

Summary
Background:
The cell membrane-derived initiators of coagulation, tissue factor (TF) and anionic phospholipid (aPL) are constitutive on the herpes simplex virus type 1 (HSV1) surface, bypassing physiological regulation. TF and aPL accelerate proteolytic activation of factor (F) X to FXa by FVIIa to induce clot formation and cell signaling. Thus, infection in vivo is enhanced by virus surface TF. HSV1-encoded glycoprotein C (gC) is implicated in this tenase activity by providing viral FX binding sites and increasing FVIIa function in solution.
Objective:
To examine the biochemical influences of gC on FVIIa-dependent FX activation.
Methods:
Immunogold electron microscopy (IEM), kinetic chromogenic assays and microscale thermophoresis (MST) were used to dissect tenase biochemistry. Recombinant TF and gC were solubilized (s) by substituting the transmembrane domain with poly-histidine, which could be orientated on synthetic unilamellar vesicles containing Ni-chelating lipid (Ni-aPL). These constructs were compared to purified HSV1 TF±/gC± variants.
Results:
IEM confirmed that gC, TF and aPL are simultaneously expressed on a single HSV1 particle where the contribution of gC to tenase activity required the availability of viral TF. Unlike viral tenase activity, the cofactor effects of sTF and sgC on FVIIa was additive when bound to Ni-aPL. FVIIa was found to bind to sgC and this was enhanced by FX. Orientation of sgC on a lipid membrane was critical for FVIIa-dependent FX activation.
Conclusions:
The assembly of gC with FVIIa/FX parallels that of TF and may involve other constituents on the HSV1 envelope with implications in virus infection and pathology.
Keywords: herpesvirus, tissue factor, coagulation factor, enzyme kinetics, enzyme mechanism
Introduction
Thrombosis and atherosclerosis are major causes of death world-wide and the incidents are increasing. These diseases have numerous well-established risk factors, which overlap [1] and have common underlying molecular bases. Viral infection is not typically considered within the etiology of thrombosis and atherosclerosis [2–4], although numerous viruses have been linked to vascular disease [5,6] and are known to affect the hemostatic system [7–14]. As an example, herpesviruses persist as lifelong infections and may contribute to vascular pathology through frequent reactivation promoting leukocyte and platelet adhesion to the vascular wall, lipid accumulation in smooth muscle cells and thrombin deposition in the periphery [15–18]. While these examples provide a retrospective clinical correlation, causal evidence was reported using herpes simplex virus 1 (HSV1) as a targeted cytolytic therapeutic for melanoma cells, which resulted in a high propensity for deep vein thrombosis [19]. Case reports have also detailed the presence of thrombi [20,21] and/or bleeding [22,23] in patients with HSV1 encephalitis. In one study examining predictors of outcome in HSV1 encephalitis, half of patients experienced acute thrombocytopenia, with ~5% reporting cerebral hemorrhage [23].
To provide a molecular explanation for the clinical evidence, our previous studies have demonstrated that members of the herpesvirus family incorporate host cell-derived coagulation cofactors during assembly of their outer envelope structure [24–26]. These are the transmembrane protein, tissue factor (TF), and anionic phospholipid (aPL), which function to accelerate the initiation and propagation of coagulation protease activation in response to vascular damage. TF is essential for life [27]. It accelerates the factor (F) VIIa-mediated proteolytic activation of FX to FXa by ~100,000-fold [28,29]. FXa subsequently produces the final protease generated during the coagulation pathway: thrombin. aPL plays a key role in accelerating coagulation protease activation by associating with FVIIa and FX via the respective γ-carboxyglutamic acid (Gla)-containing domain to enhance the assembly of the protease/substrate/cofactor complex. The strict physiological control of TF and aPL on the surface of cells is bypassed by their constitutive availability on the virus envelope surface. Coagulation enzyme activation on the virus surface and consequent induction of cell signaling pathways occur, which have pathological implications. An advantage to the virus is enhancement of the early events of infection by coagulation enzyme-mediated cell signaling, as seen in vitro [25] and in vivo [30]. When studied in cell culture, these effects on infection were facilitated by TF and the HSV1-encoded gene product, glycoprotein C (gC), both on the virus envelope.
gC is a multifunctional transmembrane glycoprotein that docks the virus to host cellular glycosaminoglycans such as heparan sulfate and chondroitin sulfate [31–33]. Evasion of host innate immunity is also aided by gC, which competes with complement protein C5b for binding sites on C3b [34,35]. gC expression on HSV1-infected endothelial cells has been linked to monocyte adhesion, possibly contributing to vascular lesion development [36]. This effect was dependent on thrombin generation and alluded to a role for gC in coagulation protease activation. Further evidence of its coagulation role was exemplified by FX binding to cell-surface gC through transgenic expression of gC in murine cells [36]. Since gC is found on the virus envelope, we investigated FX binding to purified HSV1 and showed that the presence of viral gC created FX binding sites [37]. Although a soluble form of gC contributes to FVIIa-dependent FX activation without being membrane-tethered, the activity was ~1000-fold higher when combined with the virus [38]. To explain how viruses may contribute to pathology, here we addressed the hypothesis that an aPL anchor and interactions with FVIIa and FX are integral to the gC-enhanced FX-activating complex, and that the optimal macromolecular complex involves viral TF.
Materials and Methods
Proteins and reagents.
Purified human plasma-derived FVIIa, FX and FXa, as well as corn trypsin inhibitor (CTI) were purchased from Haematologic Technologies (Essex Junction, VT). Recombinant human FVIIa (NovoSeven) was gratefully obtained from Novo Nordisk (Mississauga, ON) through an unrestricted research grant. Innovin (Dade Behring, Mississauga, ON) was the source of liposome-reconstituted purified recombinant human TF. Soluble TF (sTF-His) [39] was expressed in Escherichia coli (strain BL21-DE3) with a His6 tag and 5-amino acid spacer replacing the transmembrane and cytoplasmic domains, and was purified by nickel affinity chromatography. gC was similarly solubilized (gCΔ457t) by substituting the membrane-spanning domain with His5 in a baculovirus expression system and was purified using rabbit anti-gCΔ457t polyclonal antibody (R118) as previously reported [40][41]. Biotin-annexin V was purchased from BioLegend (San Diego, CA). Goat anti-mouse IgG Fc pre-adsorbed with 6 nm gold particles (ab105285) and goat anti-rabbit IgG H&L pre-adsorbed with 15 nm gold particles (ab27236) were purchased from Abcam (Toronto, ON). Goat anti-biotin pre-adsorbed with 10 nm gold particles (BBI Solutions) and acetylated BSA (Aurion) were commercially obtained (Cedarlane, Burlington, ON). Mouse monoclonal anti-TF (TF9–9B4) antibody was produced and purified as before [42]. Frozen pooled normal human plasma (NP) was obtained from George King Bio-Medical (Overland Park, KS). Cyanogen bromide, L-α-phosphatidylserine (Brain, Porcine, PS), L-α-phosphatidylcholine (Egg, Chicken, PC) and the nickel salt of 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid) succinyl] (DOGS-NTA-Ni) were purchased from Sigma Aldrich (St. Louis, MO). The FXa-preferred (S-2765) and broad specificity (S-2288) chromogenic substrates were from Diapharma (West Chester, Ohio).
Viruses.
HSV1 NS strain, a clinical isolate that expresses gC (gC+) and ns-1 strain, a naturally occurring HSV1 mutant that lacks the extracellular domain of gC (gC−) were passaged in African green monkey kidney cells (Vero CCL-81; ATCC, Manassas, VA) to generate initial virus stocks. To obtain viruses containing specific combinations of surface gC and/or TF, NS or ns-1 were propagated in a human melanoma cell line engineered to express TF after treatment with zeocin (A7/TF), as described [25]. Thus, HSV1/TF+/gC+, HSV1/TF+/gC−, HSV1/TF−/gC+ and HSV1/TF−/gC− viruses were produced in the same cell type. Virus concentrations were quantified by negative-staining transmission electron microscopy (EM) and the presence or absence of viral gC and TF was confirmed by western blot analysis [25]. The amount of viral TF (~150 molecules/virus particle) was determined antigenically by comparison to a purified sTF standard curve using densitometry (not shown).
Small unilamellar vesicles (SUVs).
Compositions of SUVs (mole %) were: PCPS (20% PS and 80% PC); NiPC (15% DOGS-NTA-Ni and 85% PC); and NiPCPS (15% DOGS-NTA-Ni, 20% PS and 65% PC) [43]. SUVs were prepared by sonication and differential centrifugation, as described [44]. The concentration of lipid was derived after enzymatic hydrolysis using a Phospholipids C kit (Wako, Mountain View, CA). Quantified SUVs were sized by dynamic light scattering using a Beckman Coulter N4 PLUS Particle Size Analyzer and were 41 ± 15 nm average diameter.
Chromogenic virus-mediated FX activation.
All chromogenic experiments were in HBS (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 mM NaCl, pH 7.4) with BSA (0.1%). Virus was first mixed with FX (100 nM) and FVIIa (1 nM) in 15 μL followed by addition of 5 μL of CaCl2 (calcium, 20 mM) to start the reaction. After 20 minutes at 37°C with shaking every 5 minutes, 10 μL was transferred to a 96-well plate and combined with 90 μL of S-2765 (200 μM) and EDTA (12 mM). The amount of FXa generated was read at 405 nm for 5 minutes at room temperature using a Vmax or Spectramax multi-well plate kinetic spectrophotometer (Molecular Devices, Sunnyvale, CA,). The amount of FX activated was derived from a purified FXa standard curve.
Immunogold EM.
Carbon formvar-coated nickel 400-mesh grids (Electron Microscopy Sciences, Cedarlane) were pre-treated with 10 μg/mL human IgG (tested positive for anti-HSV1 antibodies) to attenuate the deposition of HSV1 as aggregates. Purified HSV1 particles (1×108 virus particles), sTF-His (10 μg), gCΔ457t (10 μg), PCPS (200 μM) or PC only vesicles (200 μM) were adsorbed to grids for 5 minutes and then blocked with BSA (5%), fish scale gelatin (0.1%) and goat serum (1%) in low-salt HBS (LHBS; 75 mM NaCl, 20 mM HEPES, pH 7.4) for 40 minutes. Following 2 washes of LHBS, mixtures of rabbit anti-gC (R118, 1:50), mouse anti-TF (TF9–9B4, 5 μg/mL) [42] and/or biotin-annexin V (1.25 μg/mL) in LHBS and acetylated BSA (0.1%) were layered onto the grid and incubated for 1 hour. Samples probed for aPL using annexin V had CaCl2 (5 mM) included in all buffers and the corresponding control included EDTA (10 mM). After five 3-minute washes in LHBS, secondary antibody solutions of 6 nm gold-conjugated goat anti-mouse IgG (1:40), 15 nm gold-conjugated goat anti-rabbit IgG (1:160) and 10 nm gold-conjugated goat anti-biotin (1:40) in LHBS and acetylated BSA (0.1%) was added and incubated for 45 minutes. Following five 3-minute washes in LHBS, the grids were negatively stained with PTA (2%) stain pH 6.5 for 1 minute. The grids were wick-dried with filter paper and viewed on a 120 kV Hitachi H-7600 transmission electron microscope (Bioimaging Facility, University of British Columbia, Vancouver, Canada). HSV1 particles were defined as particles having gC antigen and a diameter of ~120–300 nm. The proportion of particles that express TF and/or aPL was quantified in 10 squares on the EM grid selected using a random number generator (https://numbergenerator.org/). Within each square, 5 sections were manually enumerated for a total of ~500 virus particles for each replicate.
gCΔ457t enhancement of FVIIa-mediated FX activation.
The function of gCΔ457t in FVIIa-mediated FX activation was followed by a discontinuous chromogenic assay. Innovin (1:3000) was incubated with gCΔ457t (2 μM), FX (0–100 nM) and FVIIa (0–10 nM). Calcium (5 mM) was added to initiate the reaction and the mixture was incubated at 3 °C for 20 minutes. The effect of membrane lipids on FVIIa-mediated FX activation by sTF-His or gCΔ457t was similarly monitored. PCPS, NiPC or NiPCPS (50 μM) in the presence or absence of gCΔ457t (1.2 nM), sTF-His (1 ρM), FX (100 nM), FVIIa (1 nM), or calcium (5 mM), were incubated for 20 minutes at 37°C. In all cases, the amount of FXa generated was determined by a chromogenic assay.
Equilibrium complex formation by microscale thermophoresis.
Protein complex formation between gCΔ457t and FVIIa and/or FX was assessed using microscale thermophoresis (MST), by quantifying differential mobility in a small temperature gradient due to complex formation. MST was conducted using a Monolith NT.115Pico instrument (NanoTemper Technologies, Munich, Germany). To detect thermophoretic shifts of a single species in an equilibrium mixture, one protein was covalently labeled with an NHS-linked fluorophore using the manufacturer’s kit or labeled reversibly to study solution-phase interactions through association of the poly-His tag with RED-tris-NTA. Fluorophore Alexa Fluor 647 succinimidyl ester was purchased from Invitrogen (Carlsbad, CA). His-Tag Labeling Kit RED-tris-NTA and Monolith Protein Labeling Kit RED-N-Hydroxysuccinimide (RED-NHS) 2nd Generation was from NanoTemper Technologies. Based on the protein concentration determined by colorimetric assays (BCA; Thermo Fisher Scientific, Waltham, MA) and the red fluorescent probe extinction coefficient (239,000 and 195,000 cm−1 M−1 for Alexa Fluor 647 and NanoTemper Red at 650 nm, respectively), the labeling stoichiometries of proteins used here were 1.4, 2.1, and 0.9 for Alexa Fluor 647-labeled gCΔ457t, Alexa Fluor 647-labeled sTF-His, and NanoTemper RED-labeled FVIIa (FVIIa-R), respectively. The labeled protein function was comparable to unlabeled in FX chromogenic assays in the presence of sTF-His or gCΔ457t.
Microscale thermophoresis measurements were performed at 25°C with medium MST power and varying LED power to ensure sufficient fluorescence signal. Standard and Premium Capillaries for MST experiments were from NanoTemper. In addition to using BSA as a carrier, premium capillaries were specially treated to minimize protein adsorption on the capillary wall. Capillaries were cut in half using a zirconium blade and nitrogen gas was purged through the capillaries to remove debris. Cut and uncut capillaries gave identical results. For solution phase binding, gCΔ457t (200 nM) was mixed with the RED-tris-NTA fluorescent probe (3.5 nM) for 30 minutes at room temperature before the addition of FX or FVIIa in HBS/BSA (0.05%)/Tween-20 (0.05%)/calcium (5 mM). To probe for membrane-bound gCΔ457t interactions, FVIIa-R (2 nM) was titrated with gCΔ457t on PCPS, NiPC or NiPCPS (50 μM) vesicles in HBS/BSA (0.1%)/calcium (5 mM). Benzamidine (2 mM) was also included to inhibit protease activity. Equilibrium dissociation constants (Kd values) were derived by fitting data to either a conventional binding isotherm equation based on conservation of mass or the Hill equation [45,46].
sTF-His or gCΔ457t binding to FVIIa and SUV.
Binding of sTF-His or gCΔ457t to either FVIIa or SUVs were assessed using continuous chromogenic assays monitoring FXa generation [43,47]. For SUV binding, varying concentrations of SUVs were mixed in HBS containing calcium (5 mM), FVIIa (100 ρM), and either gCΔ457t (120 nM) or sTF-His (100 ρM). For FVIIa binding, sTF-His or gCΔ457t was titrated in HBS with calcium (5 mM), FVIIa (10 ρM) and SUVs (100 μM). After 10 minutes of pre-incubation at 25°C, FX (30 nM) and S-2765 (200 μM) was added to start the reaction. The amount of FXa generated was immediately monitored spectrophotometrically at 405 nm for 20 minutes.
Kinetics of FX activation.
To derive Michaelis-Menten kinetic parameters Vmax and Km describing FVIIa function, initial rates of FX activation were measured as reported [43] with minor modifications. Reaction mixtures in HBS contained calcium (5 mM), FVIIa (500 ρM), and either gCΔ457t (10 nM) or sTF-His (10 ρM) with NiPCPS (100 μM). These experiments were conducted at concentrations of cofactor that would saturate the FVIIa. Varying concentrations of FX were added to initiate the reaction at 37°C. Over the course of 10 minutes, 20 μL aliquots of the reaction mixture were added to 50 μL stop buffer (Mes-NaOH (40 mM), pH 5.8, EDTA (12 mM), NaCl (50 mM), Triton X-100 (0.25%) and Antifoam C (0.012%) at 4°C. The stopped reactions were rapidly warmed to room temperature, and the amount of FXa generated was determined by the addition of S-2765 (200 μM), calcium (5 mM) and Tricine-NaOH (0.6 M), pH 8). A standard curve was derived using FXa to convert mOD to nM FXa.
Plasma clotting.
Virus-induced coagulation was monitored using an ST4 coagulation analyzer (Diagnostica Stago). In a final reaction volume of 75 μL, 12.5 μL of virus and 12.5 μL of HBS was added to 37.5 μL of NP or congenital FVIII-deficient human plasma. Pre-warmed magnetic beads were added to the cuvette and incubated for 1 minute at 37°C. Clotting was initiated with 12.5 μL of calcium (60 mM). To inhibit contact pathway initiation, CTI was pre-incubated with the NP for 5 minutes at 37°C. For gCΔ457t-mediated plasma clotting, PCPS or NiPCPS (50 μM final concentration) was incubated with gCΔ457t (1 μM) and calcium (10 mM) for 5 minutes before adding to NP to initiate clotting.
Statistical analyses.
Data are presented as mean ± SEM when 3 or more replicates were performed. Otherwise, mean ± SD was used for controls showing evident effect and a n = 2 was reported. The effect of gCΔ457t on virus-mediated FX activation was analyzed by unpaired Student’s t-test. FVIIa amidolytic activity data was analyzed by one-way ANOVA. Statistical analysis using t-tests and 1-way ANOVA were performed using Excel software. Fits of MST data and their confidence intervals were derived using quadratic binding isotherm equations written on MathWorks MATLAB® software version 2018b (MathWorks, Natick, MA, USA).
Results
TF, aPL and gC are simultaneously available on the HSV1 surface.
To investigate the basis of FX activation on HSV1, the assumption that TF, aPL and gC are simultaneously available on a single virus particle was directly examined by multi-label immunogold EM. Fig. 1A shows in representative electron micrographs that single HSV1 particles carry TF, aPL and gC. Fig. 1B confirmed that the gC and TF antibodies, and the aPL probe (i.e., annexin V) were specific to the individual components by using irrelevant isotype controls on HSV1 or by chelating residual calcium for annexin V. Additional controls using recombinant gCΔ457t, sTF-His, PC and PCPS vesicles further confirmed specificity (Fig. S1). Virus particles had various appearances due to variable penetrance of negative staining into the envelope, and differential visualization of the virus capsid. Quantification of gold labeling revealed that ~10% of gC-bearing HSV1 particles also had detectable TF and aPL (Fig. 1C). Of the remaining gC-positive HSV1 particles, ~20% also had detectable TF while another ~20% had aPL. These images verify that the virus surface contains combinations of procoagulant cofactors.
Fig. 1.
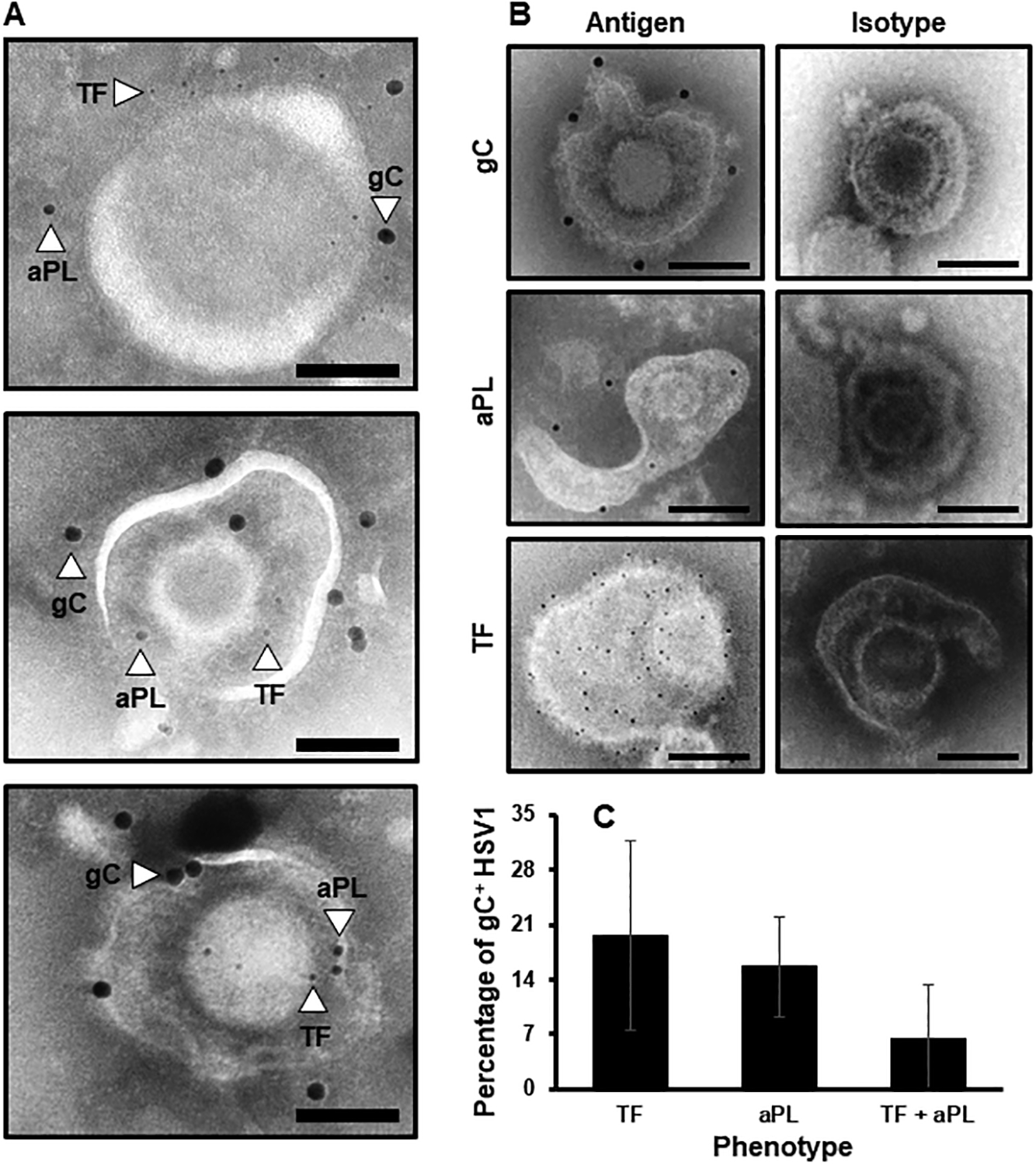
TF, aPL and gC are simultaneously available on the HSV1 surface. (A) Representative immunogold electron micrographs concurrently identifying the HSV1 marker, gC (15 nm gold bead), aPL (10 nm bead) and TF (6 nm gold bead). (B) Individual controls for specificity using gold-labeled irrelevant isotype antibody controls for gC and TF or biotinylated-annexin V in the absence of calcium. (Scale bars = 100 nm. n = 3). (C) The proportion of gC-positive HSV1 particles that express TF, aPL or TF and aPL was determined by immunogold EM. (n = 2 ± SD).
Viral gC enhances HSV1-initiated FX activation when viral TF is present.
To dissect the FX-activating mechanism on the HSV1 surface, the effect of purified HSV1/TF+/gC+, HSV1/TF+/gC−, HSV1/TF−/gC+ or HSV1/TF−/gC− on FVIIa function was compared using a chromogenic assay. Compared to the fully competent HSV1/TF+/gC+ (Fig. 2A), both TF-deficient viruses had <2% FVIIa-mediated FX activation (Fig. 2B). The presence of gC on the TF-deficient virus had no measurable enhancement on FX activation (Fig. 2B). The small concentration-dependent increase in FX activation due to HSV1/TF−/gC− suggested that additional constituents such as envelope aPL contribute to FX activation. Interestingly, the presence of viral gC and viral TF enhanced the activation of FX ~2.5-fold relative to HSV1/TF+/gC−(Fig 2A). When gCΔ457t was added to 1 × 105 vp/μl of purified HSV1 variants, FX activation was further increased on TF-bearing virus (Fig. 2C). HSV1/TF+/gC+ exhibited a greater enhancement by gCΔ457t compared to HSV1/TF+/gC−. Addition of gCΔ457t to TF-deficient virus had no measurable effect on FX activation (Fig. 2D). Thus, viral TF is required for a contribution of gC or gCΔ457t to FVIIa activity mediated by purified HSV1. FX activation in the absence of FX, FVIIa or calcium was negligible (Fig. 2E) demonstrating that purified virus has insignificant intrinsic hydrolytic activity toward S-2765.
Fig. 2.
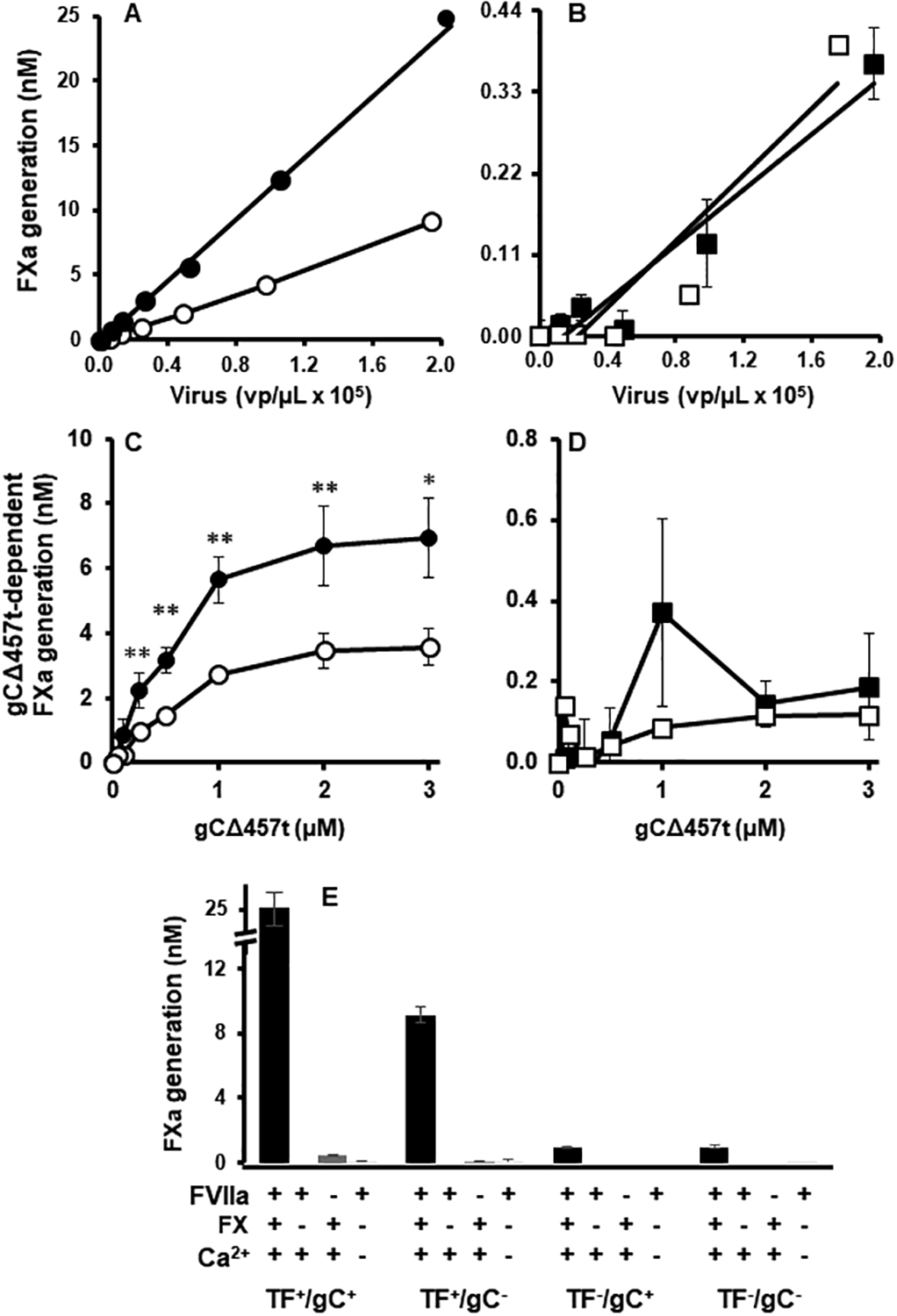
gC enhances HSV1-initiated FX activation when viral TF is present. (A and B) Purified FX (100 nM) and FVIIa (10 nM) were combined with HSV1/TF+/gC+ (●), HSV1/TF+/gC- (○), HSV1/TF-/gC+ (◼) and HSV1/TF-/gC- (◻) viruses in the presence of calcium (5 mM) and FXa generation was followed using the chromogenic substrate S-2765. (C and D) FX activation by HSV1 was followed as in (A and B) and the effect of adding purified gCΔ457t to the panel of HSV1 (1 × 105 vp/μL) was monitored. (E) FX activation with 1 × 105 vp/μL HSV1/TF+/gC+, HSV1/TF+/gC-, HSV1/TF-/gC+ and HSV1/TF-/gC- with (first bar) or without FX, FVIIa or calcium (following bars in order). (A - D: n = 4 ± SEM, * P < 0.05, ** P < 0.01; E: Virus and no FVIIa: n = 4 ± SEM, no FX or no Ca2+: n = 2 ± SD; error bars may be smaller than the size of symbols).
gCΔ457t enhances FVIIa-mediated FX activation on vesicles.
To exclude other virus- or host-encoded factors associated with the envelope of HSV1, gCΔ457t was added to lipidated full-length TF and aPL (Innovin). gCΔ457t enhanced FX activation by FVIIa with saturable dependence on FVIIa (Fig. 3A) or FX (Fig. 3B). Under these conditions, the addition of gCΔ457t increased maximal activation by ~2-fold.
Fig. 3.
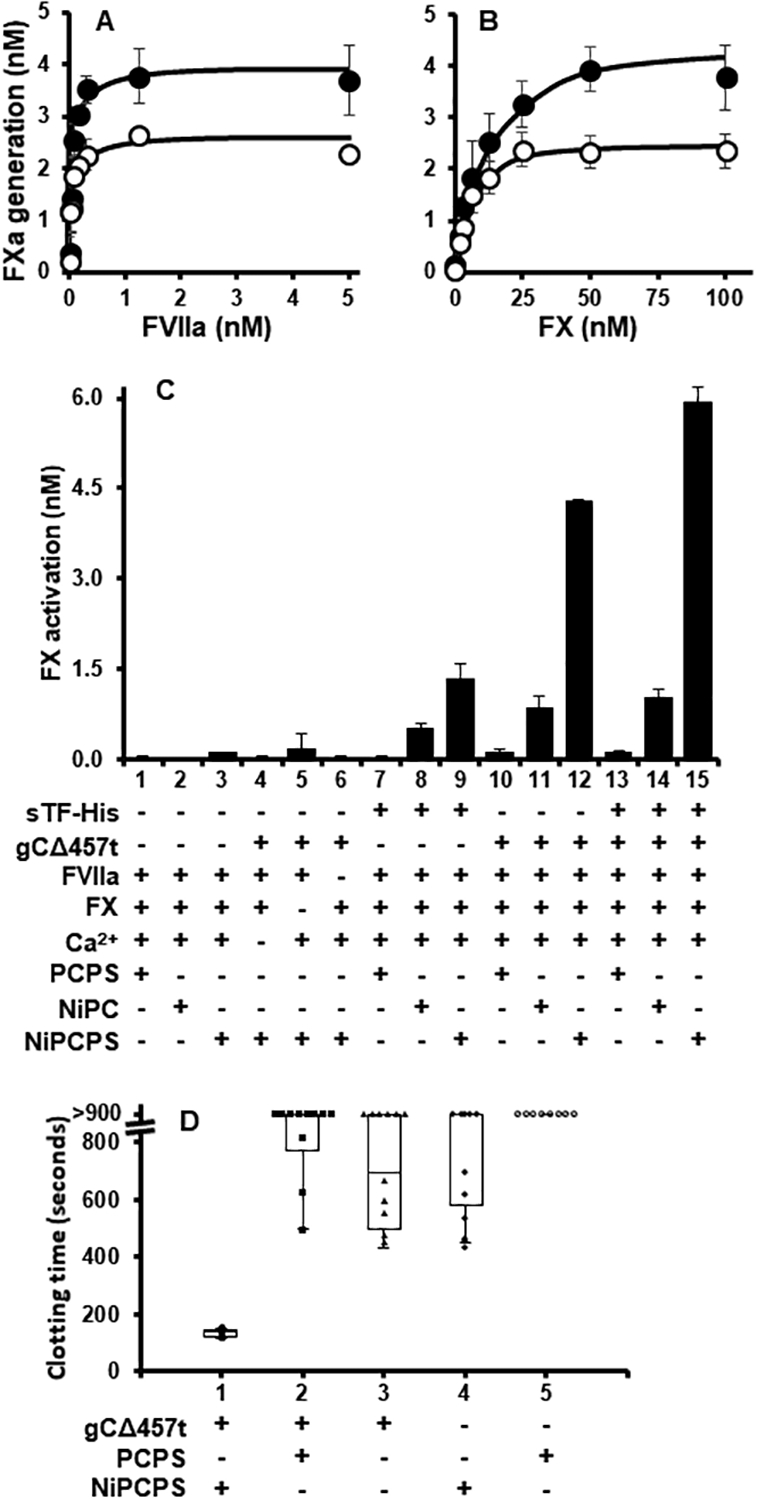
gCΔ457t enhances FVIIa-mediated FX activation. (A) FX activation by relipidated full-length TF (1:3000, Innovin) was followed chromogenically in the presence (●) or absence (○) of gCΔ457t (2 μM) at constant FX (100 nM, A) or FVIIa (10 nM, B) and titrating either FVIIa (A) or FX (B). (C) FX activation was monitored by variably assembling tenase constituents on PCPS, NiPC or NiPCPS (50 μM) in the presence or absence of gCΔ457t (1.2 nM), sTF-His (1 ρM), FX (100 nM), FVIIa (1 nM), or calcium (5 mM) with 20 min incubation at 37°C. (D) Normal pooled human plasma clotting time was monitored after initiation by gCΔ457t (1 μM), 50 μM PCPS or NiPCPS, and calcium (10 mM). (A – C: n = 3 ± SEM; D: n = 3, box plot of pooled replicates; error bars may be smaller than the size of symbols.)
sTF-His is known to enhance FX activation by FVIIa optimally when DOGS-NTA-Ni is incorporated into an aPL-containing membrane [43], where DOGS-NTA-Ni facilitates membrane-binding of sTF-His and the aPL enables Gla-domain-dependent membrane-binding of FVIIa and FX. Thus, the respective contributions toward increasing the activity of FVIIa by gCΔ457t were evaluated by comparing vesicles of various lipid composition, PCPS, NiPC and NiPCPS. Significant FX activation by FVIIa was not observed in the absence of sTF-His or gCΔ457t (Fig. 3C, bars 1–3). Expression of gCΔ457t (Fig. 3C, bars 4–6) or sTF-His (not shown) activity was dependent on FVIIa, FX and calcium ions. As a positive control for the anticipated activity of the three vesicle compositions, FX activation was shown to be enhanced by sTF-His in the following order NiPCPS > NiPC >> PCPS (Fig. 3C, bars 7–9). Interestingly, gCΔ457t also enhanced FVIIa activity and followed the same lipid composition dependence as sTF-His, where combined binding of the His-tagged cofactor and Gla-containing proteins facilitated by NiPCPS was optimal (Fig. 3C, bars 10–12). To explore the apparent synergistic effect of gC and TF on the virus (Fig. 2), sTF-His was combined with gCΔ457t (Fig. 3C, bars 13–15). Unlike the HSV1 variant experiments, the effect of gCΔ457t on sTF-His was additive in these purified protein experiments when the cofactor/enzyme/substrate complex was assembled on NiPCPS (i.e., bar 9 + bar 12 = bar 15).
Further demonstrating a gCΔ457t cofactor effect on FVIIa, incubation with NP shortened clotting times in the presence of NiPCPS (Fig. 3D, bar 1). With the substitution of NiPCPS for PCPS (Fig. 3D, bar 2) the majority of samples did not form a clot during the experimental time (15 min). Similarly, gCΔ457t without lipids or the absence of gCΔ457t with either PCPS or NiPCPS yielded sporadic clotting with the majority not forming a clot (Fig. 3D, bars 3 – 5, respectively). Of note, the NP contained antibodies that recognize HSV1 and gCΔ457t (Fig. S2), accredited to the prevalence of HSV1 within the general adult population, which may have affected gCΔ457t clotting activity.
FX enhances the interaction between gCΔ457t and FVIIa.
The His-tag of gCΔ457t was non-covalently conjugated to fluorescent RED-tris-NTA and monitored by MST to follow interactions with FX (Fig. 4A) or FVIIa (Fig. 4B) in the absence of vesicles. Although an interaction between gCΔ457t and FVIIa or FX was demonstrable, both were relatively weak as the binding isotherms did not approach saturation at the highest available concentration of either ligand. These binding isotherms could not be fit to a binding model with statistical confidence. While the solution-phase enhancement of FVIIa activity was independent of labelling the gCΔ457t with RED-tris-NTA via the poly-His tag, it could not be used to follow membrane-associated complex assembly because it quantitatively blocked NiPCPS-binding (not shown).
Fig. 4.
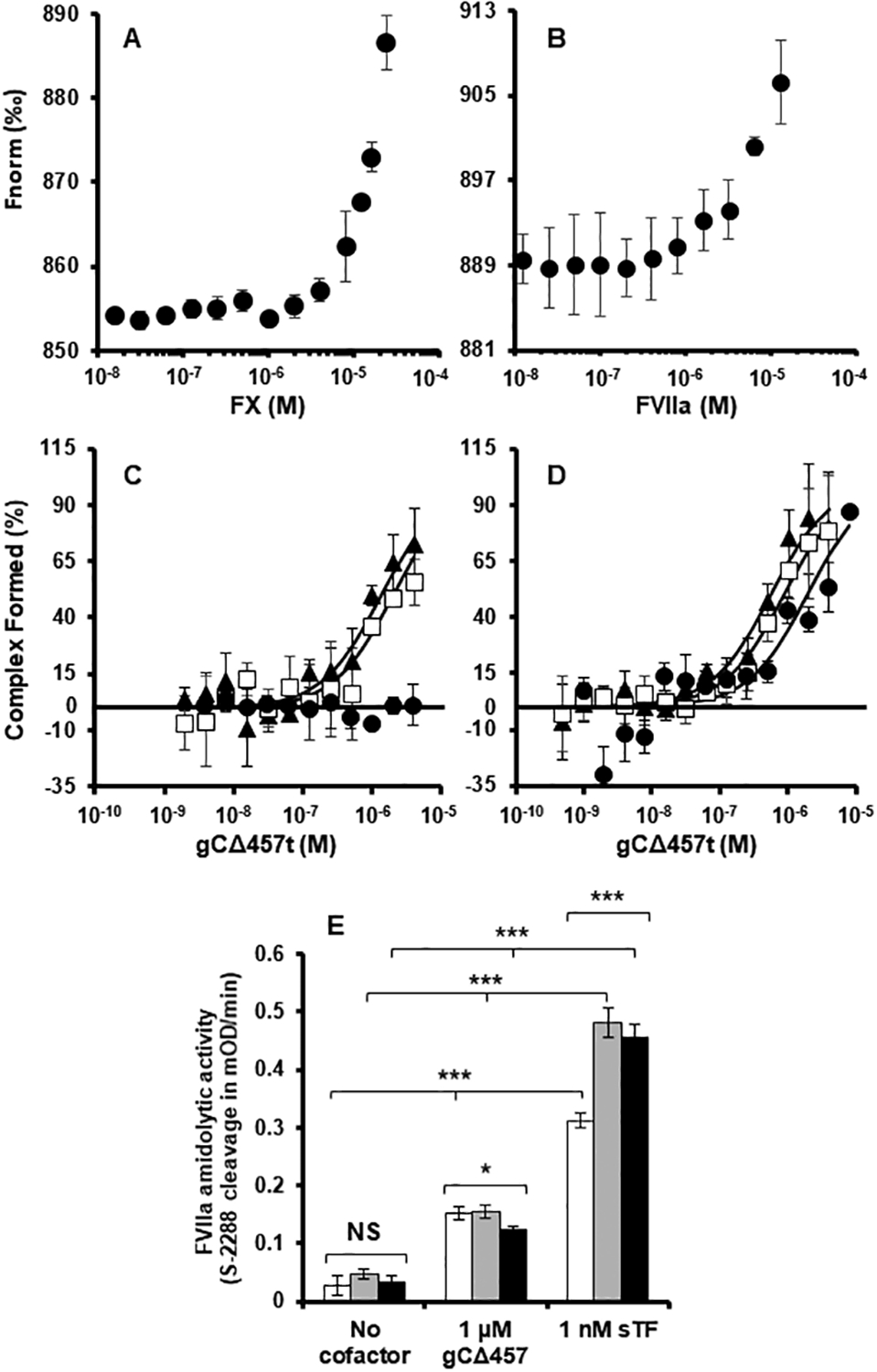
FX enhances the interaction between gCΔ457t and FVIIa. MST traces demonstrating weak associations of (A) FX and (B) FVIIa with gCΔ457t non-covalently labeled with RED-tris-NTA in the absence of membrane associations. (C) MST was also used to follow gCΔ457t binding to fluorescent FVIIa-R (2 nM) in the presence of PCPS (●), NiPC (◻) or NiPCPS (▲) (50 μM) and benzamidine to inhibit FVIIa. (D) Similar to (C) but with the inclusion of FX (30 nM). (E) Effects on FVIIa catalytic activity was assessed by following FVIIa (2 nM) cleavage of chromogenic substrate S-2288 in the presence of gCΔ457t (1 μM) or sTF-His (1 nM) with calcium (5 mM) and PCPS (white bars), NiPC (grey bars) or NiPCPS (black bars) (50 μM). (All graphs: n ≥ 3 ± SEM). P values are provided as determined by 1-way ANOVA. * P < 0.05, *** P < 0.001, P ≥ 0.05 was not significant (NS).
To follow the effects of membrane orientation and the presence of FX on incorporation of FVIIa into the gCΔ457t/FX complex, the FVIIa was fluorophore-labeled via amine coupling (FVIIa-R). As measured by MST, the dissociation constant (Kd) of ~1 nM (Table 1) was ~100-fold higher than the reported Kd value for FVIIa binding to sTF-His/NiPCPS that was derived using a FXa product-linked chromogenic assay (~10 ρM) [43]. To reconcile this discrepancy, FX was included in the MST studies to mimic the conditions of the reported chromogenic method. In the presence of excess benzamidine to prevent proteolysis, FX enhanced sTF-His/NiPCPS-binding to FVIIa-R by ~30-fold (Kd = ~30 ρM; Fig. S3), which was consistent with the previous report.
Table 1.
FVIIa binding to cofactors determined by MST in the presence or absence of FX.
| No FX | With FX | ||
|---|---|---|---|
| Ligand | Lipid | Kd (μM) (95% CI) | Kd (μM) (95% CI) |
| sTF-His | NiPCPS | 8.2 × 10−4 (8.6 × 10−5 – 1.5 × 10−3) | 3.0 × 10−5 (8.0 × 10−6 – 5.2 × 10−5) |
| gCΔ457t | NiPC | 2.0 (1.3 – 2.8) | 0.9 (0.7 – 1.0) |
| gCΔ457t | NiPCPS | 1.3 (0.8 – 1.7) | 0.6 (0.2 – 0.9) |
| gCΔ457t | PCPS | >13.0 | 2.2 (0.9 – 3.5) |
| gCΔ457t | None | >12.0 | 2.9 (1.5 – 4.2) |
gCΔ457t assembled onto NiPC or NiPCPS exhibited greater affinity for FVIIa-R compared to vesicles without DOGS-NTA-Ni (Fig. 4C). This interaction was only moderately affected by the presence of PS, implying that a FVIIa-R-membrane interaction may not be important; the Kd values were 1.6 and 0.8 μM for NiPC or NiPCPS, respectively (Table 1). Binding of gCΔ457t to FVIIa-R in the presence of PCPS was detectable in the absence of DOGS-NTA-Ni but was not quantifiable (Kd >13 μM) and consistent with the concentration range used for gCΔ457t binding to FVIIa in the absence of vesicles (Fig. 4B). In the presence of NiPC or NiPCPS, the addition of FX (Fig. 4D) increased the gCΔ457t-FVIIa-R affinity ~2-fold (Kd values of 1.0 and 0.4 μM, respectively) indicating that Gla-dependent associations are not required for this enhancement. gCΔ457t emulated the FVIIa-sTF-His binding enhancement by FX, but this was modest in comparison to the ~30-fold observed for the latter (Table 1). Omission of DOGS-NTA-Ni reduced the affinity for FVIIa-R binding to gCΔ457t in the presence of FX and PCPS (Kd = 2.7 μM).
Direct functional effects conferred to the FVIIa catalytic site by gCΔ457t was monitored by following chromogenic substrate cleavage (S-2288). Fig. 4E shows that at 1 μM gCΔ457t, FVIIa-mediated cleavage of S-2288 was enhanced. This result was independent of DOGS-NTA-Ni or PS availability. sTF-His also enhanced S-2288 cleavage by FVIIa but, unlike gCΔ457t, was further enhanced in the presence of DOGS-NTA-Ni. gCΔ457t was less efficient than sTF-His at directly affecting FVIIa amidolytic activity by ~3700-fold on a molar basis (Fig. 4E).
gCΔ457t- and sTF-His-binding to Ni-chelating vesicles and FVIIa are similar by kinetics.
To understand the mechanism of gCΔ457t cofactor function in our model system further, the His-tag equilibrium with NiPCPS and NiPC was determined by measuring the kinetically-linked activation of FX. The availability of PS (Fig. 5A) in NiPCPS increased the maximal FX activation rate by >15-fold for both sTF-His and gCΔ457t compared to NiPC. sTF-His and gCΔ457t had similar apparent binding affinities (Fig. 5B) for either NiPCPS or NiPC (Kd,app = 48 and 34 μM, respectively), as summarized in Table 2. A role for PS in membrane binding of sTF-His and gCΔ457t was implied by an ~2.5-fold increase of the Kd,app values when PS was omitted from the Ni-chelating vesicle.
Fig. 5.
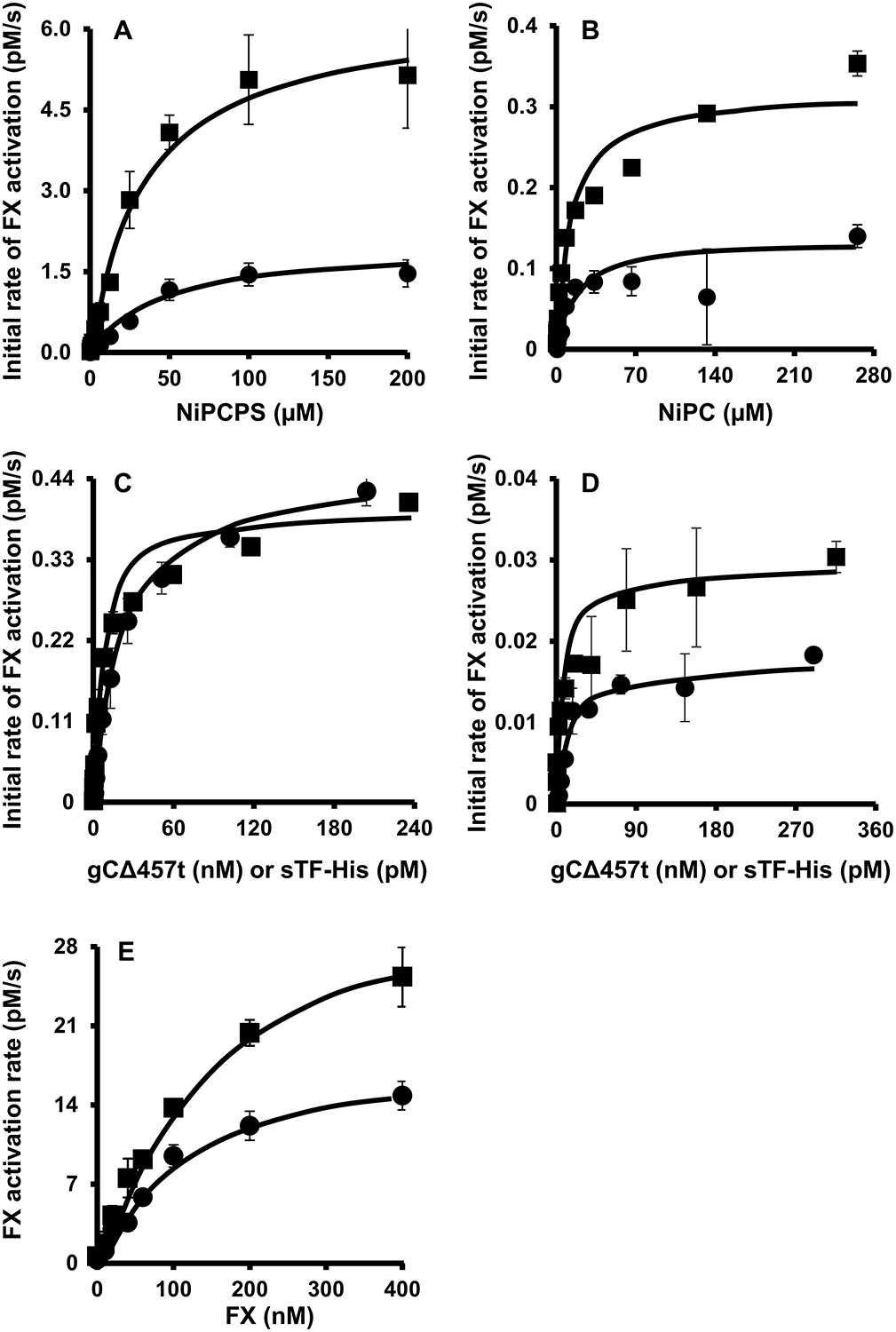
gCΔ457t- and sTF-His-binding to Ni-chelating vesicles and FVIIa are similar by kinetics. FX (30 nM) activation by FVIIa (100 ρM) was followed in the presence of gCΔ457t (120 nM, ●) or sTF-His (100 ρM, ◼), calcium (5 mM) and titrated with (A) NiPCPS or (B) NiPC. Binding of cofactors to FVIIa was followed in a similar manner except FVIIa (10 ρM), FX (30 nM), calcium (5 mM) and NiPCPS (50 μM, C) or NiPC (50 μM, D), were titrated with gCΔ457t (●) or (◼) sTF-His. E. The kinetic parameters of FXa generation by FVIIa (Table 3) was determined by deriving the initial FX activation rate by a limiting concentration of FVIIa (10 ρM) in the presence of gCΔ457t (120 nM, ●) or sTF-His (100 ρM, ◼) with FX, calcium (5 mM) and NiPCPS (100 μM). (All graphs: n = 3 ± SEM).
Table 2.
Equilibrium binding constants derived from Figures 5A and 5B determined by linked chromogenic activity.
| Nickel binding | FVIIa binding | ||
|---|---|---|---|
| Ligand | Lipid | Kd,app (μM) (95% CI) | Kd,app (nM) (95% CI) |
| sTF-His | NiPCPS | 33.8 (23.0 – 44.4) | 7.0 × 10−3 (4.4 × 10−3 – 9.6 × 10−3) |
| sTF-His | NiPC | 13.4 (5.6 – 21.2) | 8.9 × 10−3 (2.9 × 10−3 – 1.5 × 10−4) |
| gCΔ457t | NiPCPS | 48.0 (26.0 – 70.0) | 21.5 (18.3 – 24.7) |
| gCΔ457t | NiPC | 19.1 (7.4 – 30.7) | 18.0 (8.7 – 27.3) |
The binding of FVIIa to sTF-His or gCΔ457t in the presence of NiPCPS (Fig. 5C) or NiPC (Fig. 5D) was measured enzymatically by limiting the detection of FXa generation to the concentration of FVIIa. Whereas both the gCΔ457t/NiPCPS and sTF-His/NiPCPS pairs exhibited similar Vmax values (Fig. 5C), reactions involving sTF-His/NiPC yielded a Vmax value that was two-fold greater than those with gCΔ457t/NiPC (Fig. 5D), implying an effect of PS on the number of gCΔ457t-FVIIa binding sites. NiPCPS-tethered sTF-His had a binding affinity (Table 2) consistent with earlier reports addressing FVIIa-binding to membrane-inserted full-length TF (Kd,app ~10 ρM). This effect was independent of the presence of PS. The Kd,app describing the interaction of gCΔ457t with FVIIa was ~20 nM, which was also not influenced by the presence of PS.
With both lipid and FVIIa binding affinities known, the concentration of the lipid/cofactor/enzyme complex can be estimated and, consequently, the Michaelis-Menten kinetics were derived (Fig. 5E). The Km for FX activation by FVIIa in the presence of either sTF-His or gCΔ457t was ~0.15 μM (Table 3). The turnover number (kcat) for FX conversion was moderately higher in the presence of sTF-His (3.8 sec−1) compared to gCΔ457t (2.5 sec−1). Nonetheless, sTF-His and gCΔ457t had comparable catalytic efficiencies (kcat/Km) with a nominal 1.4-fold difference. Therefore, a large part of the differential efficacy of sTF-His versus gCΔ457t is attributed to their relative affinity for FVIIa.
Table 3.
Kinetic effects of gCΔ457t compared to sTF on FX activation.
| Cofactor | Lipid | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|---|
| sTF-His | NiPCPS | 0.16 ± 0.02 | 3.8 ± 0.5 | 23.6 ± 1.4 |
| gCΔ457t | NiPCPS | 0.14 ± 0.02 | 2.5 ± 0.3 | 16.6 ± 1.0 |
Discussion
Here we demonstrated that the coagulation tenase cofactor function of HSV1-encoded gC is analogous to that of TF using solubilized recombinant forms, gCΔ457t and sTF-His (respectively), in well-defined biochemical studies. Similar to full-length TF and sTF-His [43][48], gCΔ457t required orientation on a membrane as shown by enhanced activity in the presence of vesicles that contained Ni-chelating lipid to facilitate association of the C-terminal poly-His. Direct binding to gCΔ457t of the tenase substrate, FX, was demonstrated, which confirmed previous studies [37]. The protease subunit of tenase, FVIIa, was shown for the first time to also associate with gCΔ457t, which acted like a receptor for FVIIa. The gCΔ457t-FVIIa interaction was enhanced by FX. Thus, gC is a constituent of a ternary protein complex that is localized to a membrane surface by the availability of substrate, like the TF-dependent tenase.
In addition to the Ni-chelating lipid that anchors gCΔ457t to the membrane, the presence of PS was important for FVIIa-dependent FX activation. The FX-mediated high affinity FVIIa assembly into the gCΔ457t/NiPCPS tenase required PS, which may in part be explained by the intrinsic affinity of FX for PS-containing vesicles being much greater than FVIIa (Kd ~0.2 μM and ~15 μM, respectively) [29,30] and a consequent pseudo-receptor effect. Thus, the PS contribution to the overall gCΔ457t/FVIIa/FX tenase complex formation is predominantly attributed to FX, presumably via the Gla-domain [49]. In contrast, PS provided minimal contributions to FVIIa binding to gCΔ457t indicating that the Gla-domain-dependent association of FVIIa with PS is a relatively unimportant variable in cofactor/enzyme complex formation. These observations parallel the mechanism of sTF-His [43,50]. gCΔ457t/FVIIa complex formation was enhanced considerably by FX even in the absence of membrane, demonstrating a Gla-independent aspect of viral tenase assembly. Thus, the utility of vesicles comprised of Ni-chelating lipid and PS combinations has revealed congruent characteristics for the various protein-membrane interactions in gCΔ457t/FVIIa/FX and sTF-His/FVIIa/FX.
The mixture of gCΔ457t and sTF-His with saturating FVIIa, FX and NiPCPS resulted in the generation of FXa consistent with the sum of the gCΔ457t and sTF-His individually measured. To evaluate the tenase function of full-length gC and TF within the full complement of the intricate environment of the virus envelope surface, our unique virus panel of HSV1/TF+/gC+, HSV1/TF+/gC− and HSV1/TF−/gC+ were compared. Interestingly, the additive effect with purified proteins was not observed on the HSV1 surface, where FXa generation was enhanced by gC only when TF or purified sTF-His was also available. This synergistic effect on gC by TF may imply a direct gC-TF interaction. Since four methods (i.e. MST, crosslinking, non-denaturing electrophoresis and ELISA) failed to detect such an association either on the virus or between purified proteins (data not shown), an additional host or virus-encoded constituent(s) may be involved in the macromolecular FX activating complex on the virus surface.
Since FX enhances the gCΔ457t/FVIIa interaction, it follows that the association of FVIIa with gCΔ457t was ~10-fold weaker when determined by MST in the absence of FX compared to the kinetically linked FX-activation method. A possible contributor to the higher Kd was the effect of amine modification of FVIIa to produce FVIIa-R. However, this was unlikely a contributor since FVIIa-R retained tenase activity comparable to unlabeled FVIIa where the fluorophore stoichiometry per FVIIa was carefully limited to ~1:1. As an additional precaution during MST equilibrium measurements, benzamidine prevented proteolytic activity without altering gCΔ457t binding to FVIIa (data not shown). TF-FVIIa binding measurements by others have reported a wide range of Kd values, because of variable contributions of FX and aPL depending on experimental conditions and the limited sensitivity of some approaches that required protein concentrations in vast excess over Kd [48]. Surface plasmon resonance using relipidated TF yielded a Kd in the ρM range [48] that was very similar to that obtained by enzymatic assays or fluorescence anisotropy [47,51,52]. However, the TF/FVIIa/aPL complex did not dissociate to completion over the course of the measurement, potentially compromising the accuracy of Kd derivation as recognized by the authors. We show a high affinity recruitment of FVIIa (~30 ρM) into the sTF-His/FX complex on a NiPCPS membrane measured consistently by either MST or enzymatic assays, supporting the importance of membrane orientation- and FX-dependent macromolecular assembly and resolving any literature discrepancy.
Our work has revealed a novel interaction between virus-encoded gC, and the initiating coagulation protease FVIIa, that requires FX and aPL for optimal complex formation. This mechanism parallels that of the physiological hemostatic trigger TF and implies molecular mimicry. The role of gC in enhancing FX activation, even in the absence of TF, is a probable contributor to viral infection [30] and to haemostatic abnormalities correlating to atherosclerotic and thrombotic propensity [5,6][15–18,21].
Addendum
BL designed and conducted experiments, analyzed data and wrote the manuscript; MS prepared specialized reagents, assisted in experimental design, analyzed data and edited the manuscript; FR assisted in experimental design; JM provided specialized reagents, assisted in experimental design, analyzed data, and edited the manuscript; EP directed the project, designed experiments and co-wrote the manuscript.
Supplementary Material
Essentials.
Host tissue factor (TF) and viral glycoprotein C (gC) combine to trigger clotting on oral herpes
Like TF, gC binds FX and enhances FVIIa-dependent FX activation
gC also imitates TF by binding FVIIa, which is enhanced by FX
gC requires orientation on a lipid membrane to function as a FVIIa cofactor
Acknowledgements
The technical excellence of V. Smith (University of British Columbia) was invaluable. Drs. J. Kizhakkedathu, F. Duong and M. Roberge are acknowledged for generously providing access to equipment (University of British Columbia). We thank the UBC Bioimaging Facility for TEM access. The baculovirus expression vector containing gCΔ457t, antibody (R118) and HSV1 strains were a kind gift from Drs. G. Cohen and R. Eisenberg (University of Pennsylvania). E. coli strain BL21-DE3 and zirconium blades were graciously provided by Drs. Natalie Strynadka and Liam Worrall (University of British Columbia). This work was supported by the Canadian Institutes of Health Research grant 273985 (EP), Heart and Stroke Foundation of Canada G-19-0026524 (EP) and National Institutes of Health R35 HL135823 (JM). BL was supported by a Centre for Blood Research Graduate Award Program Grant.
Footnotes
Disclosure of Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Småbrekke B, Rinde LB, Hindberg K, Hald EM, Vik A, Wilsgaard T, Løchen M-L, Njølstad I, Mathiesen EB, Hansen J-B, Brækkan S. Atherosclerotic Risk Factors and Risk of Myocardial Infarction and Venous Thromboembolism; Time-Fixed versus Time-Varying Analyses. The Tromsø Study. Zeller T, editor. PLoS One 2016; 11: e0163242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson FAJ, Spencer FA. Risk factors for venous thromboembolism. Circulation 2003; 107: I9–16. [DOI] [PubMed] [Google Scholar]
- 3.Gregson J, Kaptoge S, Bolton T, Pennells L, Willeit P, Burgess S, Bell S, Sweeting M, Rimm EB, Kabrhel C, Zöller B, Assmann G, Gudnason V, Folsom AR, Arndt V, Fletcher A, Norman PE, Nordestgaard BG, Kitamura A, Mahmoodi BK, et al. Cardiovascular Risk Factors Associated With Venous Thromboembolism. JAMA Cardiol 2019; 4: 163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus 2011; 9: 120–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yonemitsu Y. Viruses and vascular disease. Nat Med 1998; 4: 253. [DOI] [PubMed] [Google Scholar]
- 6.Morre S, Stooker W, Lagrand W, van den Brule AJC, Niessen H. Microorganisms in the aetiology of atherosclerosis. J Clin Pathol 2000; 53: 647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funahara Y, Sumarmo, Wirawan R. Features of DIC in dengue hemorrhagic fever. Bibl Haematol 1983; 201–11. [DOI] [PubMed] [Google Scholar]
- 8.Srichaikul T, Nimmannitya S. Haematology in dengue and dengue haemorrhagic fever. Baillieres Best Pract Res Clin Haematol 2000; 13: 261–76. [DOI] [PubMed] [Google Scholar]
- 9.Chuansumrit A, Chaiyaratana W. Hemostatic derangement in dengue hemorrhagic fever. Thromb Res 2014; 133: 10–6. [DOI] [PubMed] [Google Scholar]
- 10.Ambrosino P, Tarantino L, Criscuolo L, Nasto A, Celentano A, Di Minno MND. The risk of venous thromboembolism in patients with hepatitis C. A systematic review and meta-analysis. Thromb Haemost 2016; 116: 958–66. [DOI] [PubMed] [Google Scholar]
- 11.Kiefer EM, Shi Q, Hoover DR, Kaplan R, Tracy R, Augenbraun M, Liu C, Nowicki M, Tien PC, Cohen M, Golub ET, Anastos K. Association of hepatitis C with markers of hemostasis in HIV-infected and uninfected women in the women’s interagency HIV study (WIHS). J Acquir Immune Defic Syndr 2013; 62: 301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Violi F, Ferro D, Basili S, Artini M, Valesini G, Levrero M, Cordova C. Increased rate of thrombin generation in hepatitis C virus cirrhotic patients. Relationship to venous thrombosis. J Investig Med 1995; 43: 550–4. [PubMed] [Google Scholar]
- 13.Bibas M, Biava G, Antinori A. HIV-Associated Venous Thromboembolism. Mediterr J Hematol Infect Dis 2011; 3: e2011030–e2011030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garlassi E, Carli F, Orlando G, Guaraldi G, Menozzi M, Zona S, Berti A, Rossi E, Roverato A, Palella F. Premature Age-Related Comorbidities Among HIV-Infected Persons Compared With the General Population. Clin Infect Dis 2011; 53: 1120–6. [DOI] [PubMed] [Google Scholar]
- 15.Vercellotti GM. Effects of viral activation of the vessel wall on inflammation and thrombosis. Blood Coagul Fibrinolysis 1998; 9 Suppl 2: S3–6. [PubMed] [Google Scholar]
- 16.Etingin OR, Hajjar DP. Evidence for cytokine regulation of cholesterol metabolism in herpesvirus-infected arterial cells by the lipoxygenase pathway. J Lipid Res 1990; 31: 299–305. [PubMed] [Google Scholar]
- 17.Span AH, van Dam-Mieras MC, Mullers W, Endert J, Muller AD, Bruggeman CA. The effect of virus infection on the adherence of leukocytes or platelets to endothelial cells. Eur J Clin Invest 1991; 21: 331–8. [DOI] [PubMed] [Google Scholar]
- 18.Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, Esmon CT, Jacob HS. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci U S A 1990; 87: 7095–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrington KJ, Andtbacka RH, Collichio F, Downey G, Chen L, Szabo Z, Kaufman HL, European Medicines Agency. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: subanalysis of the Phase III OPTiM trial. Onco Targets Ther Dove Medical Press; 2016; 9: 7081–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leite J, Ribeiro A, Goncalves D, Sargento-Freitas J, Trindade L, Duque V. Cerebral Venous Thrombosis as Rare Presentation of Herpes Simplex Virus Encephalitis. Case Rep Infect Dis 2019; 2019: 7835420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Görek A, Akçay Ş, Ibiş OA, Atar I, Eyüboǧlu FÖ, Gorek A, Akcay S, Ibis OA, Atar I, Eyuboglu FO. Herpes simplex virus infection, massive pulmonary thromboembolism, and right atrial thrombi in a single patient: Case report. Heart Lung 2007; 36: 148–53. [DOI] [PubMed] [Google Scholar]
- 22.Li JZ, Sax PE. HSV-1 encephalitis complicated by cerebral hemorrhage in an HIV-positive person. AIDS Read 2009; 19: 153–5. [PubMed] [Google Scholar]
- 23.Singh TD, Fugate JE, Hocker S, Wijdicks EFM, Aksamit AJ, Rabinstein AA. Predictors of outcome in HSV encephalitis. J Neurol 2016; 263: 277–89. [DOI] [PubMed] [Google Scholar]
- 24.Sutherland MR, Raynor CM, Leenknegt H, Wright JF, Pryzdial EL. Coagulation initiated on herpesviruses. Proc Natl Acad Sci U S A 1997; 94: 13510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutherland MR, Ruf W, Pryzdial ELG. Tissue factor and glycoprotein C on herpes simplex virus type 1 are protease-activated receptor 2 cofactors that enhance infection. Blood 2012; 119: 3638–45. [DOI] [PubMed] [Google Scholar]
- 26.Pryzdial EL, Wright JF. Prothrombinase assembly on an enveloped virus: evidence that the cytomegalovirus surface contains procoagulant phospholipid. Blood 1994; 84: 3749–57. [PubMed] [Google Scholar]
- 27.Aasrum M, Prydz H. Gene targeting of tissue factor, factor X, and factor VII in mice: their involvement in embryonic development. Biochemistry (Mosc) 2002; 67: 25–32. [DOI] [PubMed] [Google Scholar]
- 28.Nakagaki T, Foster DC, Berkner KL, Kisiel W. Initiation of the extrinsic pathway of blood coagulation: evidence for the tissue factor dependent autoactivation of human coagulation factor VII. Biochemistry 1991; 30: 10819–24. [DOI] [PubMed] [Google Scholar]
- 29.Mackman N. The role of tissue factor and factor VIIa in hemostasis. Anesth Analg 2009; 108: 1447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutherland MR, Simon AY, Shanina I, Horwitz MS, Ruf W, Pryzdial ELG. Virus envelope tissue factor promotes infection in mice. J Thromb Haemost 2019; 17: 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altgärde N, Eriksson C, Peerboom N, Phan-xuan T, Moeller S, Schnabelrauch M, Svedhem S, Trybala E, Bergstro T, Bally M, Altgarde N, Eriksson C, Peerboom N, Phan-xuan T, Moeller S, Schnabelrauch M, Svedhem S, Trybala E, Bergstrom T, Bally M. Mucin-like Region of Herpes Simplex Virus Type 1 Attachment Protein Glycoprotein C (gC) Modulates the Virus-Glycosaminoglycan Interaction. J Biol Chem 2015; 290: 21473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mardberg K, Trybala E, Tufaro F, Bergstrom T. Herpes simplex virus type 1 glycoprotein C is necessary for efficient infection of chondroitin sulfate-expressing gro2C cells. J Gen Virol 2002; 83: 291–300. [DOI] [PubMed] [Google Scholar]
- 33.Rux AH, Lou H, Lambris JD, Friedman HM, Eisenberg RJ, Cohen GH. Kinetic Analysis of Glycoprotein C of Herpes Simplex Virus Types 1 and 2 Binding to Heparin, Heparan Sulfate, and Complement Component C3b. Virology 2002; 332: 324–32. [DOI] [PubMed] [Google Scholar]
- 34.Friedman HM, Cohen GH, Eisenberg RJ, Seidel CA, Cines DB. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 1984; 309: 633–5. [DOI] [PubMed] [Google Scholar]
- 35.Hung SL, Peng C, Kostavasili I, Friedman HM, Lambris JD, Eisenberg RJ, Cohen GH. The interaction of glycoprotein C of herpes simplex virus types 1 and 2 with the alternative complement pathway. Virology 1994; 203: 299–312. [DOI] [PubMed] [Google Scholar]
- 36.Etingin OR, Silverstein RL, Friedman HM, Hajjar DP. Viral activation of the coagulation cascade: molecular interactions at the surface of infected endothelial cells. Cell 1990; 61: 657–62. [DOI] [PubMed] [Google Scholar]
- 37.Livingston JR, Sutherland MR, Friedman HM, Pryzdial ELG. Herpes simplex virus type 1-encoded glycoprotein C contributes to direct coagulation factor X-virus binding. Biochem J 2006; 393: 529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutherland MR, Friedman HM, Pryzdial ELG. Herpes simplex virus type 1-encoded glycoprotein C enhances coagulation factor VIIa activity on the virus. Thromb Haemost 2004; 92: 947–55. [DOI] [PubMed] [Google Scholar]
- 39.Nuzzio KM, Watt ED, Boettcher JM, Gajsiewicz JM, Morrissey JH, Rienstra CM. High-Resolution NMR Studies of Human Tissue Factor. PLoS One 2016; 11: e0163206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tal-Singer R, Peng C, Ponce De Leon M, Abrams WR, Banfield BW, Tufaro F, Cohen GH, Eisenberg RJ. Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J Virol 1995; 69: 4471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willis SH, Peng C, Leon MP, Nicola AV, Rux AH, Cohen GH, Eisenberg RJ. Expression and Purification of Secreted Forms of HSV Glycoproteins from Baculovirus-Infected Insect Cells. Methods Mol Med 1998; 10: 131–56. [DOI] [PubMed] [Google Scholar]
- 42.Morrissey JH, Fair DS, Edgington TS. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb Res 1988; 52: 247–61. [DOI] [PubMed] [Google Scholar]
- 43.Waters EK, Morrissey JH. Restoring full biological activity to the isolated ectodomain of an integral membrane protein. Biochemistry 2006; 45: 3769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins DL, Mann KG. The interaction of bovine factor V and factor V-derived peptides with phospholipid vesicles. J Biol Chem 1983; 258: 6503–8. [PubMed] [Google Scholar]
- 45.Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun 2010; 1: 100. [DOI] [PubMed] [Google Scholar]
- 46.Scheuermann TH, Padrick SB, Gardner KH, Brautigam CA. On the acquisition and analysis of microscale thermophoresis data. Anal Biochem 2016; 496: 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neuenschwander PF, Morrissey JH. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J Biol Chem 1994; 269: 8007–13. [PubMed] [Google Scholar]
- 48.Sen P, Neuenschwander PF, Pendurthi UR, Rao LVM. Analysis of factor VIIa binding to relipidated tissue factor by surface plasmon resonance. Blood Coagul Fibrinolysis 2010; 21: 376–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem 1991; 266: 2158–2166. [PubMed] [Google Scholar]
- 50.Huang Q, Neuenschwander PF, Rezaie AR, Morrissey JH. Substrate recognition by tissue factor-factor VIIa. Evidence for interaction of residues Lys165 and Lys166 of tissue factor with the 4-carboxyglutamate-rich domain of factor X. J Biol Chem 1996; 271: 21752–7. [DOI] [PubMed] [Google Scholar]
- 51.Petersen LC, Albrektsen T, Hjorto GM, Kjalke M, Bjorn SE, Sorensen BB. Factor VIIa/tissue factor-dependent gene regulation and pro-coagulant activity: effect of factor VIIa concentration. Thromb Haemost 2007; 98: 909–11. [PubMed] [Google Scholar]
- 52.Waxman E, Ross JB, Laue TM, Guha A, Thiruvikraman SV, Lin TC, Konigsberg WH, Nemerson Y. Tissue factor and its extracellular soluble domain: the relationship between intermolecular association with factor VIIa and enzymatic activity of the complex. Biochemistry 1992; 31: 3998–4003. [DOI] [PubMed] [Google Scholar]
- 53.Longstaff C, Thelwell C, Williams SC, Silva MMCG, Szabó L, Kolev K. The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: kinetic and microscopic studies. Blood 2011; 117: 661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCallum CD, Hapak RC, Neuenschwander PF, Morrissey JH, Johnson AE. The location of the active site of blood coagulation factor VIIa above the membrane surface and its reorientation upon association with tissue factor. A fluorescence energy transfer study. J Biol Chem 1996; 271: 28168–75. [DOI] [PubMed] [Google Scholar]
- 55.Neuenschwander PF, Morrissey JH. Deletion of the membrane anchoring region of tissue factor abolishes autoactivation of factor VII but not cofactor function. Analysis of a mutant with a selective deficiency in activity. J Biol Chem 1992; 267: 14477–82. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.