first demonstrated by Nishiuchi and Sakakibara for the synthesis of α-GI and its disulfide isomers using Boc chemistry (Scheme 1A),123 and has been applied to the syntheses of many other peptides using both Boc and Fmoc chemistry.157
Scheme 1.

A Regioselective off-resin folding strategy for the synthesis of α-GI using S-Acm in combination with the acid-labile S-Meb protecting group by Boc chemistry.1 B Semi-directed off-resin folding strategy for the synthesis of ω-MVIID using S-Acm in combination with the acid-labile S-Trt protecting group by Fmoc chemistry.2 C Regioselective off-resin folding strategy for the synthesis of ω-MVIIA using S-Acm in combination with an acid-labile S-Trt and S-Mob protecting groups by Fmoc chemistry.3
The S-Acm protection strategy can also be applied to the synthesis of conotoxins containing three disulfide bonds by focusing disulfide bond formation into the desired path. This has been demonstrated by the semidirected synthesis of ω-MVIID (Scheme 1B).158 Random oxidation of the first two disulfide bonds formed predominantly a single isomer, which was followed by the directed formation of the third disulfide bond using iodine-mediated oxidation of two remaining S-Acm protected cysteine residues. In a more selective approach, the S-Mob pair was introduced in addition to the S-Acm and S-Trt protection, which allowed consecutive formation of all three disulfide bonds, illustrated in the synthesis of ω-MVIIA by Fmoc chemistry (Scheme 1C).159
The one-pot synthesis of α-SI employed the combination of the S-Meb and S-tBu protecting groups, where the different temperature dependence of these two groups under DMSO/TFA/anisol oxidation conditions was exploited.160 Cysteine residues protected with S-tBu were rapidly converted to the corresponding disulfide bond at room temperature, whereas S-Meb cysteine residues remained largely intact. Subsequent heating of the solution led to deprotection and oxidation of the S-Meb groups, yielding the second disulfide bond (Scheme 2A).160,161 This approach was successfully combined with the S-Acm/S-Trt combination to allow selective formation of an α-SI dimer containing four disulfide bonds (Scheme 3).162 This double one-pot oxidation strategy with appreciable yields was the first example of using four distinct protecting groups to produce conotoxin analogues. Despite the utility of such a procedure, it is restricted by the nature of amino acid residues in the sequence, particularly by tryptophan and methionine, which are known to undergo irreversible oxidation if left unprotected under these conditions.163 This was evident in the synthesis of α-ImI in an one-pot procedure, where no detectable amounts of native α-ImI were recovered.161 Alternatively, S-tBu was also used in combination with S-Acm cysteine derivatives for the synthesis of α-GI, where upon reductive cleavage of the S-tBu protection the first disulfide bond was formed by air oxidation, which was then followed by iodine oxidation of the S-Acm groups (Scheme 2B).164 S-Acm and S-tBu have also different stability towards iodine, a feature that was explored in the synthesis of α-GI, where the first disulfide bond was formed by direct oxidation of the S-Acm pair followed by S-tBu oxidation via the chlorosilane/sulfoxide procedure (Scheme 2C).165 Although the first disulfide was formed correctly, treatment with chlorosilane/sulfoxide resulted in the non-native and not desired bead isomer and the native isomer could only be recovered by reshuffling with Cys / Tris buffer.
Scheme 2.

A Regioselective off-resin folding strategy for Fmoc chemistry of α-SI and α-GI using S-tBu and S-Meb with one-pot disulfide formation at different temperatures.4,5 B Off-resin folding strategies of α-GI using S-tBu and S-Acm groups. C Unsuccessful regioselective off-resin folding strategy for α-GI trying to exploit the S-tBu stability to iodine.
Scheme 3. Regioselective off-resin folding strategy for the synthesis of an α-SI dimer by Fmoc chemistry utilizing S(tBu), S(Trt), S(Meb) and S(Acm) for orthogonal disulfide bond formation.6 .

5.4.2. Polymer-supported oxidation
A variety of polymer-supported oxidants have been explored including hydrocarbon-based resins,166 agarose-bound folding chaperones,167 PEG-PS (Polyethylene glycol-polystyrene), Sephadex, controlled pore glass168 and more recently, cross-linked ethoxylate acrylate resin (Clear) preloaded with Ellman’s reagent (ClearOx).169 Commercially available ClearOx resin has been continuously used in conotoxin synthesis, where folding occurs upon immobilization of the reduced peptide with the on-resin disulfide, generating a peptide-resin disulfide intermediate, that is released again upon its intramolecular disulfide bond formation.169 The low loading of the resin takes advantage of the pseudodilution effect, favoring intramolecular disulfide bond formation. This makes it an attractive alternative for scale-up and high-throughput folding, since the amount of solvent is significantly reduced without problems of oligomerisation. ClearOx resin has been successfully applied to fold the α-SI and α-GI as well as to the regioselective two-step oxidation procedure forming a non-native isomer using the xanthyl (Xan) and Acm groups.166,170 More recently, the application of ClearOx was successfully extended to three disulfide-bridged conotoxins, namely μ-SIIIA, μ-KIIIA and ω-GVIA, showing improvements compared to standard solution oxidations.171
5.4.3. Native chemical ligation
Considering the disulfide-rich nature of conotoxins it is surprising that NCL has not been exploited more than it is currently the case. One reason for this is the small size of conotoxins, yet NCL certainly has the potential to be used in a variety of applications, including incorporation of handles, overcoming difficult sequences or for combinatorial assembly. The later was demonstrated in the efficient production of chimeras of ω-CVID and ω-MVIIC to evaluate the contributions of the N- and C-terminal segments to activity.172 NCL was also employed to synthesize δ-PVIA covalently bound to its propeptide to study the involvement of propeptides in the folding process.173 Last but not least, NCL plays an important role in the synthesis of cyclic versions of conotoxins that possess improved enzymatic stability, a feature that will be discussed in more detail in section 5.6.
5.5. On-resin approaches
5.5.1. Recent successes and fundamental problems with this approach
The principle of on-resin strategies is to form one or multiple disulfide bridges while the peptide is still anchored to the solid support. On-resin approaches are operationally more convenient, save time-intensive purification steps and avoid large excess of oxidation solvents due to the pseudodilution effect, which simulates high dilution thus favoring intramolecular disulfide bond formation. Especially in view of creating conotoxin libraries for SAR or drug discovery purposes, on-resin folding becomes an essential feature. Current limitations to the on-resin approaches are often poor recovery caused by undesired polymerization or disulfide interaction with the resin,174 in addition to the lack of uniform oxidative folding conditions or strategies to control multiple disulfide bond formation. For example, in the on-resin synthesis of α-SI, closure of the smaller loop before the larger loop was important to obtain the desired isomer, while in solution either oxidation order was successful.174 In addition, disulfide bond formation on a solid support can often be sluggish due to steric hindrance caused by side chain protecting groups, and recovery yields are often lower than compared with in solution methods.174,175 Microwave-assisted on-resin disulfide bond formation may improve synthesis and folding, and was successfully employed on the synthesis of α-MII providing increased yields compared with other on-resin cyclization methods.176 Scheme 4 depicts three examples of orthogonal on-resin oxidation strategies that have been successfully carried out on conotoxins, yet in all cases, in-solution folding was more efficient in both, yields and recovery of the desired isomer.
Scheme 4.

A Regioselective on-resin folding strategy using S-Fm in combination with S-Acm for the synthesis of α-GI by Boc-SPPS.7 B and C Regioselective on-resin folding strategy for α-SI employing the S(Tmob) and S(Xan) groups in combination with the S(Acm) protecting group.8,9
The base-labile S-Fm group in combination with the S-Acm group was utilized using Boc chemistry for the synthesis of α-GI (Scheme 4A).177 In this approach the base-labile S-Fm group was removed on-resin using piperidine, followed by S-Acm deprotection by iodine. Although earlier reports suggested that disulfide bonds are unstable under highly acid conditions, a careful selection of scavengers for the HF cleavage yielded the intact isomer. The Barany lab studied different orthogonal methodologies for the synthesis of α-SI by Fmoc chemistry (Scheme 4B and 4C).174,175 The initial approach included deprotection of the S-Tmob groups without significant peptide chain loss from the resin, followed by the formation of the first disulfide bond via oxidation with CCl4-Et3N in NMP, with the second cysteine pair still protected with Acm group (Scheme 4B). Oxidation of S-Acm to form the second disulfide bond was carried out with Tl(TFA)3 in DMF with anisole as the scavenger. Overall yields of the monomeric conotoxin were as high as 14% with additional oligomeric material retained on the solid support. Alternatively, the orthogonal combination of S-Xan and S-Acm can be used, which was illustrated in the directed on-resin (and in solution) synthesis of all three possible isomers of α-SI (Scheme 4C).
Whereas there is certainly room for improvement for on-resin disulfide bond formation, one has to remember that when it comes to conotoxin library design, low yields can be sacrificed for higher screening efficiency. Once a hit has been isolated and characterized, in-solution folding might be the better option for scale-up and further optimization or SAR studies.
5.5.2. High-throughput synthesis via combinatorial peptide library generation
Cone snails themselves utilize a combinatorial approach to diversify and optimize their venom. Their cell machinery facilitates hypermutations of individual loop residues while maintaining a rigid and highly conserved disulfide-bond framework that gives rise to rigid three-dimensional structures optimized to interact with different receptor classes. Combinatorial chemical synthesis of conotoxin libraries has been more problematic with the main limitation being efficient folding and identification of the correct bioactive isomer. Nevertheless, harvesting non-native disulfide bond isomers can also lead to active analogues with interesting pharmacological properties. Thus, the non-native ribbon form of α-AuIB was initially found to be more active than its globular counterpart and further studies disclosed that these two isomers differ in their inhibitory mechanisms with the ribbon isomer inhibiting only low-sensitivity α3β4 nAChRs competitively, whereas the globular form inhibits α3β4 nAChRs irrespectively of receptor stoichiometry by a non-competitive mechanism.104,178 In the case of α-BuIA, the non-native and inactive ribbon isomer is structurally better defined than the active native form and one might speculate that the ribbon isomer also acts on a target that has yet to be identified.179
Drug discovery programs in venom peptide biotechnology companies take advantage of such non-native activities and a methodology was developed employing a safety catch acid-labile (SCAL) amide linker enabling random DMSO oxidation during linker cleavage that yielded conotoxin disulfide-bond mixtures that can be directly screened against a wide variety of targets.180 The SCAL linker was also employed in the generation of α-conotoxin libraries where it facilitated on-resin selenocysteine directed folding, allowing chain assembly, deprotection and folding to be performed in parallel to yield pure libraries of pure ribbon or globular isomers.136
Alternatively, positional scanning synthetic combinatorial libraries (PS-SCL) can be utilized, which are mixture-based libraries providing rapid means to acquire information around each position within a chemical framework. This led to the largest conotoxin library to date, a library generated employing a multistep synthetic combinatorial approach for the purpose of improving the activity and selectivity profile of α-ImI.84 In this study, a range of substitution mutants was generated in order to produce α-ImI analogues with superior potency towards α7 nAChR subtypes. A PS-SCL was constructed around three residues important for activity to give a total of 10 648 possible combinations, which was followed up by another two rounds of optimization driven by biological activity. Finally, in the third round, 96 analogues were synthesized of which three analogues were found to display a greater than 10-fold increase in activity towards the α7 subtype compared to native α-ImI. Considering these recent advances in accessing large numbers of correctly folded and modified α-conotoxin analogues, it may be concluded that the initial problems of low-throughput synthesis of conotoxins with one and two disulfide bonds have been solved, thereby facilitating efficient SAR studies and optimization of potency and selectivity.
5.6. Cysteine isosteres
5.6.1. Carba, lactam, thioether and selenocysteine bridges
Despite the importance in stabilizing structure, disulfide bonds are inherently unstable under reducing conditions. Reduction or scrambling of disulfide-rich peptide therapeutics by thiol oxidoreductases or other thiol-containing agents, such as serum albumin or glutathione, have the potential to decrease their effectiveness as pharmacological agents in vivo.150 Significant progress has been made in disulfide bond engineering to improve the stability against such degradation.150,181
The α-conotoxins have proven to be a particularly good model system for such disulfide bond mimetics due to their rigid three-dimensional structure, size, diversity, and ease of synthesis and pharmacological characterization.178 For example, replacement of a disulfide bridge in α-ImI by an unsaturated dicarba bridge resulted in both cis and trans isomers, with one displaying no biological activity, while the other retained significantly reduced activity.182 Reduction of the Cys3 to Cys12 disulfide bond in α-ImI had no effect on its affinity and its overall structure was quite similar to the native peptide. However, its rigid conformation was lost to a certain degree with an overall backbone rmsd value of 1.49 Å compared to 0.78 Å for native α-ImI.183 Exchange of a single disulfide bond in α-SI by a lactam bridge resulted in either 60-70 fold or complete loss of activity, depending on which disulfide bond was modified.184
More promising was the development of thioether, selenoether and diselenide mimetics. Initial backbone thioether replacement of one disulfide bond in α-GI resulted in a 260-800 fold loss of activity of the two isomers obtained,185 which can most likely be contributed to the shorting of the loop by deletion of one sulfur atom. Direct isosteric replacement of the disulfide bonds by a CH2-S group was introduced in α-conotoxin α-ImI, where both disulfide bonds were systematically substituted by redox-stable cystathionine thioethers.186 Regioselective thioether formation was achieved on-resin via substitution of a γ-chloro-homoalanine by an intramolecular cysteine thiol to generate a hybrid thioether/disulfide as well as a dual cystathionine analogue.186 NMR analysis showed that they had homologous structures to the native peptide. One of the hybrid isomers displayed identical activity to the native peptide, and the other two analogues showed a modest 3-fold decrease in activity.
Similar to the cystathionine analogues, a comprehensive structural and functional study of a wide range of α-conotoxins containing selenocysteine replacements illustrated that such a modification had no significant impact on torsion angles, activity or receptor subtype selectivity of this class of peptides.136,150 X-ray analysis at 1.4 Å resolution of selenoconotoxin α-PnIA showed that the diselenide bond was 0.3 Å longer than the disulfide bond (2.03 Å) with torsion angles of 93.9° and 83.1°, respectively. In addition, it was shown that the increased hydrophobicity and surface exposure of the diselenide bond had a small beneficial effect on the activity in some of the analogues.136
5.7. Selenoconotoxins
5.7.1. Novel chemical properties of selenocysteine
Selenocysteine (Sec) is referred as the 21st proteinogenic amino acid due to its genetically controlled bioincorporation and importance in many organisms.187–189 Sec is often found in enzymatic active sites, where its known function is either acting as a nucleophile, a metal ligand, or a redox element.190,191 Selenium has been investigated as a substitute to sulfur extensively mainly due to its similarity in physico-chemical properties. Chemical synthesis and structural and pharmacological analysis of a wide range of selenoanalogues of bioactive peptides confirmed its isosteric character to its sulfur homologs. It is particularly interesting for X-ray crystallography since it significantly facilitates the phasing problem, and it avoids the lengthy and problematic heavy atom screening procedure.192–194 NMR analysis can also take advantage of the nuclear spin of I=1/2 of one of its isomers (77Se),195,196 a feature that has already been exploited in a few studies.197,198
Despite selenium and sulfur being neighboring chalcogens, Sec exhibits distinct chemical properties when compared to cysteine. These include higher nucleophilicity,199–201 better leaving group character,202 higher susceptibility to nucleophilic attack,203,204 and higher acidity.199,205,206 In early pKA determination studies selenocysteine exhibited lower pKA values than cysteine (pKA (Sec) = 5.24-5.63, pKA (Cys) = 8.25),199,205,206 suggesting that at physiological pH the Sec residue will be present largely in its reactive anionic form, the selenolate, while the cysteine residue would remain largely protonated. This has been confirmed in a more recent study, where the pKA of two selenocysteine residues was determined within the short peptide, vasopressin, to be 3.3 and 4.3, emphasizing that selenoproteins must have a distinct function and activity other than their cysteine homologs.197 Another feature of diselenide bonds is their lower redox potential, which not only procures an increased resistance in reducing environments, but also allows preferential formation of diselenide over disulfide bonds, a property that has been exploited in the folding of conotoxins.136,207,208
5.7.2. Directed folding of conotoxin
The concept of preferred diselenide formation over disulfide bond formation, originally described by Moroder et al.,209 was employed for conotoxin synthesis and ultimately shown to be true for a wide range of conotoxins.136,207 Pairs of complementary cysteine residues were replaced with selenocysteine residues and diselenide formation occurred rapidly, even under low pH. Once the diselenide bridge was formed, the other disulfide bonds formed accelerated and at lower pH when compared to the all-cysteine homologs.136 This methodology was also applied on non-native isomer production, where folding was significantly improved, however complete regioselectivity could not be achieved (observation of some selenylsulfide formation). The overall structures of the selenoconotoxins were comparable with their respective native conotoxins and functional studies showed that the selenocysteine incorporated peptides exhibited similar or enhanced potency.136,150,208 This intriguing increase in potency in some of the analogues is proposed to be due to the hydrophobic nature of the diselenide bond. Plasma stability was also enhanced significantly for all the selenocysteine analogues tested.94,136 This approach can also be used orthogonally with earlier mentioned thiol-protecting groups, enabling folding of more complex peptides containing three or more disulfide bridges.
To date, the directed folding approach using selenocysteine has been applied to the α-, μ-, and ω-conotoxin classes,136,150,207,208,210,211 yet the strategy can in principle be extended to all disulfide bond containing peptides (and many other peptides have been synthesized).212 Its compatibility with solid-support chemistry enables the generation of libraries thus accelerating drug discovery efforts as well as SAR studies. During a selectivity optimization project on the three-disulfide bond conotoxin μ-KIIIA incorporation of a diselenide bond in combination with depletion of a disulfide bond simplified its synthesis dramatically without compromising its bioactivity.211 This facilitated rapid positional scanning yielding an improved selectivity mutation in blocking Nav1.2 over Nav1.4 subtype. Additionally, its stability against scrambling makes diselenides attractive for drug development, since thiols present in the body (eg. serum albumin or glutathione) cannot deactivate such compounds.136,150 The orthogonal character of diselenide formation adds to the repertoire of controlled regioselective folding and it is expected that different Sec protecting groups will be developed in the future, further expanding this toolbox.
5.8. Cyclic conotoxins
5.8.1. N to C cyclization, including regiocontrol of disulfide bonds, enzyme stability and oral availability
The short half-life of peptides is a major hurdle in drug development and numerous studies have investigated means to extend the half-life of conotoxins in vivo.89,90 Although disulfide-rich peptides (in particular cystine knot motifs) that possess rigid secondary structures display improved stability compared to unstructured peptides, exopeptidases can break down peptides rapidly via C- and N-terminal access. Backbone N – C cyclization is an elegant approach to prevent such degradation and can easily be carried out via NCL owing to the cysteine-rich nature of the conotoxins. In this method, the N and C-termini of the peptide are joined directly or by the addition of short amino acid linkers to cyclize the peptide backbone. The linkers are chosen based on the distances between termini and their orientation so as to ensure that the overall structure of the reengineered conotoxins is not compromised. Cyclization also stabilizes the structure of peptides by decreasing the conformational energy of the unfolded state213 and can introduce additional rigidity that can protect against endopeptidases.214
Studies exemplifying the advantages of cyclization have been conducted on both α- and χ-conotoxins.90,215–217 Three cyclic analogues of α-MII comprising of five, six or seven residue linker sequences were synthesized. The six- and seven-residues linker analogues of α-MII retained the structural and biological features of the native peptide, while significantly increasing their plasma stability. By contrast, the five-residue linker analogue displayed structural perturbations resulting in loss of biological activity.89 In another study, a cyclic analogue of χ-MrIA, containing a two-residue linker joining the ends of the native peptide sequence was synthesized (Figure 7) and structural and functional analysis revealed that the cyclic analogue maintained the native peptide structure and equivalent biological activity.90,218 Furthermore, it had substantially increased resistance to trypsin proteolysis. This was an interesting observation as the trypsin cleavage site was distant from the site of cyclization, confirming that backbone cyclization can also improve stability against endopeptidases.90
Figure 7. Structural representations of linear χ-MrIA (top) and cyclic χ-MrIA (bottom).2 .

Both the peptides have very similar structures. The β-sheets are in blue, the loop and turn regions are in purple. The residues used to link N and C-termini of χ-MrIA are labeled and highlighted in orange. The disulfide bonds in green are shown in a ball-and-stick representation. The structures were visualized using PyMol.1
The linker length can influence the relative abundance of the possible disulfide isomers during random oxidation, in some cases leading to non-native isomers, as demonstrated in a study on cyclic α-ImI analogues.216 This minor synthetic drawback was overcome using a selective orthogonal cysteine protecting scheme.216 It would be of interest to see if this isomeric control could also be achieved via the selenocysteine strategy with advantage of the rapid in situ folding and suppression of scrambling. A direct comparison between different disulfide bond mimetics and N-to-C-terminal cyclization showed that backbone cyclization yielded significantly higher stability compared to disulfide bond mimetics.181 The cyclization approach also has wider applications as it can be used on any protein or peptide with termini no more than ~ 20 Å apart. For such cases, linkers up to seven amino acids long can be used.217,219
To date the most impressive cyclic conotoxin analogue is a cyclic α-Vc1.1 analogue that is being developed for the treatment of neuropathic pain. Although the preclinical development of a linear synthetic version of α-Vc1.1 was earlier discontinued, the more stable cyclic version displayed analgesic activity in rats when delivered orally.215 The use of cyclization to engender a peptide with oral activity is an exciting breakthrough that has the potential to overcome a long-standing bioavailability problem generally associated with peptide-based drugs.
6. Dissecting structure-activity relationships
6.1. Conotoxin discovery
In the post-genomic era, understanding ligand-protein interactions remains a priority to better understand the molecular basis of biological processes, particularly those associated with human disease. The function of many proteins is modulated by a complex network of interactions with associated proteins and/or endogenous ligands. Mapping these interactions on three-dimensional structures is expected to provide a framework for the design of better drugs. Conotoxins are a unique class of venom peptides that have high affinity and selectivity for many functionally critical membrane proteins, characteristics that make them valuable for target validation and drug development.220 To expand their potential as templates for peptidomimetic development, a deep understanding of the specific interactions made with their targets is required to facilitate rational design and associated lead optimization.
The high affinity interaction of conotoxins with their targeted receptors is thought to be mainly driven by shape complementarity (e.g. concept of lock-and-key), hydrophobic contacts and electrostatic potentials.221 While the final complex toxin-receptor is optimally stabilized by a more extensive network of short-range attractive forces (hydrophobic interactions, hydrogen bonds, aromatic stacking, electrostatic and Van der Waals interactions), the first steps of the formation of this complex are predominantly stabilized by long-range electrostatic interactions between the protein and the ligand.222 Favorable electrostatic interactions are produced by complementary charge distribution between binding partners, which in turn dictate the overall association rate.223 Conotoxins often contain ionizable amino acids, whose ionization state influences their physicochemical properties and net electrostatic potential. The distribution of charges throughout the threedimensional structure of conotoxin thus contributes to the on-rate of binding through these long-range electrostatic effects, as well as contributing to the selectivity profile of each ligand through shorter-range complementary interactions including salt bridge and cation-π interactions.221
6.2. Positional scanning
SAR studies integrating crystallographic and/or NMR three-dimensional structures (see section 4) with the results from alanine scanning along the conotoxin sequence, have revealed the position and nature of critical residues contributing to target binding and subtype selectivity. These residues often involve charged and polar amino acids on the peptide surface, with the disulfide bonds often largely buried in the molecule, suggesting that hydrophobic effects contribute to the folding of disulfide rich conotoxin.69 In general, the relative contributions of electrostatic and hydrophobic interactions to the energy of association and dissociation appear to be dependent on the type of interaction. For example, electrostatic interactions appear more important for ion channel “pore blockers”,224 while conotoxins having a lock-and-key type interaction, like the α-conotoxins, rely on a more diverse combination of interactive forces.3 In the next section, we detail the better-defined examples of conotoxin-receptor interactions, with a particular emphasis on the contribution of charge complementarity and electrostatic potentials to binding.
6.3. ω-Conotoxin SAR
ω-Conotoxins isolated from the venom of piscivorous cone snails are among the most potent antagonists of voltage-gated calcium channels (VGCCs) known to date.26 The high potency of ω-MVIIA combined with its high selectivity for the mammalian N-type calcium channel (Cav2.2) was instrumental in the identification of this receptor as a spinal pain target.225 This remarkable property led to the development of this ω-conotoxin as a novel type of analgesic (Prialt), the first marine drug approved by the FDA.77 Although highly efficacious, dose-limiting side effects have limited the market for Prialt, possibly due to on-target effects on neuronal pathways outside the ascending pain pathway and uncharacterized off-target effects.226 Attempts to design small and orally available molecules, non-peptidic mimics of ω-conotoxins, have mostly been unsuccessful so far, most likely due to their reduced size which limits receptor interaction and receptor selectivity, resulting in an increased side effect profile when delivered peripherally.227
The three-dimensional structures of ω-GVIA, ω-MVIIA (both Cav2.2 selective) and ω-MVIIC (Cav2.1 228-230 selective) have been determined using NMR spectroscopy.228–230 Remarkably, all three ω-conotoxins display a similar backbone conformation built around an inhibitory cystine knot motif (see section 4), despite divergent primary sequences (Figure 8).231 Since these conotoxins discriminate among closely related targets (VGCCs), it appears that shape complementarity might play an important role in determining subtype specificity. However, ω-TxVIIA isolated from the molluscivorous Conus textile, also possesses the typical inhibitory cystine knot motif with an overall similar shape to piscivorous ω-conotoxins, but has no detectable activity at mammalian VGCCs.232 Mapping the electrostatic potentials on its three-dimensional surface revealed dramatic differences in the distribution of charges compared to ω-MVIIA, ω-MVIIC and ω-GVIA (Figure 8). ω-TxVIIA is devoid of positively charged residues, which results in a hydrophobic, negatively charged molecule compared to the strongly positively charged surface of piscivorous ω-conotoxins. This is consistent with the expected “pore blocking” effect of ω-conotoxins, which presumably have evolved a positively charged electrostatic surface that mimics the Ca++ ions transported by this channel. Indeed, the external vestibule of VGCCs with its negatively charged residues has been identified as the likely binding site for ω-conotoxins, allowing long-range attractive force to orientate the positively charged ω-conotoxins.
Figure 8. Structures and electrostatic surfaces of ω-conotoxins.

Important residues identified through SAR studies are indicated on the left panels. ω-MVIIA, ω-MVIIC and ω-GVIA target mammalian voltage-gated calcium channels, whereas ω-TxVIIA is a mollusc-selective toxin. Obvious differences in electrostatic potentials likely account for the different pharmacologies.
ω-MVIIA, ω-MVIIC and ω-GVIA SAR studies have revealed a highly conserved dyad of critical residues (Tyr13 and Lys2 in ω-MVIIA) that has been identified in other animal toxins that target VGCCs.228,233–235 Although the aromatic moiety, its orientation, and the hydroxyl group of Tyr13 appear critical for high affinity binding to Cav2.2,233,234,236 the positive charge of Lys2 is not as essential as originally anticipated.237 Indeed, Lys2 is involved in intramolecular stabilization of the three-dimensional structure, as alanine replacement shows increased loop flexibility. In support of a non-direct interaction of Lys2 with the receptor, the side chain of this positively charged residue in ω-GVIA was shown to be tolerant to lengthening and shortening. A specific interaction with the receptor usually implies an optimum distance between partners, and variations around this length are expected to result in significant decrease in affinity.
Furthermore, residues in intercysteine loops 2 and 4 also contribute to ω-conotoxin affinity and selectivity.228,238,239 Affinity appears mainly driven by loop 4 residues R21 in ω-MVIIA and R17, Y22 and K24 in ω-GVIA, while the residue at position 10 in loop 2 seems to influence subtype selectivity.239 Indeed, the residue at position 10 is consistently a lysine in ω-conotoxins with high affinity for Cav2.1, but is replaced with an arginine or hydroxy-proline in Cav2.2-selective peptides.78,230 Interestingly, an arginine in position 10 reduces recovery from Cav2.2 block for both ω-MVIIA and ω-CVID, suggesting that the effect of this residue on subtype selectivity could be due to altered recovery characteristics.240 Together with Lys2 and Tyr13, this key residue at position 10 forms a highly positively charged face of ω-conotoxins (Figure 8). More subtle electrostatic differences are also visible on the surface of ω-MVIIA when compared to ω-MVIIC and ω-GVIA that could contribute to the observed differences in subtype selectivity. Thus the pharmacophore of ω-conotoxins responsible for high affinity, subtype-selective inhibition of Cav2 is becoming increasingly clear. It remains to be seen if our current understanding can be successfully applied to the design of orally active ω-conotoxin peptidomimetics that are analgesic, although early attempts have shown some promise.227,241,242
6.4. μ-Conotoxin SAR
As part of their prey-capture strategy, cone snails have also evolved a diverse range of peptides that modulate voltage gated sodium channels (VGSCs), including the μ, μO, δ and ι-conotoxins. In this section, we focus on the best studied and most promising in terms of drug development, the μ-conotoxins that target TTX-sensitive VGSCs. Of the nine subtypes of VGSCs (Nav1.1-Nav1.9) found in mammals, several are therapeutically relevant, including Nav1.7 and Nav1.8 for the treatment of acute and chronic pain. While all characterized μ-conotoxins show potent inhibition of the skeletal muscle Nav1.4 and brain Nav1.2 channels, none to-date have been found to selectively target therapeutically relevant subtypes. The three dimensional structures of several μ-conotoxins show a conserved central alpha-helical motif, which is reminiscent of the structures of neuronal nicotinic antagonists α-conotoxins (Figure 9). Accordingly, the recently described μ-CnIIIC was found to inhibit VGSCs as well as neuronal nAChRs (α3β2>α4β2>α7).243 Mapping the electrostatic potentials on the surface of three-dimensional structures of μ-conotoxins revealed a strong net positive charge (Figure 9). Although ω-conotoxins also display an overall positive charge, μ-conotoxins have a different fold and distribution of positive charges, with the arginine and lysine residues distributed in a “ring” protruding out from the core of the molecule. Since μ-conotoxins are known pore blockers, such a charge orientation appears well-suited to interact with the ring of negatively charged residues associated with the selectivity filter of VGSCs. Recently, the crystal structures of bacterial NavAb and NavRh confirmed that glutamate side chains do indeed line the pore vestibule, creating a strong long-range electrostatic attraction to positively charged molecules including Na+ ions and μ-conotoxins.244,245
Figure 9. Structures and electrostatic surfaces of μ-conotoxins.

Important residues identified through SAR studies are indicated on the left panels. μ-SmIIIA, μ-GIIIA, μ-PIIIA and μ-TIIIA target Nav1.4 > Nav1.2 mammalian voltage-gated calcium channels, whereas μ-SIIIA and μ-KIIIA have shorter sequences and display a reverse selectivity (Nav1.2> Nav1.4).
The venom of C. geographus provided the first μ-conotoxins investigated and the μ-GIIIA-C were found to potently target skeletal muscle Nav1.4.246 Other μ-conotoxins have now been isolated from a range of piscivorous species, including C. tulipa (μ-TIIIA),247 C. striatus (μ-SIIIA and μ-SIIIB),248,249 C. kinoshitai (μ-KIIIA),248 C. purpurascens (μ-PIIIA)250 and C. stercusmuscarum (μ-SmIIIA).251 The pharmacophore of these μ-conotoxins has been mainly mapped to Arg13 and Arg14 in loop 2, and for instance, the replacement of Arg13 in μ-GIIIA dramatically reduced the affinity for Nav1.4.252 This particular residue has been demonstrated to interact directly with two glutamate residues in domain I of the channel.253 Recent simulations of μ-PIIIA binding to the bacterial sodium channel NavAb predict subnanomolar affinity, demonstrating that these bacterial channels will be useful models for studying μ-conotoxin-VGSC interactions.254 Unexpectedly, the simulations demonstrated that docked μ-PIIIA could adopt different binding orientations or alternative binding modes in the channel. Although these docking poses need to be confirmed experimentally, this result suggests that pseudo-symmetry of the μ-conotoxins might allow different electrostatic complexes to form. With the six basic residues potentially able to plug the pore, the overall net charge of μ-conotoxins appears essential for the high affinity binding to VGSCs.
The natural primary sequence variations that exist between μ-SIIIA and μ-SIIIB (Arg14>Lys14 and Asp15>Gly15, respectively) offer some insights into the possible engineering of neuronal selective μ-conotoxins. Indeed, μ-SIIIA preferentially inhibits neuronal sodium channel (Nav1.2) over the muscle sodium channel subtype (Nav1.4), whereas μ-SIIIB shows the reverse selectivity.255 However, this appears contradictory to the μ-[E15A]-TIIIA analogue having enhanced affinity for the Nav1.2 subtype.247 Interestingly, extensions to the N- and C-termini of μ-SIIIA and μ-SIIIB can increase selectivity for Nav1.2 over Nav1.4, without significantly compromising rat neuronal affinity.237 The smaller μ-conotoxins μ-KIIIA and μ-SIIIA have a different pharmacophore centered around a helical motif in loops 2 and 3.255 In contrast to most of the larger μ-conotoxins, they show a preference for the Nav1.2 over Nav1.4 subtype. In particular, much attention has been directed to μ-KIIIA since it was shown to be analgesic after systemic administration.256 The most promising analogue, μ-KIIIA-[R14A], preferentially blocks Nav1.7 over Nav1.2 and Nav1.4.257 Very recently, different disulfide bond isomers of μ-KIIIA were shown to potently block the Nav1.4 subtype, with surprisingly a non-native fold being more potent.258 This result further supports the net charge of μ-conotoxins being the main driver of its binding to VGSCs, and offers insight into novel engineering opportunities. While VGSCs comprising only the α subunit are fully functional, they are often associated with auxiliary subunits in vivo, which modulate the pharmacology of these channels. For instance, the co-expression of Nav β subunit with α subunit markedly modifies the kinetics of interaction of μ-conotoxins with VGSCs, complicating the interpretation of in vitro experimental results.259
6.5. α-Conotoxin SAR
Nicotinic acetylcholine receptors are divided into two distinct classes: the muscle nAChRs and the neuronal nAChRs.260 The adult muscle nAChR is made of 2α, δ, γ and β subunits, and is the major neurotransmitter receptor at the neuromuscular junction, representing a target of choice for many paralyzing toxins.261 Not surprisingly given their key physiological roles, nAChRs are targeted by a number of venomous animals to facilitate prey capture and/or defensive strategies. Most Conus species venom investigated to date contain at least one α-conotoxin that inhibits these ligand gated ion channels.262 In fact, α-conotoxins represent the largest group of venom peptides isolated from cone snail venoms, and appear to have evolved early in the radiation of this genus.65 As a result of their high affinity and exquisite selectivity, many α-conotoxins have significantly contributed to the pharmacological characterization of the various subtypes of nAChRs both in vitro and in vivo.263 The conotoxin-nAChR complex is also one of the best understood toxin-protein interactions, owing to decades of biochemical and structural studies.3
The overall structure (Figure 10) of each nAChR subunit comprises an extracellular N-terminus (the ligand binding domain), four transmembrane domains, an intracellular loop, and an extracellular C-terminus.260 A low resolution (4 Å) structure of the muscle nAChR determined by cryo-electron microscopy264 and crystallographic structures of bacterial homologues are now available to define the overall structure of these membrane proteins.265–267 However, the crystal structure of the acetylcholine binding protein (AChBP), a soluble homologue of the ligand binding domain of nAChRs,268 provided the first detailed view of the ligand binding domain. AChBP, first isolated from the mollusc Lymnaea stagnalis, is a soluble pentameric protein that binds to all prototypic nAChR ligands.269 It has become the established model for the ligand-binding domain of ligand-gated ion channels. Since this initial discovery, AChBPs have also been characterized from other snails, including Aplysia californica, Bulinus truncatus and recently from the annelid worm Capitella teleta.270–272 The structures of these AChBPs solved by crystallography are all superimposable, consistent with a strong conservation of structure and function, despite high primary sequence variations. The binding site for competitive agonists and antagonists is formed in a small pocket lined by aromatic residues at the interface of two α subunits or at the interface of α and β subunits.268 AChBP has also been co-crystallized with several α-conotoxins, and therefore the corresponding binding site and molecular determinants for conotoxin-nAChR interactions are well understood.270,273,274
Figure 10. 4Å resolution structure of torpedo acetylcholine receptor (left) and crystal structure of AChBP, which is homologues to the extracellular nAChR domain (right).

Left: 4Å resolution structure of torpedo acetylcholine receptor showing the extracellular, transmembrane and intracellular domains.3 The receptor is made of five subunits (each subunit is shown in a different color). Right: 2.2Å resolution crystal structure of AChBP, which is homologues to the extracellular (ligand binding) nAChR domain.4 This protein is a pentamer with five identical subunits surrounding the channel pore. The β-sheets are shown in green and α-helices in pink.
α-Conotoxins that bind to muscle nAChR are among the smallest venom peptides known, yet they exhibit high affinity and remarkable selectivity. The first characterized conotoxin was α-GI isolated from Conus geographus, which acts as a potent antagonist of the muscle-type nAChR in both binding assays and animal isolated tissues.31 An alanine walk along the sequence of α-GI revealed a crucial role of Arg9 for both high potency and selectivity for the muscle-type nAChRs.275,276 Remarkably, these α-conotoxins can also distinguish between non-equivalent binding sites within the same receptor.277 Indeed, α-MI isolated from Conus magus preferentially binds to the α/δ interface with a > 10 000 fold higher affinity over the α/γ interface in the mouse receptor, whereas it displays the opposite selectivity in nAChRs found in the electric organ of the Torpedo ray.278,279 The binding affinity of some α-conotoxins is even influenced by the 5th subunit, as observed for the binding of α-AuIB to α3β4α3β4α3 vs. α3β4α3β4β4.280
The three-dimensional structures of several muscle-selective α-conotoxins have been determined by NMR, and show a superimposable backbone conformation, with the highly conserved Pro and Tyr residues providing key binding determinants (Figure 11). Pairwise interactions have been identified between α-conotoxins and the muscle nAChR, indicating that hydrophobic interactions stabilize the complex.281 However, electrostatic interactions were also shown to play an important role in the binding of α-conotoxins to muscle nAChR. For example, an arginine at position 9 was responsible for α-GI selectivity at α/δ vs. α/γ interfaces, while α-SI from Conus striatus has a proline at this position and did not discriminate between these two interfaces.276 Furthermore, addition of a positively charged residue at the C-terminus increases affinity for the muscle nAChR. Mapping the electrostatic potentials to the surface of α-GI, α-SI and α-CnIA reveals a conserved hydrophobic side (seen as a white and red patch in Figure 11), and complementary side that is largely positively charged. This “two-faced” α-conotoxin structure fits well with the binding site properties of the muscle nAChR (Figure 12). The principal side (α1 subunit) of the receptor is mainly hydrophobic, whereas the complementary side is highly negatively charged. Therefore, in contrast to ω- and μ-conotoxins, which are pore blockers, α-conotoxins instead fit tightly into their binding site nested between two nAChR subunits. This lock-and-key type of interaction282 explains the high affinity observed for such small ligands with low contact surface area.
Figure 11. Structures and electrostatic surfaces of muscle α-conotoxins.

Important residues identified through SAR studies are indicated on the left panels. α-GI, α-SI and α-CnIA target muscle nicotinic receptors.
Figure 12. Structures and electrostatic surfaces of neuronal α-conotoxins.

Important residues identified through SAR studies are indicated on the left panels. 4/7 Conotoxins α-A10L-TxIA, α-PnIB, α-MII and α-Vc1.1 target AChBP, α7, α3β2 and α9α10 nAChRs, whereas 4/3 conotoxins α-ImI and α-RgIA target α3β2 and α9α10 nAChRs, respectively.
Although a muscle nAChR selective antagonist is an obvious weapon to paralyze prey, the neuronal active α-conotoxins have proved most interesting. Indeed, they can distinguish among different subunit arrangements and therefore represent valuable research tools to study the properties of nAChR subtypes, their distribution in native tissues, and may have potential as leads to new therapeutics. From the 12 neuronal subunits known, only a few form functional homopentamers (α7 and α9), with most native nAChRs having a subunit stoichiometry that comprises combinations of different α- and β-subunits, e.g. α3β2 or α6α4β2β3.263 A number of nAChR subtypes have therapeutic potential. For instance, abnormal functions of α6β2β3 have been linked to neurodegenerative diseases283 and the activation of α7 nAChR subtype has also been implicated in cancer.284 The main nAChR subtype present in the brain is composed of α4 and β2 subunits, and is responsible for the nicotine addiction.285 Only α-conotoxin α-GID and α-MII show submicromolar affinity for α4β2 nAChR, though these peptides have nanomolar affinity for α3β2 and α7 nAChRs.286,287 Therefore, like several other human nAChRs, this subtype remains orphan of any selective α-conotoxin. A recent study has shown that only two residues in α4 subunit prevent the binding of α-conotoxins, and in principle, the α4β2 binding pocket can accommodate such ligands, since the double mutant [R185I/P195Q]α4β2 could allow high affinity binding of α-TxIA.288 Interestingly, an alanine scan along the α-GID sequence revealed that the majority of mutations have 10-fold or complete loss of activity compared to the native peptide.289 α-GID has an unusual 4 residue N-terminal tail, which appears critical for activity at α4β2 but not α3β2 or α7. Therefore, engineering α-conotoxins with N-terminal extensions might provide a new strategy to achieve α4β2 selectivity.
Overall, it seems that many α-conotoxins have naturally evolved to selectively target α7 and α3β2 nAChRs, suggesting that these subtypes, or close homologues of these, have critical roles in their prey physiology.290,291 More surprisingly, α-conotoxins that are selective for the unusual α9α10 nAChR have also been discovered, revealing a potential role of this subtype in pain.292 Of particular interest, α-conotoxins α-PnIB, α-MII, α-Vc1.1, α-A10L-TxIA, α-ImI and α-RgIA have been the subject of extensive structure-activity studies,80,273,288,293–300 and their mode of interaction with nAChR provides the best experimental data for understanding α-conotoxin subtype selectivity (Figure 11). α-PnIB together with α-PnIA have been isolated from the venom of C. pennaceus 301 and were shown to have an unusual sulfotyrosine at position 15.302 Interestingly, α-PnIA and α-PnIB differ only at two positions (10 and 11) but their nAChR selectivity is shifted dramatically from α3β2 to α7, respectively. A hydrophobic side chain (Leu) at position 10 was found critical for the α7 preference.296,297 Analysis of molecular surface electrostatics of α3β2 and α7 nAChR homology models reveals that this lengthy hydrophobic side chain can be accommodated in a unique hydrophobic cavity on the complementary (–) face of α7, which is smaller in α3β2 mainly because of the presence of the bulky side chain of β2-Phe117 (Figure 12).282 An analogue of α-PnIA co-crystallized with AChBP confirmed the predominant hydrophobic interaction between the two partners.270 AChBP has also been used as a bait to screen cone snail venoms, and all Conus species investigated showed nicotinic activity.273 α-TxIA was pulled out of C. textile crude venom using this strategy, despite being expressed at very low levels.273 α-TxIA has very high affinity for AChBP (1 nM) and α3β2 (2 nM), but is at least 200-fold less active at α7 (400 nM) nAChRs. An α-A10L-TxIA analogue showed improved affinity at α7 (10-fold) while retaining the same binding properties at AChBP and α3β2. This more potent analogue was cocrystallized with AChBP and showed an overall similar binding mode compared to the α-PnIA variant except for a 20° rotation around Pro7. This tilt in the orientation of α-A10L-TxIA was due to a specific and critical salt bridge between R5 and D195. Interestingly, this ionic bond was also important for the interaction with α7 but not α3β2 nAChRs, providing some molecular basis for nAChR selectivity.
α-MII from C. magus has the highest reported affinity for the α6* containing nAChR subtypes (e.g. α6α3β2).286,298 This peptide was used to show that α6* nAChRs are down-regulated following long term nicotine exposure in rats.303,304 Radiolabelled and fluorescent α-MII also helped to identify that α6* nAChRs are involved in dopamine release in the striatum and down regulation in brain regions is affected by Parkinson’s disease.305 Similarly to α-A10L-TxIA and α-PnIB, α-MII has a hydrophobic patch on its surface (made of P6, V7 and L10) (Figure 13). However, the critical hydrophobic residue present at position 9 of α7-selective α-conotoxins is replaced with a His residue in α-MII, which likely explains its pH-dependence and selectivity for α3 and/or α6 containing nAChRs.273 Interestingly α-Vc1.1, an analgesic α-conotoxin isolated from C. victoriae, uses Ser4 and Asn9 to bind with high affinity to α9α10.294 This is consistent with the rather hydrophilic nature of the α9α10 nAChR binding site (Figure 12) and the recent identification of Thr59 as key determinant for the high potency of α-Vc1.1 in rat.306 α-Vc1.1 has an order of magnitude lower affinity for the human receptor, which has a hydrophobic Ile at this position.306 This structure activity data is expected to help in the design of ligand selective for the human α9α10 subtype, which is involved in immune responses and pain.292 Design of an N–C cyclized form of α-Vc1.1 that retained analgesic properties and was orally bioavailable showed that α-conotoxins can provide a suitable template for novel therapeutic development.215 However, since GABAB has also been proposed to be the pain target of α-Vc1.1, the exact mechanism through which α-Vc1.1 achieves analgesia is still a matter of conjecture.307
Figure 13. Structures and electrostatic surfaces of nAChR subtypes.

Top left panel shows the “lock and key” mechanism of interaction as seen in the α-A10L-TxIA / AChBP complex. Top right panel emphasises on the principal and complementary subunits, between which conotoxins need to fit. Bottom panel compares the molecular surfaces of the binding site in AChBP, α7, α3β2, α9α10 and muscle nAChRs.
The smaller α-conotoxins α-ImI and α-RgIA also target neuronal nAChRs. α-ImI was isolated from the venom of the vermivorous C. imperialis, and was initially found to be α7 selective.308 However, α-ImI was later shown to inhibit α3β2 with a 10-fold higher potency compared to α7 nAChR.309 To date, all SAR studies have been performed on the α7 subtype where residues Asp5, Pro6 and Arg 7 in the first loop and Trp10 in the second loop were shown to interact with the α7 nAChR.80 Thermodynamic mutant cycle analysis was used to determine several pairwise interactions, including a major contribution between Arg7 and Tyr195.299 Based on these distance constraints, docking solutions using nAChR homology models and an NMR structure of α-ImI were obtained that satisfied most experimental data.310,311 The precise interaction of α-ImI within the ACh binding pocket was eventually solved with the crystal structure of the α-ImI-AChBP complex.274,312 The structure of α-ImI bound to AChBP was found to align well with that of the α-PnIA variant bound to AChBP, suggesting that this particular orientation allows favorable interactions between the ligand and the receptor. Recently, a molecular dynamics simulations study concluded that the affinity of α-ImI for α7 was mostly driven by Van der Waals and non-polar desolvation energies, as well as confirming that electrostatic interactions were critical for selectivity.313
α-RgIA was originally cloned from C. regius, another worm-hunting species, and shown to be the most selective α9α10 ligand to date.314 The composition of the first loop of α-RgIA is identical to α-ImI, but the second loop is highly divergent. As determined for α-ImI, residues Asp5, Pro6 and Arg7 in the first loop are important for binding to both α9α10 and α7, while the analogue α-Y10W-RgIA had no effect.293 In contrast to α-ImI, replacement of the residue in position 9 by an alanine in α-RgIA demonstrated the critical role of Arg9 for specific binding to α9α10. Similar to α-Vc1.1, α-RgIA was also shown to be analgesic in a rat model of neuropathic pain,292 and a N–C cyclic version of α-RgIA retained analgesic activity and had increased stability in human plasma,315 again highlighting the potential of α-conotoxin templates for future drug development.
In conclusion, the interaction of α-conotoxins with the various nAChR subtypes relies on a lock-and-key binding mechanism in contrast to the pore blockers μ- and ω-conotoxins. Hydrophobic contacts anchor α-conotoxins deep into the ACh binding site, with complementary interactions (H-bonds and salt-bridges) tuning the selectivity. The accumulated wealth of structural and pharmacological information, together with future additional co-crystal structures, homology modeling and docking simulations, are expected to provide exciting new opportunities for the development of subtype selective nAChR inhibitors.
6.6. Conotoxin mimetics
Differences in conotoxin loop sizes result in remarkable changes in target receptor selectivity and potency. Several loop size truncations are observed in nature among α-conotoxins. This family has thus been further classified into sub-families based on the number of residues in loop 1 and 2 (Table 3). α-Conotoxins that belong to the 3/5 subfamily, isolated from fish hunting cone snails, are found to exclusively block the muscle type nAChRs. This subfamily has a generic sequence - CCXPACG(K/R)XYSC.316 The 4/3, 4/4, 4/6, 4/7 subfamilies have 4 residues in loop 1 and 3-7 residues in loop 2 respectively. These peptides generally have conserved Ser and Pro residues in loop 1 as mentioned earlier. They predominantly antagonize various neuronal nAChRs subtypes and are found in fish, worm and mollusc hunting cone snails. The novel α-4/5 subfamily conotoxin ca1.1 was discovered and successfully synthesized; it did not antagonize any of the α7, α3β2, α3β4, α4β2, α9α10 and muscle nAChR subtypes tested, and its target receptor remains to be identified. As the peptide was also quite flexible in solution, a high resolution NMR structure could not be obtained.317
Aside from naturally loop size variations, truncation mutagenesis studies have been carried out on a number of conotoxins. These have been undertaken to define the relationship between loop size and conotoxin structure and function. Among these, a novel “molecular pruning” approach was employed to evaluate affects of truncation on α4/7-conotoxin α-[A10L]PnIA.318 Loop 2 of this peptide was progressively truncated to give α-4/4[A10L]PnIA and α-4/3[A10L]PnIA analogs. Interestingly, the 4/4 truncation mutant retained activity and did not show any significant structural changes whereas the 4/3 truncated loop analogue lost both activity and structure. This sequential shortening of loop 2 seems to have caused conformational instability as demonstrated by more facile disulfide bond scrambling.318
Disulfide bonds play a very important role in conotoxin structure and function, and several disulfide bond deletion studies have revealed their significance. The removal of individual disulfide bridges in ω-MVIIA reduced binding affinity by from 68-5200 fold at the VGCCs and CD analysis showed significant differences in secondary structure compared to the native fold.319 Similar sequential replacement of cysteine pairs with serine residues in ω-GVIA disrupted its structure and rendered the analogues inactive.142 Hence, for the ω-conotoxins studied, the approach of minimizing conotoxin structures has yet to lead to high potency molecules. In α-conotoxins however, removal of one of the two disulfide bonds in α-GI eliminated both structural integrity and bioactivity320 while the single disulfide bond deficient analogues of α-ImI retained complete activity.183 Thus, the results of disulfide bond deletion seem to be very much dependent on the properties of the individual conotoxin.
As the number of disulfide bonds increases, folding of the synthetic peptide to the native conformation continues to be a significant challenge (see section 5). Hence, identification of the minimum structure required to maintain activity would be an immense advantage.321 This strategy was employed on μ-KIIIA, which induces potent analgesia in mice on systemic administration by acting on Nav1.2 and Nav1.4 voltage gated sodium channels (VGSCs).256 Disulfide bond deficient analogues with one pair of the native disulfide bond connectivities between either C1-C9, C2-C15 or C4-C16 removed at a time, were synthesized. The first disulfide bridge deletion analogues with respective Cys residues 1 and 9 replaced by alanine residues, showed similar structure to native μ-KIIIA without loss of biological activity. The on-rate for these analogues increased at both Nav1.2 and Nav1.4 subtypes. The off-rates, however, were significantly larger when compared to the native peptide.322 The order of preference for the various VGSC subtypes remained unchanged. The second disulfide bridge deletion analogue showed a substantial decrease in activity while the third disulfide bridge deletion mutant essentially lost all activity.322 Structural analysis showed that removal of first disulfide bond did not affect the integrity of the helical region. However, removal of the second or third disulfide bonds disrupted the α-helix indicating that this motif is important for both conotoxin structure and function. The study also showed that these two disulfide bonds are essentially enough to maintain native characteristics. Thus the active ‘core’ structure of μ-KIIIA is a loop one truncated analogue with only two disulfide bonds. While it maintains activity akin to the native peptide, it is also more readily synthesized.322 Further peptidomimetics were also employed in this study, where non-essential N-terminal serine residues were substituted with backbone spacers to give [desC1]KIIIA[S3/4Aopn,C9A] analogue (Aopn standing for 5-amino-3-oxo-pentanoic acid).321 It is important to note that this peptidomimetic analogue of μ-KIIIA could still block VGSCs and retained analgesic activity in the inflammatory pain mouse model. Thus, this approach successfully minimized the bioactive conformation of a three-disulfide-bridged μ-conotoxin μ-KIIIA.323 Based on these observations, future SAR studies could be conducted by grafting the μ-KIIIA pharmacophore on to helical peptidomimetic scaffolds such as peptoids, β-,γ- or β/γ peptides, lactam bridge helix stabilization or other synthetic non-peptide mimetics. Similar studies on adding backbone spacers to disulfide-deficient ω-GVIA diminished its potency.324,325 Thus, in general, conotoxin peptidomimetic studies have only had limited success in maintaining both potency and selectivity.
7. Conotoxins as Drug Candidates
Although less than 0.1% of the venom repertoire has been pharmacologically characterized so far, a few Conus peptides have reached preclinical/clinical development.76,326 Prialt, a synthetic version of the ω-conotoxin ω-MVIIA was the first conotoxin to gain FDA approval in 2004 for treatment of chronic pain.6,327 Its introduction onto the market not only demonstrated the therapeutic potential of conotoxins but also stimulated more interest from biotechnology companies into conotoxin research. Some of the conotoxins currently in clinical/pre-clinical trials include an analog of the χ-conotoxin χ-MrIA (Xen2174), which non-competitively inhibits noradrenaline transporter and is undergoing phase II clinical trials as a treatment for neuropathic pain.328 Other ω-conotoxins in the pipeline included ω-CVID (also called leconotide, AM336 and CNSB004) and ω-GVIA (SNX-124), which successfully completed preclinical studies, yet due to the high cytotoxic affects of these peptides observed during phase I/IIa trials, they did not proceed any further.6,329 α-Vc1.1 was also initially considered to be an attractive candidate for clinical trials due to its analgesic activity, however a number of challenges were encountered that ultimately led to it being discontinued: low stability and an inability to clearly characterize the systemic processing of the peptide were some of them, but more importantly, changes in pharmacological characteristics i.e. switch from competitive in rat to non-competitive binding in human nAChR subtypes were a major concern.330 Thus species diversity among different nAChR subtypes can itself present challenges in the pursuit of conotoxin drug therapeutic development.
Despite promising advances of conotoxins through various stages of drug development, the inability of these peptides to cross the blood-brain barrier necessitates intrathecal administration.76,330 This remains a major limiting factor to be addressed from a clinical perspective for widespread application of conotoxins as therapeutics. Improving in vivo stability and absorption will also greatly enhance their clinical success, and chemical modifications such as disulfide bond re-engineering and cyclization as described herein offer promise that this will be achieved.
Acknowledgements
We would also like to thank Max Nanis and Adam Gardner for their help with the TOC figure. National Health and Medical Research Council (NHMRC) Fellowships are held by Richard Lewis (Grant APP1019761) and David Craik (Grant APP1026501). Sébastien Dutertre was the recipient of a UQ postdoctoral fellowship. This project was supported by Australian Research Council (ARC) Grant (DP1093115 to DC), an NHMRC Project Grant (APP631457 to DC), an NHMRC Program Grant (APP569927 to RJL and PFA), and the European Union Seventh Framework Programme (FP7/2007-2013 #254897 to MM).
Notes
The authors declare no competing financial interest.
References
- (1).Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discov Today. 2010;15:40. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- (2).McGee P. Drug Discovery. 2005;8 [Google Scholar]
- (3).Dutertre S, Lewis RJ. J Biol Chem. 2010;285:13315. doi: 10.1074/jbc.R109.076596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Livett BG, Gayler KR, Khalil Z. Curr Med Chem. 2004;11:1715. doi: 10.2174/0929867043364928. [DOI] [PubMed] [Google Scholar]
- (5).Olivera BM. Annu Rev Ecol Syst. 2002;33:25. [Google Scholar]
- (6).Bingham JP, Mitsunaga E, Bergeron ZL. Chem Biol Interact. 2010;183:1. doi: 10.1016/j.cbi.2009.09.021. [DOI] [PubMed] [Google Scholar]
- (7).Endean R, Gyr P, Surridge J. Toxicon. 1977;15:327. doi: 10.1016/0041-0101(77)90015-0. [DOI] [PubMed] [Google Scholar]
- (8).Spence I, Gillessen D, Gregson RP, Quinn RJ. Life Sci. 1977;21:1759. doi: 10.1016/0024-3205(77)90156-4. [DOI] [PubMed] [Google Scholar]
- (9).Kohn AJ. Proc Natl Acad Sci USA. 1956;42:168. doi: 10.1073/pnas.42.3.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Olivera BM, Walker C, Cartier GE, Hooper D, Santos AD, Schoenfeld R, Shetty R, Watkins M, Bandyopadhyay P, Hillyard DR. Ann N Y Acad Sci. 1999;870:223. doi: 10.1111/j.1749-6632.1999.tb08883.x. [DOI] [PubMed] [Google Scholar]
- (11).Davis J, Jones A, Lewis RJ. Peptides. 2009;30:1222. doi: 10.1016/j.peptides.2009.03.019. [DOI] [PubMed] [Google Scholar]
- (12).Craig AG, Norberg T, Griffin D, Hoeger C, Akhtar M, Schmidt K, Low W, Dykert J, Richelson E, Navarro V, Mazella J, et al. J Biol Chem. 1999;274:13752. doi: 10.1074/jbc.274.20.13752. [DOI] [PubMed] [Google Scholar]
- (13).Mena EE, Gullak MF, Pagnozzi MJ, Richter KE, Rivier J, Cruz LJ, Olivera BM. Neurosci Lett. 1990;118:241. doi: 10.1016/0304-3940(90)90637-o. [DOI] [PubMed] [Google Scholar]
- (14).Maillo M, Aguilar MB, Lopez-Vera E, Craig AG, Bulaj G, Olivera BM, Heimer de la Cotera EP. Toxicon. 2002;40:401. doi: 10.1016/s0041-0101(01)00176-3. [DOI] [PubMed] [Google Scholar]
- (15).Biggs JS, Rosenfeld Y, Shai Y, Olivera BM. Biochemistry. 2007;46:12586. doi: 10.1021/bi700775p. [DOI] [PubMed] [Google Scholar]
- (16).Pisarewicz K, Mora D, Pflueger FC, Fields GB, Mari F. J Am Chem Soc. 2005;127:6207. doi: 10.1021/ja050088m. [DOI] [PubMed] [Google Scholar]
- (17).Han Y, Huang F, Jiang H, Liu L, Wang Q, Wang Y, Shao X, Chi C, Du W, Wang C. FEBS J. 2008;275:1976. doi: 10.1111/j.1742-4658.2008.06352.x. [DOI] [PubMed] [Google Scholar]
- (18).Jacobsen R, Jimenez EC, Grilley M, Watkins M, Hillyard D, Cruz LJ, Olivera BM. J Pept Res. 1998;51:173. doi: 10.1111/j.1399-3011.1998.tb01213.x. [DOI] [PubMed] [Google Scholar]
- (19).Dutertre S, Lumsden NG, Alewood PF, Lewis RJ. FEBS Lett. 2006;580:3860. doi: 10.1016/j.febslet.2006.06.011. [DOI] [PubMed] [Google Scholar]
- (20).Cruz LJ, de Santos V, Zafaralla GC, Ramilo CA, Zeikus R, Gray WR, Olivera BM. J Biol Chem. 1987;262:15821. [PubMed] [Google Scholar]
- (21).Hopkins C, Grilley M, Miller C, Shon KJ, Cruz LJ, Gray WR, Dykert J, Rivier J, Yoshikami D, Olivera BM. J Biol Chem. 1995;270:22361. doi: 10.1074/jbc.270.38.22361. [DOI] [PubMed] [Google Scholar]
- (22).Kaas Q, Westermann J-C, Craik DJ. Toxicon. 2010;55:1491. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- (23).Sharman JL, Benson HE, Pawson AJ, Lukito V, Mpamhanga CP, Bombail V, Davenport AP, Peters JA, Spedding M, Harmar AJ. Nucleic Acids Res. 2013;41:D1083. doi: 10.1093/nar/gks960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gray WR, Olivera BM, Cruz LJ. Annu Rev Biochem. 1988;57:665. doi: 10.1146/annurev.bi.57.070188.003313. [DOI] [PubMed] [Google Scholar]
- (25).Walker CS, Steel D, Jacobsen RB, Lirazan MB, Cruz LJ, Hooper D, Shetty R, DelaCruz RC, Nielsen JS, Zhou LM, Bandyopadhyay P, et al. J Biol Chem. 1999;274:30664. doi: 10.1074/jbc.274.43.30664. [DOI] [PubMed] [Google Scholar]
- (26).Terlau H, Olivera BM. Phys Rev. 2004;84:41. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- (27).Olivera BM. J Biol Chem. 2006;281:31173. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- (28).Kaas Q, Westermann JC, Halai R, Wang CK, Craik DJ. Bioinformatics. 2008;24:445. doi: 10.1093/bioinformatics/btm596. [DOI] [PubMed] [Google Scholar]
- (29).Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ. Nucleic Acids Res. 2012;40:D325. doi: 10.1093/nar/gkr886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cruz LJ, Gray WR, Olivera BM. Arch Biochem Biophys. 1978;190:539. doi: 10.1016/0003-9861(78)90308-9. [DOI] [PubMed] [Google Scholar]
- (31).Gray WR, Luque A, Olivera BM, Barrett J, Cruz LJ. J Biol Chem. 1981;256:4734. [PubMed] [Google Scholar]
- (32).Woodward SR, Cruz LJ, Olivera BM, Hillyard DR. EMBO J. 1990;9:1015. doi: 10.1002/j.1460-2075.1990.tb08204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Santos AD, McIntosh JM, Hillyard DR, Cruz LJ, Olivera BM. J Biol Chem. 2004;279:17596. doi: 10.1074/jbc.M309654200. [DOI] [PubMed] [Google Scholar]
- (34).Pi C, Liu J, Peng C, Liu Y, Jiang X, Zhao Y, Tang S, Wang L, Dong M, Chen S, Xu A. Genomics. 2006;88:809. doi: 10.1016/j.ygeno.2006.06.014. [DOI] [PubMed] [Google Scholar]
- (35).Pi C, Liu Y, Peng C, Jiang X, Liu J, Xu B, Yu X, Yu Y, Jiang X, Wang L, Dong M, et al. Biochimie. 2006;88:131. doi: 10.1016/j.biochi.2005.08.001. [DOI] [PubMed] [Google Scholar]
- (36).Biggs JS, Watkins M, Puillandre N, Ownby J-P, Lopez-Vera E, Christensen S, Moreno KJ, Bernaldez J, Licea-Navarro A, Corneli PS, Olivera BM. Mol Phylogenet Evol. 2010;56:1. doi: 10.1016/j.ympev.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu H, Bandyopadhyay PK, Olivera BM, Yandell M. BMC Genomics. 2011;12:60. doi: 10.1186/1471-2164-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu H, Bandyopadhyay PK, Olivera BM, Yandell M. BMC genomics. 2012;13:284. doi: 10.1186/1471-2164-13-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lluisma AO, Milash BA, Moore B, Olivera BM, Bandyopadhyay PK. Mar Genomics. 2012;5:43. doi: 10.1016/j.margen.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Terrat Y, Biass D, Dutertre S, Favreau P, Remm M, Stöcklin R, Piquemal D, Ducancel F. Toxicon. 2012;59:34. doi: 10.1016/j.toxicon.2011.10.001. [DOI] [PubMed] [Google Scholar]
- (41).Violette A, Leonardi A, Piquemal D, Terrat Y, Biass D, Dutertre S, Noguier F, Ducancel F, Stöcklin R, Križaj I, Favreau P. Mar Drugs. 2012;10:258. doi: 10.3390/md10020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kaas Q, Westermann JC, Craik DJ. Toxicon. 2010;55:1491. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- (43).Olivera BM, McIntosh JM, Cruz LJ, Luque FA, Gray WR. Biochemistry. 1984;23:5087. doi: 10.1021/bi00317a001. [DOI] [PubMed] [Google Scholar]
- (44).Tayo LL, Lu B, Cruz LJ, Yates JR., 3rd J Proteome Res. 2010;9:2292. doi: 10.1021/pr901032r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jakubowski JA, Kelley WP, Sweedler JV. Toxicon. 2006;47:688. doi: 10.1016/j.toxicon.2006.01.021. [DOI] [PubMed] [Google Scholar]
- (46).Gowd KH, Dewan KK, Iengar P, Krishnan KS, Balaram P. J Mass Spectrom. 2008;43:791. doi: 10.1002/jms.1377. [DOI] [PubMed] [Google Scholar]
- (47).Bhatia S, Kil YJ, Ueberheide B, Chait BT, Tayo L, Cruz L, Lu B, Yates JR, 3rd, Bern M. J Proteome Res. 2012;11:4191. doi: 10.1021/pr300312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dutertre S, Jin AH, Kaas Q, Jones A, Alewood PF, Lewis RJ. Mol Cell Proteomics. 2013;12:312. doi: 10.1074/mcp.M112.021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Moller C, Rahmankhah S, Lauer-Fields J, Bubis J, Fields GB, Mari F. Biochemistry. 2005;44:15986. doi: 10.1021/bi0511181. [DOI] [PubMed] [Google Scholar]
- (50).Dy CY, Buczek P, Imperial JS, Bulaj G, Horvath MP. Acta Crystallogr D Biol Crystallogr. 2006;62:980. doi: 10.1107/S0907444906021123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Hidaka Y, Sato K, Nakamura H, Kobayashi J, Ohizumi Y, Shimonishi Y. FEBS Lett. 1990;264:29. doi: 10.1016/0014-5793(90)80756-9. [DOI] [PubMed] [Google Scholar]
- (52).Han YH, Wang Q, Jiang H, Liu L, Xiao C, Yuan DD, Shao XX, Dai QY, Cheng JS, Chi CW. FEBS J. 2006;273:4972. doi: 10.1111/j.1742-4658.2006.05493.x. [DOI] [PubMed] [Google Scholar]
- (53).Hu SH, Gehrmann J, Alewood PF, Craik DJ, Martin JL. Biochemistry. 1997;36:11323. doi: 10.1021/bi9713052. [DOI] [PubMed] [Google Scholar]
- (54).Hill JM, Alewood PF, Craik DJ. Eur J Biochem. 2000;267:4649. doi: 10.1046/j.1432-1327.2000.01507.x. [DOI] [PubMed] [Google Scholar]
- (55).Kang TS, Jois SD, Kini RM. Biomacromolecules. 2006;7:2337. doi: 10.1021/bm060269w. [DOI] [PubMed] [Google Scholar]
- (56).Puillandre N, Koua D, Favreau P, Olivera BM, Stöcklin R. J Mol Evol. 2012;74:297. doi: 10.1007/s00239-012-9507-2. [DOI] [PubMed] [Google Scholar]
- (57).Duda TF, Jr, Lee T. PLoS One. 2009;4:e5558. doi: 10.1371/journal.pone.0005558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gilly WF, Richmond TA, Duda TF, Jr, Elliger C, Lebaric Z, Schulz J, Bingham JP, Sweedler JV. J Exp Biol. 2011;214:147. doi: 10.1242/jeb.046086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Garrett JE, Buczek O, Watkins M, Olivera BM, Bulaj G. Biochem Biophys Res Commun. 2005;328:362. doi: 10.1016/j.bbrc.2004.12.178. [DOI] [PubMed] [Google Scholar]
- (60).Duda TF, Jr, Bolin MB, Meyer CP, Kohn AJ. Mol Phylogenet Evol. 2008;49:867. doi: 10.1016/j.ympev.2008.08.009. [DOI] [PubMed] [Google Scholar]
- (61).Duda TF, Jr, Kohn AJ, Matheny AM. Biol Bull. 2009;217:292. doi: 10.1086/BBLv217n3p292. [DOI] [PubMed] [Google Scholar]
- (62).Duda TF, Jr, Palumbi SR. Proc Nat Acad Sc USA. 1999;96:6820. doi: 10.1073/pnas.96.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Conticello SG, Gilad Y, Avidan N, Ben-Asher E, Levy Z, Fainzilber M. Mol Biol Evol. 2001;18:120. doi: 10.1093/oxfordjournals.molbev.a003786. [DOI] [PubMed] [Google Scholar]
- (64).Puillandre N, Watkins M, Olivera BM. J Mol Evol. 2010;70:190. doi: 10.1007/s00239-010-9321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chang D, Duda TF., Jr Mol Biol Evol. 2012;29:2019. doi: 10.1093/molbev/mss068. [DOI] [PubMed] [Google Scholar]
- (66).Duda TF, Jr, Palumbi SR. Mol Biol Evol. 2000;17:1286. doi: 10.1093/oxfordjournals.molbev.a026412. [DOI] [PubMed] [Google Scholar]
- (67).Duda TF, Jr, Chang D, Lewis BD, Lee T. PloS One. 2009;4:e6245. doi: 10.1371/journal.pone.0006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Buczek O, Bulaj G, Olivera BM. Cell Mol Life Sci. 2005;62:3067. doi: 10.1007/s00018-005-5283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Bulaj G, Olivera BM. Antioxid Redox Signal. 2008;10:141. doi: 10.1089/ars.2007.1856. [DOI] [PubMed] [Google Scholar]
- (70).Walker CS, Jensen S, Ellison M, Matta JA, Lee WY, Imperial JS, Duclos N, Brockie PJ, Madsen DM, Isaac JTR, Olivera B, et al. Curr Biol. 2009;19:900. doi: 10.1016/j.cub.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Safavi-Hemami H, Siero WA, Gorasia DG, Young ND, Macmillan D, Williamson NA, Purcell AW. J Proteome Res. 2011;10:3904. doi: 10.1021/pr1012976. [DOI] [PubMed] [Google Scholar]
- (72).Rivera-Ortiz JA, Cano H, Marí F. Peptides. 2011;32:306. doi: 10.1016/j.peptides.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Dutertre S, Biass D, Stocklin R, Favreau P. Toxicon. 2009;55:1453. doi: 10.1016/j.toxicon.2010.02.025. [DOI] [PubMed] [Google Scholar]
- (74).Chun JBS, Baker MR, Kim DH, Leroy M, Toribo P, Bingham J-P. Toxicon. 2012;60:83. doi: 10.1016/j.toxicon.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Franco A, Pisarewicz K, Moller C, Mora D, Fields GB, Marì F. Prog Mol Subcell Biol. 2006;43:83. doi: 10.1007/978-3-540-30880-5_4. [DOI] [PubMed] [Google Scholar]
- (76).Craik DJ, Adams DJ. ACS Chem Biol. 2007;2:457. doi: 10.1021/cb700091j. [DOI] [PubMed] [Google Scholar]
- (77).Miljanich GP. Curr Med Chem. 2004;11:3029. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- (78).Nadasdi L, Yamashiro D, Chung D, Tarczy-Hornoch K, Adriaenssens P, Ramachandran J. Biochemistry. 1995;34:8076. doi: 10.1021/bi00025a013. [DOI] [PubMed] [Google Scholar]
- (79).Armishaw CJ, Jensen AA, Balle LD, Scott KC, Sorensen L, Stromgaard K. Antioxid Redox Signal. 2011;14:65. doi: 10.1089/ars.2010.3458. [DOI] [PubMed] [Google Scholar]
- (80).Quiram PA, Sine SM. J Biol Chem. 1998;273:11007. doi: 10.1074/jbc.273.18.11007. [DOI] [PubMed] [Google Scholar]
- (81).Al-Sabi A, Lennartz D, Ferber M, Gulyas J, Rivier JE, Olivera BM, Carlomagno T, Terlau H. Biochemistry. 2004;43:8625. doi: 10.1021/bi0495681. [DOI] [PubMed] [Google Scholar]
- (82).Kasheverov IE, Zhmak MN, Vulfius CA, Gorbacheva EV, Mordvintsev DY, Utkin YN, van Elk R, Smit AB, Tsetlin VI. FEBS J. 2006;273:4470. doi: 10.1111/j.1742-4658.2006.05453.x. [DOI] [PubMed] [Google Scholar]
- (83).Armishaw C, Jensen AA, Balle T, Clark RJ, Harpsoe K, Skonberg C, Liljefors T, Stromgaard K. J Biol Chem. 2009;284:9498. doi: 10.1074/jbc.M806136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Armishaw CJ, Singh N, Medina-Franco JL, Clark RJ, Scott KC, Houghten RA, Jensen AA. J Biol Chem. 2010;285:1809. doi: 10.1074/jbc.M109.071183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Pucci L, Grazioso G, Dallanoce C, Rizzi L, De Micheli C, Clementi F, Bertrand S, Bertrand D, Longhi R, De Amici M, Gotti C. FASEB J. 2011;25:3775. doi: 10.1096/fj.10-179853. [DOI] [PubMed] [Google Scholar]
- (86).Han TS, Zhang M-M, Walewska A, Gruszczynski P, Robertson CR, Cheatham TE, 3rd, Yoshikami D, Olivera BM, Bulaj G. ChemMedChem. 2009;4:406. doi: 10.1002/cmdc.200800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Carstens BB, Clark RJ, Daly NL, Harvey PJ, Kaas Q, Craik DJ. Curr Pharm Des. 2011;17:4242. doi: 10.2174/138161211798999401. [DOI] [PubMed] [Google Scholar]
- (88).Blanchfield JT, Dutton JL, Hogg RC, Gallagher OP, Craik DJ, Jones A, Adams DJ, Lewis RJ, Alewood PF, Toth I. J Med Chem. 2003;46:1266. doi: 10.1021/jm020426j. [DOI] [PubMed] [Google Scholar]
- (89).Clark RJ, Fischer H, Dempster L, Daly NL, Rosengren KJ, Nevin ST, Meunier FA, Adams DJ, Craik DJ. Proc Natl Acad Sci USA. 2005;102:13767. doi: 10.1073/pnas.0504613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Lovelace ES, Armishaw CJ, Colgrave ML, Wahlstrom ME, Alewood PF, Daly NL, Craik DJ. J Med Chem. 2006;49:6561. doi: 10.1021/jm060299h. [DOI] [PubMed] [Google Scholar]
- (91).Clark RJ, Akcan M, Kaas Q, Daly NL, Craik DJ. Toxicon. 2010 doi: 10.1016/j.toxicon.2010.12.003. [DOI] [PubMed] [Google Scholar]
- (92).MacRaild CA, Illesinghe J, van Lierop BJ, Townsend AL, Chebib M, Livett BG, Robinson AJ, Norton RS. J Med Chem. 2009;52:755. doi: 10.1021/jm8011504. [DOI] [PubMed] [Google Scholar]
- (93).Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A, Norton RS, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Chem Biol. 2007;14:399. doi: 10.1016/j.chembiol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- (94).Armishaw CJ, Daly NL, Nevin ST, Adams DJ, Craik DJ, Alewood PF. J Biol Chem. 2006;281:14136. doi: 10.1074/jbc.M512419200. [DOI] [PubMed] [Google Scholar]
- (95).de Araujo AD, Callaghan B, Nevin ST, Daly NL, Craik DJ, Moretta M, Hopping G, Christie MJ, Adams DJ, Alewood PF. Angew Chem Int Ed Engl. 2011;50:6527. doi: 10.1002/anie.201101642. [DOI] [PubMed] [Google Scholar]
- (96).Daly NL, Craik DJ. IUBMB Life. 2009;61:144. doi: 10.1002/iub.158. [DOI] [PubMed] [Google Scholar]
- (97).Marx UC, Daly NL, Craik DJ. Magn Reson Chem. 2006;44:S41. doi: 10.1002/mrc.1821. [DOI] [PubMed] [Google Scholar]
- (98).Millard EL, Daly NL, Craik DJ. Eur J Biochem. 2004;271:2320. doi: 10.1111/j.1432-1033.2004.04148.x. [DOI] [PubMed] [Google Scholar]
- (99).Muttenthaler M, Akondi KB, Alewood PF. Curr Pharm Des. 2011;17:4226. doi: 10.2174/138161211798999384. [DOI] [PubMed] [Google Scholar]
- (100).Balaji RA, Ohtake A, Sato K, Gopalakrishnakone P, Kini RM, Seow KT, Bay BH. J Biol Chem. 2000;275:39516. doi: 10.1074/jbc.M006354200. [DOI] [PubMed] [Google Scholar]
- (101).Sharpe IA, Gehrmann J, Loughnan ML, Thomas L, Adams DA, Atkins A, Palant E, Craik DJ, Adams DJ, Alewood PF, Lewis RJ. Nat Neurosci. 2001;4:902. doi: 10.1038/nn0901-902. [DOI] [PubMed] [Google Scholar]
- (102).Kang TS, Radic Z, Talley TT, Jois SD, Taylor P, Kini RM. Biochemistry. 2007;46:3338. doi: 10.1021/bi061969o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Dutton JL, Bansal PS, Hogg RC, Adams DJ, Alewood PF, Craik DJ. J Biol Chem. 2002;277:48849. doi: 10.1074/jbc.M208842200. [DOI] [PubMed] [Google Scholar]
- (104).Grishin AA, Wang C-IA, Muttenthaler M, Alewood PF, Lewis RJ, Adams DJ. J Biol Chem. 2010;285:22254. doi: 10.1074/jbc.M110.111880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Safavi-Hemami H, Gorasia DG, Steiner AM, Williamson NA, Karas JA, Gajewiak J, Olivera BM, Bulaj G, Purcell AW. J Biol Chem. 2012;41:34288. doi: 10.1074/jbc.M112.366781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Möller C, Rahmankhah S, Lauer-Fields J, Bubis J, Fields GB, Marí F. Biochemistry. 2005;44:15986. doi: 10.1021/bi0511181. [DOI] [PubMed] [Google Scholar]
- (107).Du W-H, Han Y-H, Huang F-J, Li J, Chi C-W, Fang W-H. FEBS J. 2007;274:2596. doi: 10.1111/j.1742-4658.2007.05795.x. [DOI] [PubMed] [Google Scholar]
- (108).Poppe L, Hui JO, Ligutti J, Murray JK, Schnier PD. Anal Chem. 2012;84:262. doi: 10.1021/ac203078x. [DOI] [PubMed] [Google Scholar]
- (109).Gracy J, Le-Nguyen D, Gelly JC, Kaas Q, Heitz A, Chiche L. Nucleic Acids Res. 2008;36:D314. doi: 10.1093/nar/gkm939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Heinemann SH, Leipold E. Cell Mol Life Sci. 2007;64:1329. doi: 10.1007/s00018-007-6565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Daly NL, Ekberg JA, Thomas L, Adams DJ, Lewis RJ, Craik DJ. J Biol Chem. 2004;279:25774. doi: 10.1074/jbc.M313002200. [DOI] [PubMed] [Google Scholar]
- (112).Buczek O, Wei D, Babon JJ, Yang X, Fiedler B, Chen P, Yoshikami D, Olivera BM, Bulaj G, Norton RS. Biochemistry. 2007;46:9929. doi: 10.1021/bi700797f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Miles LA, Dy CY, Nielsen J, Barnham KJ, Hinds MG, Olivera BM, Bulaj G, Norton RS. J Biol Chem. 2002;277:43033. doi: 10.1074/jbc.M206690200. [DOI] [PubMed] [Google Scholar]
- (114).Kindahl L, Sandström C, Craig AG, Norberg T, Kenne L. Can J Chem. 2002;80:1022. [Google Scholar]
- (115).McIntosh M, Cruz LJ, Hunkapiller MW, Gray WR, Olivera BM. Arch Biochem Biophys. 1982;218:329. doi: 10.1016/0003-9861(82)90351-4. [DOI] [PubMed] [Google Scholar]
- (116).Dutertre S, Croker D, Daly NL, Andersson A, Muttenthaler M, Lumsden NG, Craik DJ, Alewood PF, Guillon G, Lewis RJ. J Biol Chem. 2008;283:7100. doi: 10.1074/jbc.M706477200. [DOI] [PubMed] [Google Scholar]
- (117).Skjaerbaek N, Nielsen KJ, Lewis RJ, Alewood P, Craik DJ. J Biol Chem. 1997;272:2291. doi: 10.1074/jbc.272.4.2291. [DOI] [PubMed] [Google Scholar]
- (118).Xia Z, Chen Y, Zhu Y, Wang F, Xu X, Zhan J. BioDrugs. 2006;20:275. doi: 10.2165/00063030-200620050-00003. [DOI] [PubMed] [Google Scholar]
- (119).Pi C, Liu J, Wang L, Jiang X, Liu Y, Peng C, Chen S, Xu A. J Biotechnol. 2007;128:184. doi: 10.1016/j.jbiotec.2006.09.014. [DOI] [PubMed] [Google Scholar]
- (120).Becker S, Terlau H. Appl Microbiol Biotechnol. 2008;79:1. doi: 10.1007/s00253-008-1385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Gray WR, Luque FA, Galyean R, Atherton E, Sheppard RC, Stone BL, Reyes A, Alford J, McIntosh M, et al. Biochemistry. 1984;23:2796. doi: 10.1021/bi00307a040. [DOI] [PubMed] [Google Scholar]
- (122).Gray WR, Rivier JE, Galyean R, Cruz LJ, Olivera BM. J Biol Chem. 1983;258:12247. [PubMed] [Google Scholar]
- (123).Nishiuchi Y, Sakakibara S. FEBS Lett. 1982;148:260. doi: 10.1016/0014-5793(82)80820-x. [DOI] [PubMed] [Google Scholar]
- (124).Rivier J, Galyean R, Gray WR, Azimi-Zonooz A, McIntosh JM, Cruz LJ, Olivera BM. J Biol Chem. 1987;262:1194. [PubMed] [Google Scholar]
- (125).Nishiuchi Y, Kumagaye K, Noda Y, Watanabe TX, Sakakibara S. Biopolymers. 1986;25 Suppl:S61. [PubMed] [Google Scholar]
- (126).Merrifield RB. J Am Chem Soc. 1963;85:2149. [Google Scholar]
- (127).Doscher MS. Methods Enzymol. 1977;47:578. doi: 10.1016/0076-6879(77)47050-2. [DOI] [PubMed] [Google Scholar]
- (128).Albericio F, Grandas A, Porta A, Pedroso E, Giralt E. Synthesis. 1987;3:271. [Google Scholar]
- (129).Angeletti RH, Bonewald LF, Fields GB. Methods Enzymol. 1997;289:697. doi: 10.1016/s0076-6879(97)89071-4. [DOI] [PubMed] [Google Scholar]
- (130).Schnölzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int J Pept Protein Res. 1992;40:180. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- (131).Miranda LP, Alewood PF. Proc Nat Acad Sc USA. 1999;96:1181. doi: 10.1073/pnas.96.4.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- (133).Kent SBH. Chem Soc Rev. 2009;38:338. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- (134).Houghten RA, Pinilla C, Appel JR, Blondelle SE, Dooley CT, Eichler J, Nefzi A, Ostresh JM. J Med Chem. 1999;42:3743. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- (135).Houghten RA. Annu Rev Pharmacol Toxicol. 2000;40:273. doi: 10.1146/annurev.pharmtox.40.1.273. [DOI] [PubMed] [Google Scholar]
- (136).Muttenthaler M, Nevin ST, Grishin AA, Ngo ST, Choy PT, Daly NL, Hu S-H, Armishaw CJ, Wang C-IA, Lewis RJ, Martin JL, et al. J Am Chem Soc. 2010;132:3514. doi: 10.1021/ja910602h. [DOI] [PubMed] [Google Scholar]
- (137).Anfinsen CB. Science. 1973;181:223. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- (138).Arolas JL, Aviles FX, Chang J-Y, Ventura S. Trends in Biochem Sci. 2006;31:292. doi: 10.1016/j.tibs.2006.03.005. [DOI] [PubMed] [Google Scholar]
- (139).Benham CJ, Jafri MS. Protein Sci. 1993;2:41. doi: 10.1002/pro.5560020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Clark RJ, Daly NL, Halai R, Nevin ST, Adams DJ, Craik DJ. FEBS Lett. 2008;582:597. doi: 10.1016/j.febslet.2008.01.027. [DOI] [PubMed] [Google Scholar]
- (141).Price-Carter M, Hull MS, Goldenberg DP. Biochemistry. 1998;37:9851. doi: 10.1021/bi9803978. [DOI] [PubMed] [Google Scholar]
- (142).Flinn JP, Pallaghy PK, Lew MJ, Murphy R, Angus JA, Norton RS. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1999;1434:177. doi: 10.1016/s0167-4838(99)00165-x. [DOI] [PubMed] [Google Scholar]
- (143).Gehrmann J, Alewood PF, Craik DJ. J Mol Biol. 1998;278:401. doi: 10.1006/jmbi.1998.1701. [DOI] [PubMed] [Google Scholar]
- (144).Gething MJ, Sambrook J. Nature. 1992;355:33. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- (145).Hartl FU. Nature. 1996;381:571. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- (146).Dobson CM. Nature. 2003;426:884. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- (147).Moroder L, Musiol H-J, Götz M, Renner C. Biopolymers. 2005;80:85. doi: 10.1002/bip.20174. [DOI] [PubMed] [Google Scholar]
- (148).Akaji K, Kiso Y. In: Houben-Weyl, Synthesis of Peptides and Peptidomimetics. Goodman M, Felix A, Moroder L, Toniolo C, editors. E22b. Georg Thieme Verlag; Stuttgart: 2004. [Google Scholar]
- (149).Moroder L, Musiol HJ, Schaschke N, Chen L, Hargittai B, Barany G. In: Houben-Weyl, Synthesis of Peptides and Peptidomimetics. Goodman M, Felix A, Moroder L, Toniolo C, editors. E22a. Georg Thieme Verlag; Stuttgart: 2004. [Google Scholar]
- (150).Armishaw CJ, Daly NL, Nevin ST, Adams DJ, Craik DJ, Alewood PF. J Biol Chem. 2006;281:14136. doi: 10.1074/jbc.M512419200. [DOI] [PubMed] [Google Scholar]
- (151).Kamber B, Hartmann A, Eisler K, Riniker B, Rink H, Sieber P, Rittel W. Helv Chim Acta. 1980;63:899. doi: 10.1002/hlca.19760590510. [DOI] [PubMed] [Google Scholar]
- (152).Fujii N, Otaka A, Funakoshi S, Bessho K, Watanabe T, Akaji K, Yajima H. Chem Pharm Bull. 1987;35:2339. doi: 10.1248/cpb.35.2339. [DOI] [PubMed] [Google Scholar]
- (153).Muttenthaler M, Ramos YG, Feytens D, de Araujo AD, Alewood PF. Biopolymers. 2010;94:423. doi: 10.1002/bip.21502. [DOI] [PubMed] [Google Scholar]
- (154).Lamthanh H, Roumestand C, Deprun C, Menez A. Int J Pept Protein Res. 1993;41:85. doi: 10.1111/j.1399-3011.1993.tb00118.x. [DOI] [PubMed] [Google Scholar]
- (155).Isidro-Llobet A, Alvarez M, Albericio F. Chem Rev. 2009;109:2455. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- (156).Flemer S., Jr Mini-Rev Org Chem. 2009;6:196. [Google Scholar]
- (157).Ponsati B, Giralt E, Andreu D. Tetrahedron. 1990;46:8255. [Google Scholar]
- (158).Monje VD, Haack JA, Naisbitt SR, Miljanich G, Ramachandran J, Nasdasdi L, Olivera BM, Hillyard DR, Gray WR. Neuropharmacology. 1993;32:1141. doi: 10.1016/0028-3908(93)90008-q. [DOI] [PubMed] [Google Scholar]
- (159).Durieux JP, Nyfeler R. Pept Chem, Struct Biol, Proc Am Pept Symp, 14th. 1996:42. [Google Scholar]
- (160).Cuthbertson A, Indrevoll B. Tetrahedron Lett. 2000;41:3661. [Google Scholar]
- (161).Nielsen JS, Buczek P, Bulaj G. J Pept Sci. 2004;10:249. doi: 10.1002/psc.531. [DOI] [PubMed] [Google Scholar]
- (162).Cuthbertson A, Indrevoll B. Org Lett. 2003;5:2955. doi: 10.1021/ol035105w. [DOI] [PubMed] [Google Scholar]
- (163).Otaka A, Koide T, Shide A, Fujii N. Tetrahedron Lett. 1991;32:1223. [Google Scholar]
- (164).Atherton E, Sheppard RC, Ward P. J Chem Soc, Perkin Trans 1. 1985;10:2065. [Google Scholar]
- (165).Szabo I, Schlosser G, Hudecz F, Mezo G. Biopolymers. 2007;88:20. doi: 10.1002/bip.20616. [DOI] [PubMed] [Google Scholar]
- (166).Green BR, Bulaj G. Protein Pept Lett. 2006;13:67. doi: 10.2174/092986606774502162. [DOI] [PubMed] [Google Scholar]
- (167).Altamirano MM, Garcia C, Possani LD, Fersht AR. Nat biotechnol. 1999;17:187. doi: 10.1038/6192. [DOI] [PubMed] [Google Scholar]
- (168).Annis I, Chen L, Barany G. J Am Chem Soc. 1998;120:7226. [Google Scholar]
- (169).Darlak K, Long DW, Czerwinski A, Darlak M, Valenzuela F, Spatola AF, Barany G. J Pept Res. 2004;63:303. doi: 10.1111/j.1399-3011.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- (170).Hargittai B, Annis I, Barany G. Pept New Millennium, Proc Am Pept Symp, 16th. 2000:94. [Google Scholar]
- (171).Steiner AM, Bulaj G. J Pept Sci. 2011;17:1. doi: 10.1002/psc.1283. [DOI] [PubMed] [Google Scholar]
- (172).Hopping G, Lewis RJ, Alewood PF. Aust J Chem. 2009;62:1333. [Google Scholar]
- (173).Buczek P, Buczek O, Bulaj G. Biopolymers. 2005;80:50. doi: 10.1002/bip.20211. [DOI] [PubMed] [Google Scholar]
- (174).Munson MC, Barany G. J Am Chem Soc. 1993;115:10203. [Google Scholar]
- (175).Hargittai B, Barany G. J Pept Res. 1999;54:468. doi: 10.1034/j.1399-3011.1999.00127.x. [DOI] [PubMed] [Google Scholar]
- (176).Galanis AS, Albericio F, Grotli M. Biopolymers. 2009;92:23. doi: 10.1002/bip.21116. [DOI] [PubMed] [Google Scholar]
- (177).Alewood PF. In: Ramage R, Epton R, editors. Peptides: Proceedings of the Twenty-Fourth European Peptide Symposium; Kingswinford: Mayflower Scientific; 1996. [Google Scholar]
- (178).Muttenthaler M, Akondi Kalyana B, Alewood Paul F. Curr Pharm Des. 2011;17:4226. doi: 10.2174/138161211798999384. [DOI] [PubMed] [Google Scholar]
- (179).Jin A-H, Brandstaetter H, Nevin ST, Tan CC, Clark RJ, Adams DJ, Alewood PF, Craik DJ, Daly NL. BMC Struct Biol. 2007;7:28. doi: 10.1186/1472-6807-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Brust A, Tickle AE. J Pept Sci. 2007;13:133. doi: 10.1002/psc.825. [DOI] [PubMed] [Google Scholar]
- (181).Muttenthaler M, Andersson A, de Araujo AD, Dekan Z, Lewis RJ, Alewood PF. J Med Chem. 2010;53:8585. doi: 10.1021/jm100989w. [DOI] [PubMed] [Google Scholar]
- (182).MacRaild CA, Illesinghe J, van Lierop BJ, Townsend AL, Chebib M, Livett BG, Robinson AJ, Norton RS. J Med Chem. 2009;52:755. doi: 10.1021/jm8011504. [DOI] [PubMed] [Google Scholar]
- (183).Lamthanh H, Jegou-Matheron C, Servent D, Menez A, Lancelin JM. FEBS Lett. 1999;454:293. doi: 10.1016/s0014-5793(99)00831-5. [DOI] [PubMed] [Google Scholar]
- (184).Hargittai B, Sole NA, Groebe DR, Abramson SN, Barany G. J Med Chem. 2000;43:4787. doi: 10.1021/jm990635c. [DOI] [PubMed] [Google Scholar]
- (185).Bondebjerg J, Grunnet M, Jespersen T, Meldal M. ChemBioChem. 2003;4:186. doi: 10.1002/cbic.200390030. [DOI] [PubMed] [Google Scholar]
- (186).Dekan Z, Vetter I, Daly NL, Craik DJ, Lewis RJ, Alewood PF. J Am Chem Soc. 2011;133:15866. doi: 10.1021/ja206408q. [DOI] [PubMed] [Google Scholar]
- (187).Böck A, Forchhamer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Mol Microbiol. 1991;5:515. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- (188).Xu X-M, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. PLoS Biology. 2007;5:96. doi: 10.1371/journal.pbio.0050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Bösl MR, Takaku K, Oshima M, Nishimura S, Taketo MM. Proc Nat Acad Sc USA. 1997;94:5531. doi: 10.1073/pnas.94.11.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Stadtman TC. Annu Rev Biochem. 1996;65:83. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- (191).Johansson L, Gafvelin G, Arner ESJ. Biochim Biophys Acta, Gen Subj. 2005;1726:1. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- (192).Müller S, Senn H, Gsell B, Vetter W, Baron C, Boeck A. Biochemistry. 1994;33:3404. doi: 10.1021/bi00177a034. [DOI] [PubMed] [Google Scholar]
- (193).Sanchez JF, Hoh F, Strub MP, Strub JM, Van Dorsselaer A, Lehrer R, Ganz T, Chavanieu A, Calas B, Dumas C, Aumelas A. Acta Crystallogr D Biol Crystallogr. 2001;57:1677. doi: 10.1107/s0907444901012598. [DOI] [PubMed] [Google Scholar]
- (194).Strub M-P, Hoh F, Sanchez J-F, Strub JM, Bock A, Aumelas A, Dumas C. Structure. 2003;11:1359. doi: 10.1016/j.str.2003.09.014. [DOI] [PubMed] [Google Scholar]
- (195).Duddeck H. Prog Nucl Magn Reson Spectrosc. 1995;27:1. [Google Scholar]
- (196).Boles JO, Tolleson WH, Schmidt JC, Dunlap RB, Odom JD. J Biol Chem. 1992;267:22217. [PubMed] [Google Scholar]
- (197).Mobli M, Morgenstern D, King GF, Alewood PF, Muttenthaler M. Angew Chem, Int Ed. 2011;50:11952. doi: 10.1002/anie.201104169. [DOI] [PubMed] [Google Scholar]
- (198).Mobli M, de Araujo AD, Lambert LK, Pierens GK, Windley MJ, Nicholson GM, Alewood PF, King GF. Angew Chem Int Ed Engl. 2009;48:9312. doi: 10.1002/anie.200905206. [DOI] [PubMed] [Google Scholar]
- (199).Huber RE, Criddle RS. Arch Biochem Biophys. 1967;122:164. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- (200).Pearson RG, Sobel HR, Songstad J. J Am Chem Soc. 1968;90:319. [Google Scholar]
- (201).Hondal RJ, Nilsson BL, Raines RT. J Am Chem Soc. 2001;123:5140. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- (202).Zhong L, Arner ES, Holmgren A. Proc Nat Acad Sc USA. 2000;97:5854. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Beld J, Woycechowsky KJ, Hilvert D. Biochemistry. 2008;47:6985. doi: 10.1021/bi8008906. [DOI] [PubMed] [Google Scholar]
- (204).Koide T, Itoh H, Otaka A, Yasui H, Kuroda M, Esaki N, Soda K, Fujii N. Chem Pharm Bull. 1993;41:502. doi: 10.1248/cpb.41.502. [DOI] [PubMed] [Google Scholar]
- (205).Nygard B. Arkiv foer Kemi. 1967;27:341. [Google Scholar]
- (206).Arnold AP, Tan KS, Rabenstein DL. Inorganic Chemistry. 1986;25:2433. [Google Scholar]
- (207).Walewska A, Zhang M-M, Skalicky Jack J, Yoshikami D, Olivera Baldomero M, Bulaj G. Angew Chem Int Ed Engl. 2009;48:2221. doi: 10.1002/anie.200806085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Dantas de Araujo A, Callaghan B, Nevin ST, Daly NL, Craik DJ, Moretta M, Hopping G, Christie MJ, Adams DJ, Alewood PF. Angew Chem Int Ed Engl. 2011;50:6527. doi: 10.1002/anie.201101642. [DOI] [PubMed] [Google Scholar]
- (209).Moroder L, Besse D, Musiol H-J, Rudolph-Böhner S, Sideler F. Biopolymers. 1996;40:207. doi: 10.1002/(sici)1097-0282(1996)40:2<207::aid-bip2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- (210).Gowd KH, Yarotskyy V, Elmslie KS, Skalicky JJ, Olivera BM, Bulaj G. Biochemistry. 2010;49:2741. doi: 10.1021/bi902137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Han TS, Zhang M-M, Gowd KH, Walewska A, Yoshikami D, Olivera BM, Bulaj G. ACS Med Chem Lett. 2010;1:140. doi: 10.1021/ml900017q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Muttenthaler M, Alewood PF. J Pept Sci. 2008;14:1223. doi: 10.1002/psc.1075. [DOI] [PubMed] [Google Scholar]
- (213).Deechongkit S, Kelly Jeffery W. J Am Chem Soc. 2002;124:4980. doi: 10.1021/ja0123608. [DOI] [PubMed] [Google Scholar]
- (214).Hubbard SJ. Biochim Biophys Acta. 1998;1382:191. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- (215).Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. Angew Chem, Int Ed. 2010;49:6545. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- (216).Armishaw CJ, Dutton JL, Craik DJ, Alewood PF. Biopolymers. 2010;94:307. doi: 10.1002/bip.21360. [DOI] [PubMed] [Google Scholar]
- (217).Clark RJ, Fischer H, Dempster L, Daly NL, Rosengren KJ, Nevin ST, Meunier FA, Adams DJ, Craik DJ. Proc Nat Acad Sc USA. 2005;102:13767. doi: 10.1073/pnas.0504613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Dekan Z, Paczkowski FA, Lewis RJ, Alewood PF. Int J Pept Res Ther. 2007;13:307. [Google Scholar]
- (219).Halai R, Craik DJ. Nat Prod Rep. 2009;26:526. doi: 10.1039/b819311h. [DOI] [PubMed] [Google Scholar]
- (220).Lewis RJ, Dutertre S, Vetter I, MacDonald JC. Pharmacol Rev. 2012;64:259. doi: 10.1124/pr.111.005322. [DOI] [PubMed] [Google Scholar]
- (221).Huang X, Dong F, Zhou HX. J Am Chem Soc. 2005;127:6836. doi: 10.1021/ja042641q. [DOI] [PubMed] [Google Scholar]
- (222).Gorham RD, Jr, Kieslich CA, Morikis D. Ann Biomed Eng. 2011;39:1252. doi: 10.1007/s10439-010-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Sinha N, Smith-Gill SJ. Curr Protein Pept Sci. 2002;3:601. doi: 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- (224).Mouhat S, De Waard M, Sabatier J-M. J Pept Sci. 2005;11:65. doi: 10.1002/psc.630. [DOI] [PubMed] [Google Scholar]
- (225).Olivera BM, Miljanich GP, Ramachandran J, Adams ME. Annu Rev Biochem. 1994;63:823. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- (226).Smith HS, Deer TR. Ther Clin Risk Manag. 2009;5:521. doi: 10.2147/tcrm.s4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Schroeder CI, Smythe ML, Lewis RJ. Mol Divers. 2004;8:127. doi: 10.1023/b:modi.0000025656.79632.86. [DOI] [PubMed] [Google Scholar]
- (228).Lew MJ, Flinn JP, Pallaghy PK, Murphy R, Whorlow SL, Wright CE, Norton RS, Angus JA. J Biol Chem. 1997;272:12014. doi: 10.1074/jbc.272.18.12014. [DOI] [PubMed] [Google Scholar]
- (229).Nielsen KJ, Adams D, Thomas L, Bond T, Alewood PF, Craik DJ, Lewis RJ. J Mol Biol. 1999;289:1405. doi: 10.1006/jmbi.1999.2817. [DOI] [PubMed] [Google Scholar]
- (230).Nielsen KJ, Thomas L, Lewis RJ, Alewood PF, Craik DJ. J Mol Biol. 1996;263:297. doi: 10.1006/jmbi.1996.0576. [DOI] [PubMed] [Google Scholar]
- (231).Nielsen KJ, Schroeder T, Lewis R. J Mol Recognit. 2000;13:55. doi: 10.1002/(SICI)1099-1352(200003/04)13:2<55::AID-JMR488>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- (232).Kobayashi K, Sasaki T, Sato K, Kohno T. Biochemistry. 2000;39:14761. doi: 10.1021/bi001506x. [DOI] [PubMed] [Google Scholar]
- (233).Flinn JP, Pallaghy PK, Lew MJ, Murphy R, Angus JA, Norton RS. Eur J Biochem. 1999;262:447. doi: 10.1046/j.1432-1327.1999.00383.x. [DOI] [PubMed] [Google Scholar]
- (234).Kim JI, Takahashi M, Ogura A, Kohno T, Kudo Y, Sato K. J Biol Chem. 1994;269:23876. [PubMed] [Google Scholar]
- (235).Sato K, Park NG, Kohno T, Maeda T, Kim JI, Kato R, Takahashi M. Biochem Biophys Res Commun. 1993;194:1292. doi: 10.1006/bbrc.1993.1964. [DOI] [PubMed] [Google Scholar]
- (236).Nielsen KJ, Adams DA, Alewood PF, Lewis RJ, Thomas L, Schroeder T, Craik DJ. Biochemistry. 1999;38:6741. doi: 10.1021/bi982980u. [DOI] [PubMed] [Google Scholar]
- (237).Schroeder CI, Adams D, Thomas L, Alewood PF, Lewis RJ. Biopolymers. 2012;98:161. doi: 10.1002/bip.22032. [DOI] [PubMed] [Google Scholar]
- (238).Gandia L, Lara B, Imperial JS, Villarroya M, Albillos A, Maroto R, Garcia AG, Olivera BM. Pflugers Arch. 1997;435:55. doi: 10.1007/s004240050483. [DOI] [PubMed] [Google Scholar]
- (239).Nielsen KJ, Adams D, Thomas L, Bond T, Alewood PF, Craik DJ, Lewis RJ. J Mol Biol. 1999;289:1405. doi: 10.1006/jmbi.1999.2817. [DOI] [PubMed] [Google Scholar]
- (240).Mould J, Yasuda T, Schroeder CI, Beedle AM, Doering CJ, Zamponi GW, Adams DJ, Lewis RJ. J Biol Chem. 2004;279:34705. doi: 10.1074/jbc.M310848200. [DOI] [PubMed] [Google Scholar]
- (241).Andersson A, Baell JB, Duggan PJ, Graham JE, Lewis RJ, Lumsden NG, Tranberg CE, Tuck KL, Yang A. Bioorg Med Chem. 2009;17:6659. doi: 10.1016/j.bmc.2009.07.063. [DOI] [PubMed] [Google Scholar]
- (242).Tranberg CE, Yang A, Vetter I, McArthur JR, Baell JB, Lewis RJ, Tuck KL, Duggan PJ. Mar Drugs. 2012;10:2349. doi: 10.3390/md10102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Favreau P, Benoit E, Hocking HG, Carlier L, D’hoedt D, Leipold E, Markgraf R, Schlumberger S, Cordova MA, Gaertner H, Paolini-Bertrand M, et al. Br J Pharmacol. 2012;166:1654. doi: 10.1111/j.1476-5381.2012.01837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Nature. 2012;486:135. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J, Wang J, et al. Nature. 2012;486:130. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. J Biol Chem. 1985;260:9280. [PubMed] [Google Scholar]
- (247).Lewis RJ, Schroeder CI, Ekberg J, Nielsen KJ, Loughnan M, Thomas L, Adams DA, Drinkwater R, Adams DJ, Alewood PF. Mol Pharmacol. 2007;71:676. doi: 10.1124/mol.106.028225. [DOI] [PubMed] [Google Scholar]
- (248).Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, Smith BJ, Yoshikami D, Olivera BM. Biochemistry. 2005;44:7259. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- (249).Wang CZ, Zhang H, Jiang H, Lu W, Zhao ZQ, Chi CW. Toxicon. 2006;47:122. doi: 10.1016/j.toxicon.2005.10.008. [DOI] [PubMed] [Google Scholar]
- (250).Shon KJ, Olivera BM, Watkins M, Jacobsen RB, Gray WR, Floresca CZ, Cruz LJ, Hillyard DR, Brink A, Terlau H, Yoshikami D. J Neurosci. 1998;18:4473. doi: 10.1523/JNEUROSCI.18-12-04473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. Biochemistry. 2002;41:15388. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]
- (252).Hui K, McIntyre D, French RJ. J Gen Physiol. 2003;122:63. doi: 10.1085/jgp.200308842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Chang NS, French RJ, Lipkind GM, Fozzard HA, Dudley S., Jr Biochemistry. 1998;37:4407. doi: 10.1021/bi9724927. [DOI] [PubMed] [Google Scholar]
- (254).Chen R, Chung SH. Biophys J. 2012;102:483. doi: 10.1016/j.bpj.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Schroeder CI, Ekberg J, Nielsen KJ, Adams D, Loughnan ML, Thomas L, Adams DJ, Alewood PF, Lewis RJ. J Biol Chem. 2008;283:21621. doi: 10.1074/jbc.M802852200. [DOI] [PubMed] [Google Scholar]
- (256).Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur JR, French RJ, Gulyas J, Rivier JE, et al. J Biol Chem. 2007;282:30699. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- (257).McArthur JR, Singh G, McMaster D, Winkfein R, Tieleman DP, French RJ. Mol Pharmacol. 2011;80:573. doi: 10.1124/mol.111.073460. [DOI] [PubMed] [Google Scholar]
- (258).Tietze AA, Tietze D, Ohlenschlager O, Leipold E, Ullrich F, Kuhl T, Mischo A, Buntkowsky G, Gorlach M, Heinemann SH, Imhof D. Angew Chem Int Ed Engl. 2012;51:4058. doi: 10.1002/anie.201107011. [DOI] [PubMed] [Google Scholar]
- (259).Zhang MM, Wilson MJ, Azam L, Gajewiak J, Rivier JE, Bulaj G, Olivera BM, Yoshikami D. Br J Pharmacol. 2012;168:1597. doi: 10.1111/bph.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Corringer PJ, Le Novere N, Changeux JP. Annu Rev Pharmacol Toxicol. 2000;40:431. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- (261).Dutertre S, Lewis RJ. Biochem Pharmacol. 2006;72:661. doi: 10.1016/j.bcp.2006.03.027. [DOI] [PubMed] [Google Scholar]
- (262).McIntosh JM, Santos AD, Olivera BM. Annu Rev Biochem. 1999;68:59. doi: 10.1146/annurev.biochem.68.1.59. [DOI] [PubMed] [Google Scholar]
- (263).Nicke A, Wonnacott S, Lewis RJ. Eur J Biochem. 2004;271:2305. doi: 10.1111/j.1432-1033.2004.04145.x. [DOI] [PubMed] [Google Scholar]
- (264).Unwin N. J Mol Biol. 2005;346:967. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- (265).Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. Nature. 2009;457:111. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- (266).Hilf RJ, Dutzler R. Nature. 2008;452:375. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- (267).Hilf RJ, Dutzler R. Nature. 2009;457:115. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- (268).Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van der Oost J, Smit AB, Sixma LK. Nature. 2001;411:269. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- (269).Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, et al. Nature. 2001;411:261. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- (270).Celie PH, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, et al. Nat Struct Mol Biol. 2005;12:582. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]
- (271).Hansen SB, Talley TT, Radic Z, Taylor P. J Biol Chem. 2004;279:24197. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).McCormack T, Petrovich RM, Mercier KA, DeRose EF, Cuneo MJ, Williams J, Johnson KL, Lamb PW, London RE, Yakel JL. Biochemistry. 2010;49:2279. doi: 10.1021/bi902023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Dutertre S, Ulens C, Buttner R, Fish A, van Elk R, Kendel Y, Hopping G, Alewood PF, Schroeder C, Nicke A, Smit AB, et al. EMBO J. 2007;26:3858. doi: 10.1038/sj.emboj.7601785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. EMBO J. 2005;24:3635. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Groebe DR, Gray WR, Abramson SN. Biochemistry. 1997;36:6469. doi: 10.1021/bi970195w. [DOI] [PubMed] [Google Scholar]
- (276).Hann RM, Pagan OR, Gregory LM, Jacome T, Eterovic VA. Biochemistry. 1997;36:9051. doi: 10.1021/bi970807c. [DOI] [PubMed] [Google Scholar]
- (277).Sine SM, Kreienkamp HJ, Bren N, Maeda R, Taylor P. Neuron. 1995;15:205. doi: 10.1016/0896-6273(95)90077-2. [DOI] [PubMed] [Google Scholar]
- (278).Sine SM. J Biol Chem. 1997;272:23521. doi: 10.1074/jbc.272.38.23521. [DOI] [PubMed] [Google Scholar]
- (279).Sine SM, Bren N, Quiram PA. J Physiol. 1998;92:101. doi: 10.1016/S0928-4257(98)80145-9. [DOI] [PubMed] [Google Scholar]
- (280).Grishin AA, Wang CI, Muttenthaler M, Alewood PF, Lewis RJ, Adams DJ. J Biol Chem. 2010;285:22254. doi: 10.1074/jbc.M110.111880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Bren N, Sine SM. J Biol Chem. 2000;275:12692. doi: 10.1074/jbc.275.17.12692. [DOI] [PubMed] [Google Scholar]
- (282).Dutertre S, Nicke A, Lewis RJ. J Biol Chem. 2005;280:30460. doi: 10.1074/jbc.M504229200. [DOI] [PubMed] [Google Scholar]
- (283).Quik M, Wonnacott S. Pharmacol Rev. 2011;63:938. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Schuller HM. Curr Drug Targets. 2012;13:680. doi: 10.2174/138945012800398883. [DOI] [PubMed] [Google Scholar]
- (285).Benowitz NL. N Engl J Med. 2010;362:2295. doi: 10.1056/NEJMra0809890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. J Biol Chem. 1996;271:7522. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- (287).Nicke A, Samochocki M, Loughnan Marion L, Bansal Paramjit S, Maelicke A, Lewis Richard J. FEBS Lett. 2003;554:219. doi: 10.1016/s0014-5793(03)01161-x. [DOI] [PubMed] [Google Scholar]
- (288).Beissner M, Dutertre S, Schemm R, Danker T, Sporning A, Grubmuller H, Nicke A. Mol Pharmacol. 2012;82:711. doi: 10.1124/mol.112.078683. [DOI] [PubMed] [Google Scholar]
- (289).Millard EL, Nevin ST, Loughnan ML, Nicke A, Clark RJ, Alewood PF, Lewis RJ, Adams DJ, Craik DJ, Daly NL. J Biol Chem. 2009;284:4944. doi: 10.1074/jbc.M804950200. [DOI] [PubMed] [Google Scholar]
- (290).Jones AK, Sattelle DB. Bioessays. 2004;26:39. doi: 10.1002/bies.10377. [DOI] [PubMed] [Google Scholar]
- (291).van Nierop P, Bertrand S, Munno DW, Gouwenberg Y, van Minnen J, Spafford JD, Syed NI, Bertrand D, Smit AB. J Biol Chem. 2006;281:1680. doi: 10.1074/jbc.M508571200. [DOI] [PubMed] [Google Scholar]
- (292).Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM. Proc Natl Acad Sci USA. 2006;103:17880. doi: 10.1073/pnas.0608715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Ellison M, Feng ZP, Park AJ, Zhang X, Olivera BM, McIntosh JM, Norton RS. J Mol Biol. 2008;377:1216. doi: 10.1016/j.jmb.2008.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Halai R, Clark RJ, Nevin ST, Jensen JE, Adams DJ, Craik DJ. J Biol Chem. 2009;284:20275. doi: 10.1074/jbc.M109.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Harvey SC, McIntosh JM, Cartier GE, Maddox FN, Luetje CW. Mol Pharmacol. 1997;51:336. doi: 10.1124/mol.51.2.336. [DOI] [PubMed] [Google Scholar]
- (296).Hogg RC, Miranda LP, Craik DJ, Lewis RJ, Alewood PF, Adams DJ. J Biol Chem. 1999;274:36559. doi: 10.1074/jbc.274.51.36559. [DOI] [PubMed] [Google Scholar]
- (297).Luo S, Nguyen TA, Cartier GE, Olivera BM, Yoshikami D, McIntosh JM. Biochemistry. 1999;38:14542. doi: 10.1021/bi991252j. [DOI] [PubMed] [Google Scholar]
- (298).McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Mol Pharmacol. 2004;65:944. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- (299).Quiram PA, Jones JJ, Sine SM. J Biol Chem. 1999;274:19517. doi: 10.1074/jbc.274.28.19517. [DOI] [PubMed] [Google Scholar]
- (300).Quiram PA, McIntosh JM, Sine SM. J Biol Chem. 2000;275:4889. doi: 10.1074/jbc.275.7.4889. [DOI] [PubMed] [Google Scholar]
- (301).Fainzilber M, Hasson A, Oren R, Burlingame AL, Gordon D, Spira ME, Zlotkin E. Biochemistry. 1994;33:9523. doi: 10.1021/bi00198a018. [DOI] [PubMed] [Google Scholar]
- (302).Wolfender JL, Chu F, Ball H, Wolfender F, Fainzilber M, Baldwin MA, Burlingame AL. J Mass Spectrom. 1999;34:447. doi: 10.1002/(SICI)1096-9888(199904)34:4<447::AID-JMS801>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- (303).McCallum SE, Parameswaran N, Bordia T, McIntosh JM, Grady SR, Quik M. Mol Pharmacol. 2005;68:737. doi: 10.1124/mol.105.012773. [DOI] [PubMed] [Google Scholar]
- (304).Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. J Pharmacol Exp Ther. 2007;322:306. doi: 10.1124/jpet.107.121228. [DOI] [PubMed] [Google Scholar]
- (305).Quik M, Bordia T, Forno L, McIntosh JM. J Neurochem. 2004;88:668. doi: 10.1111/j.1471-4159.2004.02177.x. [DOI] [PubMed] [Google Scholar]
- (306).Azam L, McIntosh JM. J Neurochem. 2012;122:1137. doi: 10.1111/j.1471-4159.2012.07867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW, Mozar CA, Christie MJ. Pain. 2011;152:259. doi: 10.1016/j.pain.2010.09.007. [DOI] [PubMed] [Google Scholar]
- (308).McIntosh JM, Yoshikami D, Mahe E, Nielsen DB, Rivier JE, Gray WR, Olivera BM. J Biol Chem. 1994;269:16733. [PubMed] [Google Scholar]
- (309).Ellison M, McIntosh JM, Olivera BM. J Biol Chem. 2003;278:757. doi: 10.1074/jbc.M204565200. [DOI] [PubMed] [Google Scholar]
- (310).Dutertre S, Nicke A, Tyndall JD, Lewis RJ. J Mol Recognit. 2004;17:339. doi: 10.1002/jmr.683. [DOI] [PubMed] [Google Scholar]
- (311).Ellison M, Gao F, Wang HL, Sine SM, McIntosh JM, Olivera BM. Biochemistry. 2004;43:16019. doi: 10.1021/bi048918g. [DOI] [PubMed] [Google Scholar]
- (312).Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Proc Natl Acad Sci USA. 2006;103:3615. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Yu R, Craik DJ, Kaas Q. PLoS Comput Biol. 2011;7:e1002011. doi: 10.1371/journal.pcbi.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Ellison M, Haberlandt C, Gomez-Casati ME, Watkins M, Elgoyhen AB, McIntosh JM, Olivera BM. Biochemistry. 2006;45:1511. doi: 10.1021/bi0520129. [DOI] [PubMed] [Google Scholar]
- (315).Halai R, Callaghan B, Daly NL, Clark RJ, Adams DJ, Craik DJ. J Med Chem. 2011;54:6984. doi: 10.1021/jm201060r. [DOI] [PubMed] [Google Scholar]
- (316).Arias HR, Blanton MP. Int J Biochem Cell Biol. 2000;32:1017. doi: 10.1016/s1357-2725(00)00051-0. [DOI] [PubMed] [Google Scholar]
- (317).Sørensen L. PhD Thesis, Copenhagen University; 2007. [Google Scholar]
- (318).Jin AH, Daly NL, Nevin ST, Wang CI, Dutertre S, Lewis RJ, Adams DJ, Craik DJ, Alewood PF. J Med Chem. 2008;51:5575. doi: 10.1021/jm800278k. [DOI] [PubMed] [Google Scholar]
- (319).Price-Carter M, Hull MS, Goldenberg DP. Biochemistry. 1998;37:9851. doi: 10.1021/bi9803978. [DOI] [PubMed] [Google Scholar]
- (320).Gehrmann J, Alewood PF, Craik DJ. J Mol Biol. 1998;278:401. doi: 10.1006/jmbi.1998.1701. [DOI] [PubMed] [Google Scholar]
- (321).Han TS, Zhang MM, Walewska A, Gruszczynski P, Robertson CR, Cheatham TE, Yoshikami D, Olivera BM, Bulaj G. ChemMedChem. 2009;4:406. doi: 10.1002/cmdc.200800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Khoo KK, Feng Z-P, Smith BJ, Zhang M-M, Yoshikami D, Olivera BM, Bulaj G, Norton RS. Biochemistry. 2009;48:1210. doi: 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Zhang MM, Han TS, Olivera BM, Bulaj G, Yoshikami D. Biochemistry. 2010;49:4804. doi: 10.1021/bi100207k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI. Bioorg Med Chem. 2004;12:4025. doi: 10.1016/j.bmc.2004.05.040. [DOI] [PubMed] [Google Scholar]
- (325).Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI, Shepherd NE. Tetrahedron. 2006;62:7284. [Google Scholar]
- (326).Vetter I, Lewis RJ. Curr Top Med Chem. 2012;12:1546. doi: 10.2174/156802612802652457. [DOI] [PubMed] [Google Scholar]
- (327).Kress HG, Simpson KH, Marchettini P, Ver Donck A, Varrassi G. Pain Pract. 2009;9:338. doi: 10.1111/j.1533-2500.2009.00308.x. [DOI] [PubMed] [Google Scholar]
- (328).Brust A, Palant E, Croker DE, Colless B, Drinkwater R, Patterson B, Schroeder CI, Wilson D, Nielsen CK, Smith MT, Alewood D, et al. J Med Chem. 2009;52:6991. doi: 10.1021/jm9003413. [DOI] [PubMed] [Google Scholar]
- (329).Dutertre S, Biass D, Stocklin R, Favreau P. Toxicon. 2010;55:1453. doi: 10.1016/j.toxicon.2010.02.025. [DOI] [PubMed] [Google Scholar]
- (330).McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. Biochem Pharmacol. 2009;78:693. doi: 10.1016/j.bcp.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- (1).Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
- (2).Lovelace ES, Armishaw CJ, Colgrave ML, Wahlstrom ME, Alewood PF, Daly NL, Craik DJ. J Med Chem. 2006;49:6561. doi: 10.1021/jm060299h. [DOI] [PubMed] [Google Scholar]
- (3).Miyazawa A, Fujiyoshi Y, Unwin N. Nature. 2003;423:949. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- (4).Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Proc Natl Acad Sci USA. 2006;103:3615. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- (1).Nishiuchi Y, Sakakibara S. FEBS Lett. 1982;148:260. doi: 10.1016/0014-5793(82)80820-x. [DOI] [PubMed] [Google Scholar]
- (2).Monje VD, Haack JA, Naisbitt SR, Miljanich G, Ramachandran J, Nasdasdi L, Olivera BM, Hillyard DR, Gray WR. Neuropharmacology. 1993;32:1141. doi: 10.1016/0028-3908(93)90008-q. [DOI] [PubMed] [Google Scholar]
- (3).Durieux JP, Nyfeler R. Pept Chem, Struct Biol, Proc Am Pept Symp, 14th. 1996:42. [Google Scholar]
- (4).Cuthbertson A, Indrevoll B. Tetrahedron Lett. 2000;41:3661. [Google Scholar]
- (5).Nielsen JS, Buczek P, Bulaj G. J Pept Sci. 2004;10:249. doi: 10.1002/psc.531. [DOI] [PubMed] [Google Scholar]
- (6).Cuthbertson A, Indrevoll B. Org Lett. 2003;5:2955. doi: 10.1021/ol035105w. [DOI] [PubMed] [Google Scholar]
- (7).Alewood PF. In: Ramage R, Epton R, editors. Peptides: Proceedings of the Twenty-Fourth European Peptide Symposium; Kingswinford: Mayflower Scientific; 1996. [Google Scholar]
- (8).Munson MC, Barany G. J Am Chem Soc. 1993;115:10203. [Google Scholar]
- (9).Hargittai B, Barany G. J Pept Res. 1999;54:468. doi: 10.1034/j.1399-3011.1999.00127.x. [DOI] [PubMed] [Google Scholar]