
Figure 2. Obtaining the correct balance of π–π and non-π-based interactions in the Mpipi model.
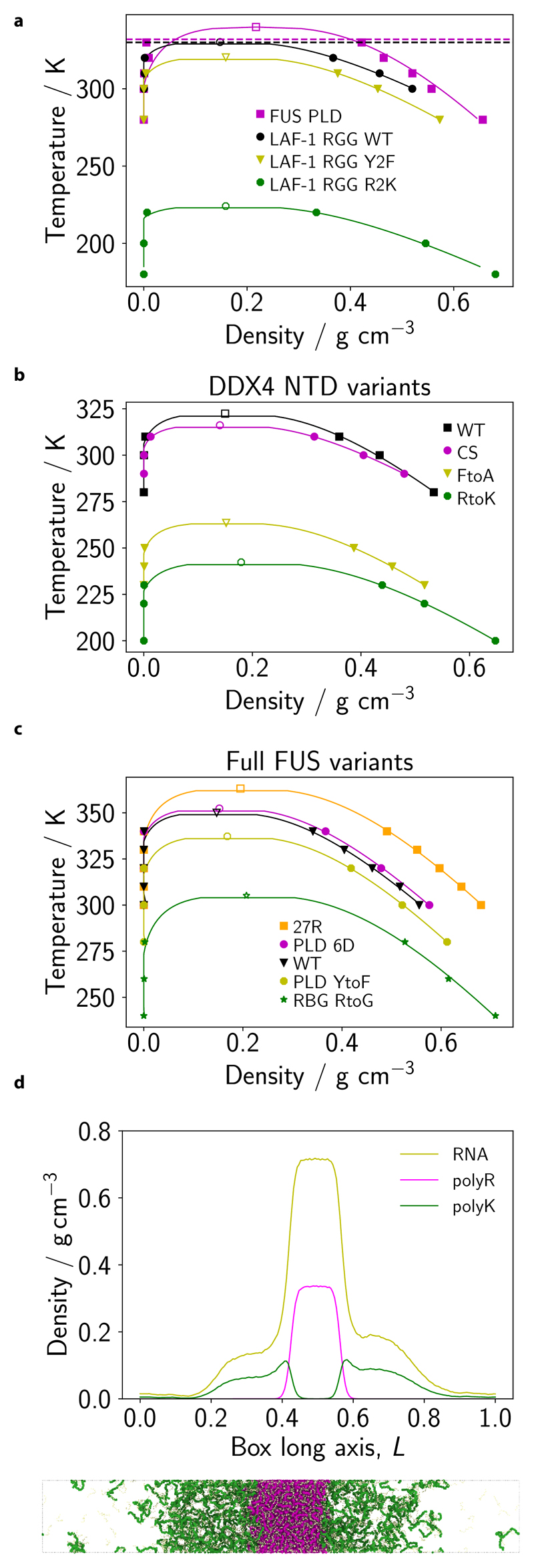
a–c PMF calculations at 150 mm NaCl salt concentration for π–π, cation–π and non-π-based interactions, respectively, as a function of the centre-of-mass (COM) distance. Statistical errors (mean±s.d.) are given as error bands, and are only just larger than the line width. They were computed via Bayesian bootstrapping of 3 independent simulations. Each pair is labelled using one-letter amino-acid codes (SI Table I). d Comparison of relative interaction strengths of selected residue pairs (SI Table I) from the PMF calculations with those implemented in the Mpipi model, relative to the Arg–Tyr (RY) interaction. Values are computed by taking the integral of the curves in a–c and the integral of the Wang-Frenkel potential only (between σ and 3σ) for the PMF and Mpipi sets, respectively; for the PMF data only the leftmost well is considered. These correspond to mean energies in the high-temperature limit. e Summary of relative interaction strengths in the Mpipi model. These relative interaction strengths include electrostatic interactions and are computed by numerically integrating Eq. (8) and normalising the result by the RY interaction strength.