

Abstract
This study was performed to investigate the compliance, safety, dosage modifications (dose reduction and/or schedule change [including permanent S‐1 withdrawal]), and clinical parameters that predict S‐1 dosage modification in gastric cancer patients receiving adjuvant S‐1 chemotherapy. One hundred and forty‐nine patients who underwent curative D2 surgery and received adjuvant S‐1 chemotherapy were enrolled. S‐1 was administered orally (40 mg/m2 twice daily on days 1–28 every 6 weeks) for 1 year. For patients unable to tolerate S‐1, the dosage was reduced or the schedule was changed to a 3‐weekly schedule of 2 weeks on treatment followed by 1 week off treatment. The planned 1‐year treatment was completed in 73.8% of patients; 69 patients required dosage modification because of toxicity. The most frequent cause of dosage modification was enterocolitis (37 patients; defined as ≥ grade 2 abdominal pain and/or ≥ grade 2 diarrhea). Most dosage modification occurred during the early cycles of treatment (within the first 3 months). Severe toxicities (≥ grade 3) included neutropenia (13.4%), abdominal pain (8.1%) and diarrhea (8.1%). In multivariate analyses, decreased relative dose intensity was related to poor disease‐free survival independent of stage, and only low creatinine clearance predicted S‐1 dosage modification. In conclusion, although adjuvant S‐1 therapy has a high compliance rate, meticulous monitoring of adverse events is required in the early period of treatment. Decreased creatinine clearance was the only factor that predicted dosage modification. In patients with creatinine clearance <50 mL/min, dosage reduction should be considered from the initiation of S‐1 treatment. (Cancer Sci 2013; 104: 123–116)
Gastric cancer (GC) is the second most common cancer in Asia, and more than half of new GC cases throughout the world occur in Eastern Asia. D2 dissection is considered the standard surgical procedure for GC in countries with a high incidence of GC, such as Korea and Japan, where there is a high volume of, and consequently much experience with, GC surgery.1, 2 However, more than 40% of patients eventually relapse after surgery and several types of adjuvant treatment have been tried to lower the recurrence rate.
The Adjuvant Chemotherapy Trial of TS‐1 for Gastric Cancer (ACTS‐GC)3, 4 showed a survival benefit of S‐1 in East Asian patients who had undergone D2 dissection for GC. S‐1 is a novel oral agent containing tegafur, a prodrug of 5‐fluorouracil (5‐FU), and two biochemical modulators of 5‐FU.5 5‐Chloro‐2,4‐dihydroxypyridine (CDHP) increases the pharmacological actions of 5‐FU by inhibiting dihydropyrimidine dehydrogenase. Potassium oxonate, which localizes to the mucosal cells of the gastrointestinal (GI) tract after oral administration, reduces the incidence of GI toxicity by suppressing the activation of 5‐FU in the GI tract.5
Data are lacking on the safety, compliance, and dosage changes during adjuvant S‐1 chemotherapy in GC patients in the real clinical practice setting; only small retrospective studies have been reported.6, 7, 8, 9 Large‐scale analyses of the compliance, adverse events, and S‐1 dosages have been reported only in GC patients receiving palliative S‐1 therapy.10 To our knowledge, most data on the use of adjuvant S‐1 treatment in GC patients have been limited to Japanese patients. As adjuvant chemotherapy aims to increase the probability of a cure from cancer, improving compliance with adjuvant treatment in cancer patients is an important issue.
The aim of this study was to investigate the toxicity profiles, compliance, and S‐1 dosage modifications (dose reduction [DR] and/or schedule change [SC]) during adjuvant S‐1 therapy in the real clinical practice setting. We also analyzed the clinical parameters to identify the factors related to DR/SC during adjuvant S‐1 therapy and survival outcomes.
Materials and Methods
Patient population
This cohort study was conducted using a GC patient cohort maintained at Seoul National University Bundang Hospital (SNUBH). The GC cohort of this study per se was a subgroup of the patient cohort with various solid tumors to which cancer patients were enrolled prospectively for a study titled “Pharmacogenetic/pharmacogenomic analyses on chemotherapeutic efficacy and toxicities in Korean cancer patients receiving chemotherapy” at SNUBH. In the study, patients who had received chemotherapy and had given consent to the study were enrolled prospectively, and their blood samples were collected for pharmacogenetic or pharmacogenomic analyses. Clinical information about the patients' demographics, tumor characteristics, applied treatments, treatment outcomes, and toxicities were also recorded. Using the database of the pharmacogenetic/pharmacogenomic study, this separate study was designed as a retrospective investigation of S‐1 dosage modification (DR and/or SC), compliance, toxicity profiles, and clinical parameters predictive of DR/SC in GC patients receiving adjuvant S‐1 chemotherapy. As the database of the pharmacogenetic/pharmacogenomic study did not include sufficient information about the dosage modifications (DR or SC) of S‐1 or the clinical courses of S‐1‐associated enterocolitis, some data were supplemented retrospectively by electronic medical chart review. This study was approved by the Institutional Review Board of SNUBH and conformed to the provisions of the 1995 Declaration of Helsinki.
Patients with gastroesophageal junction or gastric adenocarcinoma who underwent curative surgery (gastrectomy with D2 lymph node dissection) between September 2006 and March 2010 and who received adjuvant S‐1 chemotherapy were included. Pathology staging was performed using the manual of the American Joint Committee on Cancer (AJCC, 6th edition). Patients with stages II–IV(M0) were candidates for adjuvant chemotherapy; however, stage IB patients with some risk factors (i.e. N2 lymph node metastasis by the guidelines of the Japanese Gastric Cancer Association11) also received adjuvant S‐1 therapy when the patient agreed to chemotherapy. Patients had recovered fully from surgery and had adequate bone marrow, renal, and hepatic function before the initiation of S‐1 therapy. Creatinine clearance (Ccr) was calculated using the Cockcroft‐Gault formula.
Adjuvant S‐1 therapy
S‐1 was administered orally for 4 weeks, followed by a 2‐week rest period. S‐1 therapy was repeated every 6 weeks, and this interval was designated as one cycle. The duration of S‐1 chemotherapy was planned to be 1 year if there was no evidence of tumor recurrence, unacceptable adverse events, or patient refusal. S‐1 was administered twice a day, and the dose was determined relative to body surface area (BSA) as follows: 40 mg for a BSA < 1.25 m2; 50 mg for a BSA of 1.25–1.49 m2; and 60 mg for a BSA ≥ 1.5 m2. Toxicities were evaluated according to the Common Terminology Criteria for Adverse Events (CTC‐AE, version 3.0). The dose intensity (DI) was calculated as the ratio of the total dose (expressed in mg) per square meter of the patient, divided by the total treatment duration. The relative dose intensity (RDI) of S‐1 was calculated by dividing the received DI by the projected DI.
In patients who developed adverse events, the next chemotherapy cycle was delayed until the toxicities subsided to an absolute neutrophil count ≥ 1.5 × 109 per L and/or platelet count ≥ 100 × 109 per L for hematological toxicities and to ≤ grade 1 for nonhematological toxicities. The dose or treatment schedule was modified according to the toxicity profile. In principle, if a patient had a hematological or nonhematological toxicity ≥ grade 3, one level of DR was performed, from 120 to 100 mg, 100 to 80 mg, or 80 to 60 mg. If recurrent severe hematological or nonhematological toxicity (≥ grade 3) persisted despite the DR, one further level of DR was applied. If a patient was unable to tolerate 60 mg/day, the S‐1 therapy was withdrawn permanently. In patients who developed recurrent nonhematological toxicity ≥ grade 2, if the patient could withstand the full course of S‐1 without severe toxicity during the first 2 weeks, SC without DR was adjusted from a 4‐week administration followed by a 2‐week rest (6‐weekly schedule) to a 2‐week administration followed by a 1‐week rest (3‐weekly schedule). However, if sufficient S‐1 could not be administered during the first 2 weeks because of adverse events (recurrent nonhematological toxicity ≥ grade 2) despite SC to the 3‐weekly schedule, an additional level of DR was also applied.
Statistical analysis
The time to DR/SC was calculated from the date of S‐1 initiation until the first date of the DR and/or SC using the Kaplan–Meier (KM) method by censoring at the date of treatment discontinuation because of tumor recurrence, the patient's refusal, or treatment withdrawal irrespective of the S‐1‐induced toxicities. When a patient experienced a permanent S‐1 withdrawal without previous DR or SC because of toxicity, the patient was considered an ‘event’ case, and the date of permanent S‐1 withdrawal was recorded. Disease‐free survival (DFS) was defined as the interval from the date of S‐1 initiation to the date of recurrence or death from any cause. Overall survival (OS) was defined as the interval from the S‐1 initiation to the date of death from any cause. DFS and OS were also analyzed using the KM method.
Log‐rank tests were used to compare the differences in incidence of DR/SC or in survival outcomes between groups in univariate analyses. Cox proportional hazards models were used to identify the clinical parameters that predicted S‐1 dosage modification or survival outcomes in multivariate analyses. Variables with P < 0.10 in univariate analyses were included in the multivariate model, and a forward conditional method was used. Two‐sided P‐values < 0.05 were considered significant. All analyses were performed using spss for Windows, version 15.0 (SPSS Inc., Chicago, IL, USA).
Results
Patient characteristics
Between September 2006 and March 2010, 158 GC patients received adjuvant S‐1 chemotherapy. The starting dose was reduced at the physician's discretion in nine patients because of Eastern Cooperative Oncology Group Performance Status (PS) of grade 2 combined with frail general condition and old age (≥70 years) in eight patients and underlying mild neutropenia at the time of S‐1 initiation in one patient. Therefore, 149 patients were finally included in this study (Fig. 1). The baseline characteristics of the 149 patients are described in Table 1. The median age was 55 years (range 30–77), and nearly all patients (97.3%) had a PS of grade 0 or 1 at the time of S‐1 initiation. Total gastrectomy was performed in 50 patients (33.6%) and partial gastrectomy in 99 patients (66.4%). The median time from gastrectomy to chemotherapy was 29 days (range 22–70). As of July 2012, the median duration of follow‐up was 33.0 months (range, 1.0–66.0).
Figure 1.

CONSORT flow diagram. DR, dose reduction; SC, schedule change.
Table 1.
Characteristics of gastric cancer (GC) patients (n = 149) who received adjuvant S‐1 chemotherapy
| Clinical parameters | Number (%) |
|---|---|
| Sex | |
| Male | 87 (58.4) |
| Female | 62 (41.6) |
| Age | |
| <65 years | 108 (72.5) |
| ≥65 years | 41 (27.5) |
| Comorbidities | |
| Absent | 110 (73.8) |
| Present | 39 (26.2) |
| Charlson comorbidity index (median) | 0 (range, 0–5) |
| Performance status (ECOG) | |
| 0 | 62 (41.6) |
| 1 | 83 (55.7) |
| 2 | 4 (2.7) |
| Creatinine clearance (mL/min) | |
| ≥30, <50 | 17 (11.4) |
| ≥50, <80 | 87 (58.4) |
| ≥80 | 45 (30.2) |
| Primary tumor location in the stomach | |
| Upper 1/3 | 31 (20.8) |
| Middle 1/3 | 34 (22.8) |
| Low 1/3 | 51 (34.2) |
| Over 2/3 area of entire stomach | 33 (22.1) |
| Histological group | |
| Well differentiated | 1 (0.7) |
| Moderately differentiated | 52 (34.9) |
| Poorly differentiated | 69 (46.3) |
| Signet ring cell | 15 (10.1) |
| Mucinous | 8 (5.4) |
| Undifferentiated | 2 (1.3) |
| Unclassified | 2 (1.3) |
| Lauren classification | |
| Diffuse | 83 (55.7) |
| Intestinal | 54 (36.2) |
| Mixed | 11 (7.4) |
| Indeterminate | 1 (0.7) |
| Stage (AJCC 6th edition) | |
| IB | 8 (5.4) |
| II | 80 (53.7) |
| III | 57 (38.3) |
| IV (M0) | 4 (2.7) |
| Extent of gastrectomy | |
| Total gastrectomy | 50 (33.6) |
| Subtotal gastrectomy | 97 (65.1) |
| Proximal gastrectomy | 2 (1.3) |
| Type of surgical procedure | |
| Laparoscopic surgery | 71 (47.7) |
| Open surgery | 78 (52.3) |
AJCC, American Joint Committee on Cancer; ECOG, Eastern Cooperative Oncology Group.
Treatment exposure
One hundred and ten patients (73.8%) completed the planned 1‐year treatment. Ninety patients received nine cycles of S‐1, and 20 patients who had some treatment delay completed S‐1 therapy with eight cycles, based on the attending physician's discretion because nearly 1 year had already passed since the initiation of S‐1. Among the 39 patients who had not completed the 1‐year S‐1 therapy, 24 patients were permanently withdrawn from S‐1 therapy because of toxicity, and 15 patients were withdrawn for other reasons not associated with S‐1 toxicity; 10 of the 39 patients were withdrawn permanently from the S‐1 therapy without trying DR or SC because of S‐1‐induced adverse events (Fig. 1).
Relative dose intensity decreased abruptly during the initial chemotherapy periods; the mean RDIs of the first, second, third, fourth, sixth, and eighth cycles were 90.0%, 84.6%, 82.0%, 80.0%, 76.8%, and 74.6%, respectively (Fig. 2A). Treatment was continued for at least 3 months in 87% of the 149 patients, for 6 months in 80%, for 9 months in 77%, and for 12 months in 74%. Overall, median and mean RDIs during all chemotherapy cycles in all patients were 89.5% (range, 3.2–100%) and 78.0% (95% confidence interval [CI], 73.3–82.7%), respectively.
Figure 2.
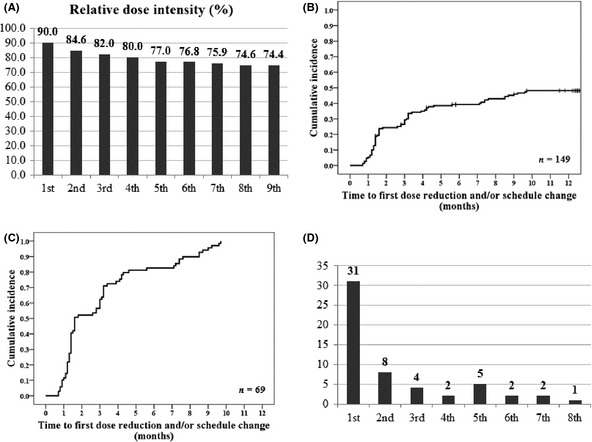
(A) Relative dose intensity of S‐1 (per cycle). (B) Time to first dose reduction and/or schedule change (in the whole population; n = 149). (C) Time to first dose reduction and/or schedule change (among patients who experienced dose modification; n = 69). (D) Frequency of S‐1‐associated enterocolitis (defined as ≥ grade 2 abdominal pain and/or ≥ grade 2 diarrhea) per cycle.
DR or SC during adjuvant S‐1 therapy
Dose reduction and/or schedule change (including permanent S‐1 withdrawal without trying DR or SC; n = 10) was applied because of S‐1‐related adverse events in 69 patients (46.3%; Fig. 1). Most DR/SC was applied during the early cycles of S‐1 therapy; the cumulative incidence rates of DR/SC were 5.4% at 1 month, 24.4% at 2 months, 29.3% at 3 months, 39.2% at 6 months, and 48.2% at 12 months (Fig. 2B). A separate analysis of the patients who had experienced a DR or SC (n = 69) showed a median time to DR/SC of 1.6 months (95% CI, 0.5–2.7 months; Fig. 2C).
Toxicity analysis
The toxicity profiles during the S‐1 therapy are shown in Table 2. Per patient analysis showed that the most common hematological toxicity was anemia (87.9%). However, the most common severe hematological toxicity (≥ grade 3) was neutropenia (13.4%). Other severe hematological toxicities except neutropenia were extremely rare. Among nonhematological toxicities (per patient analysis), diarrhea (79.2%), anorexia (77.9%), fatigue (61.1%), nausea (54.4%), and abdominal pain (48.3%) were frequently observed. Of these nonhematological toxicities, the severe adverse events (≥ grade 3) included diarrhea (8.1%), abdominal pain (8.1%), and anorexia (3.4%).
Table 2.
Toxicities developed during adjuvant S‐1 chemotherapy
| G1 n (%) | G2 n (%) | G3 n (%) | G4 n (%) | All n (%) | G3–4 n (%) | |
|---|---|---|---|---|---|---|
| Hematological toxicity (per cycle) | ||||||
| Anemia | 604 (55.1) | 89 (8.1) | 1 (0.1) | 0 (0.0) | 694 (63.3) | 1 (0.1) |
| Neutropenia | 213 (19.4) | 144 (13.1) | 20 (1.8) | 1 (0.1) | 378 (34.5) | 21 (1.9) |
| Thrombocytopenia | 61 (5.6) | 9 (0.8) | 0 (0.0) | 0 (0.0) | 70 (6.4) | 0 (0.0) |
| Hematological toxicity (per patient) | ||||||
| Anemia | 92 (61.7) | 38 (25.5) | 1 (0.7) | 0 (0.0) | 131 (87.9) | 1 (0.7) |
| Neutropenia | 40 (26.8) | 43 (28.9) | 18 (12.1) | 2 (1.3) | 103 (69.1) | 20 (13.4) |
| Thrombocytopenia | 17 (11.4) | 5 (3.4) | 0 (0.0) | 0 (0.0) | 19 (12.8) | 0 (0.0) |
| Nonhematological toxicity (per patient) | ||||||
| Hyperbilirubinemia | 48 (32.2) | 11 (7.4) | 1 (0.7) | 0 (0.0) | 60 (40.3) | 1 (0.7) |
| AST/ALT elevation | 43 (28.9) | 3 (2.0) | 1 (0.7) | 0 (0.0) | 47 (31.5) | 1 (0.7) |
| Anorexia | 77 (51.7) | 34 (22.8) | 5 (3.4) | 0 (0.0) | 116 (77.9) | 5 (3.4) |
| Nausea | 63 (42.3) | 16 (10.7) | 2 (1.3) | 0 (0.0) | 81 (54.4) | 2 (1.3) |
| Vomiting | 23 (15.4) | 4 (2.7) | 2 (1.3) | 0 (0.0) | 29 (19.5) | 2 (1.3) |
| Stomatitis | 27 (18.1) | 17 (11.4) | 2 (1.3) | 0 (0.0) | 46 (30.9) | 2 (1.3) |
| Diarrhea | 78 (52.3) | 28 (18.8) | 12 (8.1) | 0 (0.0) | 118 (79.2) | 12 (8.1) |
| Abdominal pain | 38 (25.5) | 22 (14.8) | 12 (8.1) | 0 (0.0) | 72 (48.3) | 12 (8.1) |
| Fatigue | 66 (44.3) | 23 (15.4) | 2 (1.3) | 0 (0.0) | 91 (61.1) | 2 (1.3) |
| Hand–foot syndrome | 20 (13.4) | 9 (6.0) | 0 (0.0) | 0 (0.0) | 29 (19.5) | 0 (0.0) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; G, grade.
In our study, we defined S‐1‐associated enterocolitis as ≥ grade 2 abdominal pain and/or ≥ grade 2 diarrhea. If the causal relationship between abdominal symptoms (≥ grade 2 abdominal pain and/or diarrhea) and S‐1‐related adverse events could not be excluded, all patients were considered to have developed S‐1‐associated enterocolitis. Analysis of the reasons for DR or SC in patients who had experienced dosage or schedule modifications (including drug withdrawal) because of S‐1 toxicities (n = 69) showed that enterocolitis was the most common cause (37 patients [53.6%]). Other common causes for DR/SC included weight loss in 18 patients (26.1%), anorexia in 11 patients (15.9%), fatigue in eight patients (11.6%), and neutropenia in six patients (8.7%) (Table 3).
Table 3.
Reasons for dose reduction, treatment withdrawal, or treatment schedule change (to 3‐weekly schedule)
| Adverse events | n (69 patients in total [%])a,b |
|---|---|
| Enterocolitis | 37 (53.6) |
| Abdominal pain | 29 (42.0) |
| Diarrhea | 23 (33.3) |
| Weight loss | 18 (26.1) |
| Anorexia | 11 (15.9) |
| Fatigue | 8 (11.6) |
| Neutropenia | 6 (8.7) |
| Stomatitis | 5 (7.2) |
| Infection | 3 (4.3) |
| Emesis | 2 (2.9) |
| Skin rash | 1 (1.4) |
| Hand–foot syndrome | 1 (1.4) |
| Hypoglycemia | 1 (1.4) |
| Cerebral infarct | 1 (1.4) |
The percentages and numbers do not balance because of overlap of some reasons (for dose reduction [DR], treatment withdrawal, or treatment schedule change [SC]) during the treatment.
Of the 69 patients who underwent DR, SC, or treatment withdrawal because of adverse events, DR (including 10 cases of permanent treatment withdrawal from the first) without SC was applied in 57 patients, SC to the 3‐weekly cycle without DR was applied in three patients, and both DR and SC were applied in nine patients.
S‐1‐associated enterocolitis (≥ grade 2 abdominal pain and/or ≥ grade 2 diarrhea) developed in 55 patients (36.9%). Most events of enterocolitis developed during the first cycle of S‐1 chemotherapy (Fig. 2D). Of these 55 patients, 43 experienced DR or SC. Thirty‐seven of these patients required DR or SC because of intolerable enterocolitis (abdominal pain or diarrhea) itself and six patients because of combined other intolerable toxicities rather than enterocolitis, including weight loss in four patients, fatigue in one patient, anorexia in one patient, and stomatitis in one patient; one patient had both weight loss and anorexia. Of the 55 patients with S‐1‐associated enterocolitis, 24 patients (43.6%) visited the emergency room and 19 (34.5%) were hospitalized for the management of severe enterocolitis. Computed tomography (CT) was performed in 10 of these patients. The CT findings showed bowel wall thickening in six patients (60%), ileus or bowel wall dilation in three patients (30%), and transient ascites in one patient (10%); no abnormal finding was detected in one patient.
Predictive clinical parameters for DR or SC during adjuvant S‐1 therapy
In univariate analyses using the KM method, age, sex, PS, comorbidities, type of surgery (total gastrectomy versus partial gastrectomy), and Ccr were included. Decreased Ccr was the only clinical parameter that predicted DR/SC during adjuvant S‐1 therapy (P = 0.01). There was a nonsignificant trend toward more DR/SC in female than in male patients (P = 0.099) (Table 4A and Fig. 3). In the multivariate analysis using the Cox proportional hazards model, decreased Ccr was the only independent clinical parameter that predicted DR/SC during S‐1 therapy. Compared with patients with a Ccr ≥ 80 mL/min, the hazard ratios (HRs) were 2.13 (95% CI, 1.16–3.89) in patients with 50 ≤ Ccr < 80 mL/min and 3.00 (95% CI, 1.35–6.63) in those with 30 ≤ Ccr < 50 (Table 4B).
Table 4.
(A) Univariate and (B) multivariate analyses of clinical parameters predictive of dose reduction with or without schedule modification during adjuvant S‐1 chemotherapy
| (A) Clinical parameters | n | Cumulative incidence of the first dose reduction ± schedule modification | P‐value | ||
|---|---|---|---|---|---|
| 3 months (%) | 6 months (%) | 12 months (%) | |||
| Sex | |||||
| Male | 87 | 29.3 | 36.7 | 40.6 | 0.099 |
| Female | 62 | 29.3 | 42.5 | 58.2 | |
| Age | |||||
| <65 years | 108 | 22.3 | 37.5 | 45.1 | 0.237 |
| ≥65 years | 41 | 38.1 | 43.7 | 57.8 | |
| Performance status (ECOG) | |||||
| Grade 0 | 62 | 24.3 | 34.2 | 39.2 | 0.129 |
| Grade 1 or 2 | 87 | 33.1 | 43.0 | 55.1 | |
| Comorbidities | |||||
| None | 110 | 27.7 | 37.3 | 45.3 | 0.242 |
| Yes (≥1 comorbidity) | 39 | 33.8 | 44.8 | 56.4 | |
| Surgery | |||||
| Total gastrectomy | 50 | 20.3 | 32.9 | 43.7 | 0.26 |
| Partial gastrectomy | 99 | 33.8% | 42.4 | 50.4 | |
| Creatinine clearance (Ccr, mL/min) | |||||
| Ccr ≥ 80 | 45 | 15.6 | 22.2 | 31.2 | 0.01 |
| 50 ≤ Ccr < 80 | 87 | 35.2 | 46.4 | 52.8 | |
| 30 ≤ Ccr < 50 | 17 | 36.7 | 49.3 | 79.7 | |
| (B) Creatinine clearance (Ccr, mL/min) | Hazard ratio | 95% confidence interval | P‐value |
|---|---|---|---|
| Ccr ≥ 80 | 1.00 | – | – |
| 50 ≤ Ccr < 80 | 2.13 | 1.16–3.89 | 0.014 |
| 30 ≤ Ccr < 50 | 3.00 | 1.35–6.63 | 0.007 |
ECOG, Eastern Cooperative Oncology Group.
Figure 3.

Time to first dose reduction and/or schedule change according to renal function (creatinine clearance, Ccr).
Survival analyses
Three‐year OS rate was 91.6% and 3‐year DFS rate was 84.1%. Survival outcomes according to stages were shown in Table 5. In univariate analyses related to DFS or OS, age, sex, PS, comorbidities, stage (AJCC 6th edition), type of surgery (total gastrectomy versus partial gastrectomy), Ccr and RDI (≤89.5% vs. >89.5%) were included. In multivariate analyses, considering small patient numbers with stage I or IV(M0), the stage was classified into two categories (I–II vs. III–IV[M0]).
Table 5.
(A) Univariate and (B) multivariate analyses of clinical parameters related to survival outcomes
| (A) Clinical parameters | n | Overall survival | Disease‐free survival | ||
|---|---|---|---|---|---|
| 3‐year survival rate (%) | P‐value | 3‐year disease‐free survival rate (%) | P‐value | ||
| Sex | |||||
| Male | 87 | 92.1 | 0.592 | 83.2 | 0.890 |
| Female | 62 | 90.9 | 85.0 | ||
| Age | |||||
| <65 years | 108 | 95.1 | 0.028 | 85.9 | 0.110 |
| ≥65 years | 41 | 82.2 | 79.9 | ||
| Performance status (ECOG) | |||||
| Grade 0 | 62 | 94.9 | 0.335 | 85.2 | 0.468 |
| Grade 1 or 2 | 87 | 89.4 | 83.3 | ||
| Comorbidities | |||||
| None | 110 | 94.2 | 0.101 | 86.1 | 0.276 |
| Yes (≥1 comorbidity) | 39 | 84.2 | 78.8 | ||
| Stage (AJCC 6th edition) | |||||
| I | 8 | 100.0 | <0.001 | 100.0 | <0.001 |
| II | 80 | 94.4 | 87.6 | ||
| III | 57 | 92.4 | 83.8 | ||
| IV(M0) | 4 | 25.0 | 0.0 | ||
| Surgery | |||||
| Total gastrectomy | 50 | 95.9 | 0.174 | 89.7 | 0.222 |
| Partial gastrectomy | 99 | 89.5 | 81.5 | ||
| Creatinine clearance (Ccr, mL/min) | |||||
| Ccr ≥ 80 | 45 | 95.2 | 0.124 | 85.2 | 0.426 |
| 50 ≤ Ccr < 80 | 87 | 92.3 | 83.2 | ||
| 30 ≤ Ccr < 50 | 17 | 80.8 | 82.4 | ||
| Relative dose intensity | |||||
| >89.5% | 74 | 94.1 | 0.188 | 91.6 | 0.024 |
| ≤89.5% | 75 | 89.4 | 77.3 | ||
| (B)Clinical parameters | Overall survival | Disease‐free survival | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P‐value | Hazard ratio (95% CI) | P‐value | |
| Stage (AJCC 6th edition) | ||||
| I–II | 1.0 | 0.033 | 1.0 | 0.047 |
| III–IV (M0) | 3.6 (1.1–11.5) | 2.3 (1.0–5.4) | ||
| Relative dose intensity | ||||
| >89.5% | – | – | 1.0 | 0.035 |
| ≤89.5% | – | 2.7 (1.1–6.9) | ||
| Age | ||||
| <65 years | 1.0 | 0.074 | – | – |
| ≥65 years | 2.6 (0.9–7.5) | – | ||
AJCC, American Joint Committee on Cancer; CI, confidence interval; ECOG, Eastern Cooperative Oncology Group.
In univariate analyses, both advanced stage (P < 0.001) and reduced RDI (≤89.5%; P = 0.024) were related to decreased DFS (Table 5A and Fig. 4). In multivariate analysis (Table 5B), RDI ≤89.5% was related to inferior DFS compared with RDI > 89.5% (P = 0.035; HR, 2.7; 95% CI, 1.1–6.9) independent of stage. Regarding OS, stage was the only prognostic factor and other clinical parameters had no relation in multivariate analysis (Table 5B).
Figure 4.
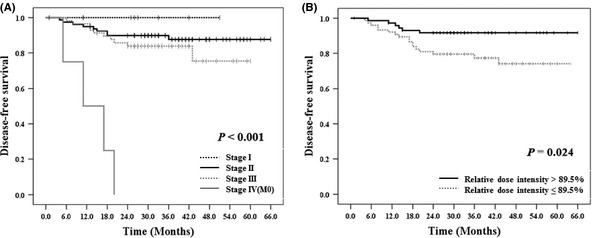
Disease‐free survival according to (A) stages and (B) relative dose intensity of S‐1.
Discussion
This study is one of the largest studies of GC patients receiving adjuvant S‐1 therapy (n = 149) after the report of ACTS‐GC3, 4 and is the first report on adjuvant S‐1 therapy outside Japan. Because our study included patients in the real clinical practice setting, we believe that the results reflect the actual reality of S‐1 adjuvant therapy.
Interestingly, 74% of patients in our study completed the 1‐year treatment, giving a rate higher than the 66% reported for the completion of the 1‐year treatment in patients enrolled in the ACTS‐GC. Three‐year OS rate was 91.6% in our study, while that was 80.1% in the S‐1 group of the ACTS‐GC. Three‐year DFS rate was 84.1% in our study, as compared with 72.2% in the S‐1 group of the ACTS‐GC. More favorable survival outcomes in our study compared to those in the ACTS‐GC might be related to less proportion of stage III or IV(M0) patients and higher compliance with 1‐year S‐1 therapy in our patient population. Our study shows that it is possible to maintain patient compliance in the daily clinical practice to the level of compliance reported in the clinical trial by appropriate measurement of S‐1‐induced adverse events. The RDI of S‐1 decreased abruptly during the early period of S‐1 treatment (18% decrease in the first 3 months versus 8% further decrease in the last 9 months, Fig. 2A). The KM curve of DR/SC (Fig. 2C) shows that the median time to DR/SC was 1.6 months (95% CI, 0.5–2.7) and that about two‐thirds (62.3%) of the DR/SC occurred in the first 3 months. In other words, most S‐1‐associated adverse events causing DR/SC developed in the first 3 months and thus dealing with those adverse events appropriately is critical to maintaining patient compliance. The toxicity profiles in this study were tolerable, as observed in the ACTS‐GC (Table 2). Neutropenia was common among the severe hematological toxicities, and abdominal pain and diarrhea were common among the severe nonhematological toxicities. Abdominal pain and diarrhea ≥ grade 3 were observed more frequently in our study than in the ACTS‐GC.
In our study, S‐1‐associated enterocolitis was defined as ≥ grade 2 abdominal pain and/or ≥ grade 2 diarrhea. We excluded grade 1 abdominal pain and/or diarrhea because we believe that they have little clinical significance and that only ≥ grade 2 symptoms affect DR/SC or drug compliance. We also intended to exclude diarrhea or abdominal pain of nonspecific causes by excluding grade 1 symptoms. However, if a causal relationship between abdominal symptoms (≥ grade 2 abdominal pain and/or diarrhea) and adverse events of S‐1 could not be excluded, all patients were considered to have S‐1‐associated enterocolitis. The CTC‐AE12, 13 defines enterocolitis as a symptom complex including abdominal pain, mucus or blood in stool, fever, ileus, and peritoneal signs; there is no comment on the development of diarrhea. However, in our study, many patients with S‐1‐induced abdominal pain also had diarrhea and thus these abdominal symptoms (abdominal pain and diarrhea) were thought to be a different presentation of one symptom complex. Therefore, we defined S‐1‐associated enterocolitis as noted above.
The most common factor that influenced DR/SC was enterocolitis (Table 3) involving 37 patients including 14 with abdominal pain only without ≥ grade 2 diarrhea, eight with diarrhea only without ≥ grade 2 abdominal pain, and 15 with both ≥ grade 2 abdominal pain and diarrhea. Enterocolitis was a large burden to patients during the S‐1 treatment because it caused frequent hospitalization and emergency room visits. In a retrospective study of GC patients receiving adjuvant S‐1 in Japan, diarrhea was one common toxicity that caused DR (32% of all toxicities that caused DR). In that report, among the patients who discontinued S‐1 therapy because of adverse events, persistent GI toxicity was the major reason, even if the grade of adverse events was not severe.7
When we compared survivals with the cut‐off value of 89.5% (median) of RDI, lower RDI (≤89.5%) was related to poor DFS in multivariate analysis independent of stage. No relation between RDI and OS may be caused by the short follow‐up duration (median, 33.0 months) or relatively small sample size in our study. Considering the relation between RDI and DFS, toxicities including enterocolitis need to be prevented and managed actively, especially during the early period of S‐1 administration. In our analysis, among patients with RDI ≤ 89.5%, RDI could not be maintained in 38.5% of patients due to the early termination of the planned 1‐year S‐1 therapy (data not shown). Therefore, as in our study, DR or SC (schedule modification to a 3‐weekly schedule) will probably be helpful for patients experiencing toxicity to maintain RDI or to complete the 1‐year treatment. Some patients who experienced toxicities were managed only with SC without DR. The 3‐weekly schedule enabled patients to have an early rest in the course of chemotherapy and to continue taking S‐1. The schedule modification may be useful for improving patient compliance without compromising RDI. In addition, it was reported that completion of 1‐year S‐1 treatment was related to improved survival in post hoc analysis of the ACTS‐GC.7, 14 As the completion of 1‐year S‐1 treatment was strongly correlated to RDI in our patient cohort (data not shown), the completion of 1‐year treatment was not included as an independent variable in our analyses considering multicollinearity.
Ccr was the only independent predictor of DR/SC (Table 4 and Fig. 3). A few reports have reported that Ccr influences DR or adverse events.7, 8, 10 Impaired renal function has been reported to influence the pharmacokinetics of S‐1 in an animal model and in patients with GC.15 Plasma clearance of CDHP and 5‐FU was retarded in proportion to the degree of renal impairment, and there was a close correlation between Ccr and plasma CDHP and 5‐FU clearance.16 In our study, HR was significantly higher in patients with Ccr < 80 mL/min than in those with Ccr ≥ 80 mL/min (Table 4B). In particular, about 80% of patients with 30 ≤ Ccr < 50 mL/min experienced DR or SC (Table 4A). This suggests that DR of S‐1 (i.e., one‐level DR) from the initiation in patients with 30 ≤ Ccr < 50 will enhance compliance by reducing toxicity.
This study has some limitations. This study was not preplanned and thus involved retrospective analyses, which may have been biased by unrecognized factors. However, because this study analyzed patients enrolled in a prospective cohort study and the clinical data on the treatments and toxicities were maintained prospectively, the possibility of bias in the process of data collection and analyses should be low. It is not clear why the frequency of severe abdominal pain and diarrhea after S‐1 therapy was higher in our study than in previous reports.3, 7 The possible contribution of ethnic differences between Korean and Japanese to the results cannot be excluded completely. There is a possibility that the symptoms on which the attending physicians focused might have influenced the reporting rate because abdominal pain and many nonhematological toxicities are subjectively perceived symptoms. Differences in renal function might be another possible reason for the higher rate of severe abdominal pain and diarrhea in our study. In our study, patients with 30 ≤ Ccr < 50 mL/min received S‐1 treatment at the conventional dose relative to BSA regardless of Ccr. Ccr was the only predictor of DR/SC in our study, and the inclusion of patients with low Ccr without consideration of the initial DR at the first cycle of S‐1 therapy might have contributed to the higher frequency of severe abdominal pain and diarrhea compared with previous studies.
In conclusion, adjuvant S‐1 chemotherapy had a high compliance rate in Korean patients and the toxicities were tolerable. Decreased RDI of adjuvant S‐1 therapy resulted in poor DFS. Because DR/SC and reduction in the RDI occurred mainly within the first 3 months, meticulous monitoring for the development of adverse events in the early period of S‐1 treatment is required. The best clinical predictor of the need for DR/SC was low Ccr. Patients with low Ccr should be monitored closely during the S‐1 therapy; in particular, dosage reduction at the initiation of adjuvant S‐1 therapy should be considered for patients with 30 ≤ Ccr < 50 mL/min.
Disclosure Statement
The authors have no conflict of interest.
(Cancer Sci, doi: 10.1111/cas.12044, 2012)
References
- 1. Wu CW, Hsiung CA, Lo SS et al Nodal dissection for patients with gastric cancer: a randomised controlled trial. Lancet Oncol 2006; 7: 309–15. [DOI] [PubMed] [Google Scholar]
- 2. Sasako M, Inoue M, Lin JT, Khor C, Yang HK, Ohtsu A. Gastric Cancer Working Group report. Jpn J Clin Oncol 2010; 40 (Suppl. 1): i28–37. [DOI] [PubMed] [Google Scholar]
- 3. Sakuramoto S, Sasako M, Yamaguchi T et al Adjuvant chemotherapy for gastric cancer with S‐1, an oral fluoropyrimidine. N Engl J Med 2007; 357: 1810–20. [DOI] [PubMed] [Google Scholar]
- 4. Sasako M, Sakuramoto S, Katai H et al Five‐year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S‐1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011; 29: 4387–93. [DOI] [PubMed] [Google Scholar]
- 5. Shirasaka T. Development history and concept of an oral anticancer agent S‐1 (TS‐1): its clinical usefulness and future vistas. Jpn J Clin Oncol 2009; 39: 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakuma K, Hosoya Y, Arai W et al Alternate‐day treatment with S‐1 in patients with gastric cancer: a retrospective study of strategies for reducing toxicity. Int J Clin Oncol 2010; 15: 166–71. [DOI] [PubMed] [Google Scholar]
- 7. Iwasa S, Yamada Y, Fukagawa T et al Management of adjuvant S‐1 therapy after curative resection of gastric cancer: dose reduction and treatment schedule modification. Gastric Cancer 2011; 14: 28–34. [DOI] [PubMed] [Google Scholar]
- 8. Aoyama T, Yoshikawa T, Hayashi T et al Risk factors for 6‐month continuation of S‐1 adjuvant chemotherapy for gastric cancer. Gastric Cancer 2012; doi: 10.1007/s10120-012-0158-1 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 9. Aoyama T, Yoshikawa T, Watanabe T et al Safety and feasibility of S‐1 adjuvant chemotherapy for gastric cancer in elderly patients. Gastric Cancer 2012; 15: 76–82. [DOI] [PubMed] [Google Scholar]
- 10. Yamanaka T, Matsumoto S, Teramukai S, Ishiwata R, Nagai Y, Fukushima M. Safety evaluation of oral fluoropyrimidine S‐1 for short‐ and long‐term delivery in advanced gastric cancer: analysis of 3758 patients. Cancer Chemother Pharmacol 2008; 61: 335–43. [DOI] [PubMed] [Google Scholar]
- 11. Japanese Gastric Cancer A . Japanese Classification of Gastric Carcinoma ‐ 2nd English Edition. Gastric Cancer 1998; 1: 10–24. [DOI] [PubMed] [Google Scholar]
- 12. CTCAE, Cancer Therapy Evaluation Program: Common terminology criteria for adverse events, version 3.0, DCTD, NCI, NIH, DHHS, 2003. [Cited 5 August 2011.] Available from URL: http://ctep.cancer.gov
- 13. CTCAE, Common Terminology Criteria for Adverse Events version 4.0, 2010. [Cited 5 August 2011.] Available from URL: http://evs.nci.nih.gov/ftp1/CTCAE/About.html
- 14. Sakuramoto S, Kikuchi S, Watanabe M. Efficacy of S‐1 (oral fluoropyrimidine) for gastric cancer. The Medical Frontline (Saishin Igaku) 2009; 64: 1075–80. [Google Scholar]
- 15. Fujita K, Yamamoto W, Endo S et al CYP2A6 and the plasma level of 5‐chloro‐2, 4‐dihydroxypyridine are determinants of the pharmacokinetic variability of tegafur and 5‐fluorouracil, respectively, in Japanese patients with cancer given S‐1. Cancer Sci 2008; 99: 1049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ikeda M, Furukawa H, Imamura H et al Pharmacokinetic study of S‐1, a novel oral fluorouracil antitumor agent in animal model and in patients with impaired renal function. Cancer Chemother Pharmacol 2002; 50: 25–32. [DOI] [PubMed] [Google Scholar]