

Abstract
This randomized phase II study was intended to identify the optimal dose of TAS‐108, a novel steroidal antiestrogen, for the treatment of breast cancer in postmenopausal Japanese women. The potential clinical effects of TAS‐108 on the uterus, bone, serum lipids, and hormones were also investigated. Postmenopausal women with hormone receptor‐positive metastatic breast cancer who had previously received one or two endocrine therapies were randomly assigned to one of the three possible dose levels of TAS‐108 (40, 80 or 120 mg/day). Oral TAS‐108 was given daily, and the efficacy and safety of the three doses were evaluated. A total of 97 patients (33, 32, and 32 in the 40‐, 80‐, and 120‐mg groups, respectively) were treated with TAS‐108. The clinical benefit rate was 30.3% for the 40‐mg, 25.0% for the 80‐mg, and 25.0% for the 120‐mg group. The 40‐mg group achieved the prespecified target threshold. TAS‐108 at all dose levels was well tolerated and appeared to have no harmful effects in terms of the variables examined in this study. We conclude that the optimal dose of TAS‐108 among the three doses is 40 mg, once daily, for further studies. JAPIC Clinical Trials Information number: Japic CTI – 121754.
Aromatase inhibitors have been widely used as first‐line endocrine therapeutic agents for postmenopausal patients with HR‐positive breast cancer and also adjuvant therapy in postmenopausal women with early breast cancer.1, 2 However, tamoxifen showed equivalent disease‐free survival compared with AIs in patients with low tumor values of Ki‐67 in an adjuvant trial,3 and it has also been reported that tamoxifen holds potential for sequential treatment of postmenopausal patients with MBC progressing after AI treatment.4 Therefore, tamoxifen still remains an important treatment option in HR‐positive breast cancer. However, due to its estrogen‐like effects on the uterus, tamoxifen has been associated with the risk of developing endometrial cancer,5 which has been an important motivating factor in the development of new types of antiestrogen with different pharmacologic profiles.
A novel steroidal antiestrogenic compound, TAS‐108 binds strongly to ERα and ERβ with a mechanism of action unlike tamoxifen,6 and in humans is mainly metabolized by CYP3A4 enzymes in the liver.7 TAS‐108 shows pure antagonistic activity as it blocked both the N‐terminal AF‐1 and C‐terminal AF‐2 transactivation functions of ERα, abolished the recruitment of co‐activators, but promoted the recruitment of co‐repressors and allowed normal DNA binding. Additionally, TAS‐108 has shown antagonistic effects on a mutant ERα reported to have a tamoxifen‐resistant phenotype and preliminarily shown to have antitumor activity against tamoxifen‐ and AI‐resistant cell lines.8 TAS‐108 has also shown fewer estrogenic effects on the uterus than tamoxifen in animal models.6 Furthermore, a preclinical study suggested possible positive effects of TAS‐108 on BMD.9
Several phase I studies were carried out in the USA involving postmenopausal healthy women and MBC patients.10, 11 In these studies, TAS‐108 was well tolerated at all doses of 40–160 mg, and showed possible antitumor activity.
Two phase I studies of TAS‐108 in Japan in 12 postmenopausal healthy women12 and 15 MBC patients,13 involving doses of 40, 80, or 120 mg showed a favorable safety profile, and encouraging antitumor activity in the MBC group. However, these studies were not designed to establish the optimal dose of TAS‐108.
The present multicenter study was carried out to evaluate both the efficacy and safety of three different TAS‐108 doses, and subsequently to identify the optimal dose of TAS‐108 for further studies in postmenopausal Japanese patients with MBC. Considering long‐term use, especially in the adjuvant setting, the effect on aspects not directly related to cancer would be especially important for administration in postmenopausal women. We also investigated the potential clinical impact of TAS‐108 on the uterus, bone, serum lipids, and hormones.
Patients and Methods
Study design and treatment
In this multicenter, randomized, non‐blinded phase II study carried out in Japan, patients were randomly assigned to receive oral TAS‐108 with a daily dose of either 40, 80 or 120 mg (in units of 40 mg tablets; Taiho Pharmaceutical, Tokyo, Japan) after the first daily meal for 24 weeks or until disease progression, development of unacceptable toxicity, or withdrawal of consent. Patients with favorable response (CR, PR, or SD) at 24 weeks could continue the treatment. Two stratification factors: response to prior endocrine treatment, and presence of visceral metastasis, were used to balance the patient populations among the three dose groups at randomization.
At baseline, a full medical history was taken and a physical examination carried out. Patients also underwent clinical laboratory tests, examination of vital signs, electrocardiogram, and PS evaluation. At 2 and 4 weeks after initiating drug intake, and subsequently every 4 weeks, evaluations were carried out, including physical examination, toxicity assessment, and clinical laboratory tests. Endometrial thickness was measured by transvaginal (transabdominal) ultrasonography at baseline and every 24 weeks during treatment. Lumbar spine (L2–L4) BMD was assessed by dual energy X‐ray absorptiometry at baseline and after 24 weeks of treatment. Serum hormones (E2, FSH, prolactin, thyroid‐stimulating hormone, cortisol, testosterone, and sex hormone‐binding globulin), serum lipid (apolipoprotein A‐I and B) and BMMs (serum osteocalcin and I‐CTP) were assessed at baseline and at regular intervals (measured at SRL Medisearch, Tokyo, Japan). All blood tests including for serum lipids (total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, and triglycerides), were carried out on specimens obtained before the first daily meal.
Eligibility criteria
Postmenopausal HR‐positive women aged 20–80 years with histologically or cytologically proven, locally advanced or MBC, were eligible for the study if they appeared suitable for endocrine therapy. The postmenopausal status was defined as being amenorrheic for at least 1 year (for patients aged 50 years or over), being amenorrheic for at least 1 year and with both serum E2 and FSH levels in the postmenopausal range, or being amenorrheic due to radiotherapy for at least 3 months with both E2 and FSH levels in the postmenopausal range (for patients aged under 50 years). All patients had to have at least one progressive target lesion after one or two different endocrine therapies. Patients could have received one prior chemotherapy regimen, unless it had been given as the most recent prior treatment. Patients who had had only adjuvant endocrine therapy were eligible if they had relapsed during therapy or <6 months from the completion or discontinuation of the therapy. Other inclusion criteria included: adequate organ function; a predicted life expectancy of >3 months; PS of 2 or less on the Zubrod scale.
Patients were ineligible if they had allergies to steroid preparations; abnormal vaginal bleeding at the start of the treatment; past serious thromboembolism; current serious complication(s); active double cancer; inflammatory breast cancer, lung metastasis with cancer‐related lymphangitis, brain metastasis with any symptoms, and widespread liver metastasis.
The study was approved by the institutional review board of each participating center. Written informed consent was obtained from all patients.
Efficacy and safety assessments
Tumor response assessments were carried out at baseline and at 8‐week intervals. The response was assessed according to the Response Evaluation Criteria in Solid Tumors criteria.14 For CR and PR, the response had to be confirmed more than 4 weeks after the first date when a response was documented. The efficacy results were reviewed and determined by the independent CEC. Retrospective analyses for the response to TAS‐108 were carried out to explore the subgroup, including patients who had tamoxifen‐ or AI‐resistant tumors, defined as patients who had: (i) previously failed to respond to the most recent prior treatment for advanced disease; (ii) progressed following response to treatment; and (iii) relapsed either on adjuvant therapy or within 6 months from the completion of adjuvant therapy. Adverse events were graded according to the Common Terminology Criteria for Adverse Events, version 3.0.
Statistical considerations
The primary end‐point was the CBR at 24 weeks, defined as the percentage of eligible patients who achieved a CR, PR, or SD for at least 24 consecutive weeks. The secondary end‐points included ORR (CR or PR) and TTP. The secondary end‐points also included safety and effects on ET, BMD, BMMs, serum lipids, and hormone levels.
We considered 35% as a clinically meaningful CBR and that that would be the expected CBR of TAS‐108 in the study population, whereas a 10% CBR would be considered poor and lacking promise for future development. Sample size was estimated to ensure both appropriate precision for CBR estimation in all evaluable patients and sufficient statistical power to reject the null hypothesis with adjusted significance level for multiple comparisons. At least a total of 84 evaluable patients, 28 in each dose group, were required to carry out binomial tests for P1 = 35% and P0 = 10% in each dose group with 2.5% family‐wise one‐sided type‐I error level with 80% statistical power in each test. For CBR estimation in total, and assuming a 5% drop‐out rate, a total of 96 patients, 32 in each dose group, were planned to be enrolled in the study.
The Kaplan–Meier method was used to estimate TTP, which was defined as the time from first drug administration to disease progression. The 98.3% exact binomial CI was estimated for CBR and ORR in each dose group.
In order to explore the potential clinical impact of TAS‐108 on the uterus, bone, serum lipids, and hormones, we carried out non‐parametric analysis. Bone mineral density and BMMs were assessed in patients with no bone metastasis at baseline. Because of uncertainty regarding the asymptotic normality of changes in variables, the Wilcoxon signed‐rank test was used to assess the significance of changes from baseline within each dose group with a value <0.05 considered as statistically significant.
Results
Patient population
A total of 98 patients were enrolled at 34 centers in Japan (Appendix I). One patient randomized to the 120‐mg group was censored and did not receive any TAS‐108 treatment due to not having had endocrine therapy as the most recent prior treatment. The treated patient population therefore comprised 97 patients who were fully assessable for efficacy and safety; 33 in the 40‐mg group, and 32 each in the 80‐mg and 120‐mg groups. The baseline characteristics of the patients were well balanced among the three dose groups (Table 1). The study population included 14 patients with metastatic disease refractory to prior tamoxifen treatment and 70 were refractory to prior AI treatment.
Table 1.
Patient characteristics at baseline
| Characteristics | Dose group | Total (n = 97) | ||
|---|---|---|---|---|
| 40 mg (n = 33) | 80 mg (n = 32) | 120 mg (n = 32) | ||
| Median age, years (range) | 63.0 (44–80) | 63.0 (50–74) | 58.5 (48–78) | 62.0 (44–80) |
| Performance status, n (%) | ||||
| 0 | 28 (85) | 27 (84) | 28 (88) | 83 (86) |
| 1 | 5 (15) | 5 (16) | 4 (13) | 14 (14) |
| 2 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Body mass index (median), kg/m2 | 23.2 | 23.0 | 22.1 | 22.7 |
| Prior treatment, n (%)a | ||||
| Endocrine therapy regimens | ||||
| 1 | 19 (58) | 13 (41) | 21 (66) | 53 (55) |
| 2 | 14 (42) | 19 (59) | 11 (34) | 44 (45) |
| Chemotherapy regimens | ||||
| 0 | 21 (64) | 28 (88) | 23 (72) | 72 (74) |
| 1 | 12 (36) | 4 (13) | 9 (28) | 25 (26) |
| Sites of metastasis, n (%) | ||||
| Soft tissue | 20 (61) | 23 (72) | 19 (59) | 62 (64) |
| Bone | 12 (36) | 15 (47) | 17 (53) | 44 (45) |
| Visceral | 23 (70) | 23 (72) | 23 (72) | 69 (71) |
| Other | 2 (6) | 0 (0) | 4 (13) | 6 (6) |
| Receptor status, n (%)b | ||||
| ER+/PgR+ | 22 (67) | 20 (63) | 19 (59) | 61 (63) |
| ER+/PgR− | 11 (33) | 9 (28) | 11 (34) | 31 (32) |
| ER−/PgR+ | 0 (0) | 2 (6) | 1 (3) | 3 (3) |
| ER−/PgR− | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HER2+ | 4 (12) | 4 (13) | 7 (22) | 15 (15) |
| HER2− | 24 (73) | 23 (72) | 23 (72) | 70 (72) |
| HER2 unknown | 5 (15) | 5 (16) | 2 (6) | 12 (12) |
| Disease‐free interval, n (%) | ||||
| <2 years | 5 (15) | 5 (16) | 5 (16) | 15 (15) |
| ≥2 years | 21 (64) | 22 (69) | 17 (53) | 60 (62) |
Counting a case treated with adjuvant endocrine therapy/chemotherapy as one regimen, if it had relapsed either during therapy or within 6 months of completion of therapy.
Hormone receptor status (estrogen receptor [ER]/progesterone receptor [PgR]) was determined by each study site.
Efficacy
The CBR determined by CEC was 30.3% (98.3% CI, 13.3–52.4) in the 40‐mg group, 25.0% (98.3% CI, 9.5–47.2) in the 80‐mg group, and 25.0% (98.3% CI, 9.5–47.2) in the 120‐mg group, respectively (Table 2). The 40‐mg group exceeded the target threshold of 10% in its lower limit of CI. The CEC‐determined ORR was 9.1% (98.3% CI, 1.3–27.9) in the 40‐mg group, 9.4% (98.3% CI, 1.3–28.6) in the 80‐mg group, and 6.3% (98.3% CI, 0.4–24.3) in the 120‐mg group, respectively (Table 2). The median TTP was 4.6 months in the 40‐mg group, 3.7 months in the 80‐mg group, and 3.6 months in the 120‐mg group (Table 2). The Kaplan–Meier curve of TTP is shown in Figure 1.
Table 2.
Efficacy results
| Dose group | |||
|---|---|---|---|
| 40 mg (n = 33) | 80 mg (n = 32) | 120 mg (n = 32) | |
| Response | |||
| CBR, % | 30.3 | 25.0 | 25.0 |
| 98.3% CI | 13.3–52.4 | 9.5–47.2 | 9.5–47.2 |
| ORR, % | 9.1 | 9.4 | 6.3 |
| 98.3% CI | 1.3–27.9 | 1.3–28.6 | 0.4–24.3 |
| CR, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| PR, n (%) | 3 (9.1) | 3 (9.4) | 2 (6.3) |
| SD ≥ 24 weeks, n (%) | 7 (21.2) | 5 (15.6) | 6 (18.8) |
| SD < 24 weeks, n (%) | 15 (45.5) | 14 (43.8) | 13 (40.6) |
| PD, n (%) | 8 (24.2) | 10 (31.3) | 11 (34.4) |
| Time to progression, months | |||
| Median | 4.6 | 3.7 | 3.6 |
| 95% CI | 3.6–5.4 | 2.1–5.7 | 1.9–5.6 |
CBR, clinical benefit rate; CI, confidence interval; CR, complete response; ORR, objective tumor response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Figure 1.
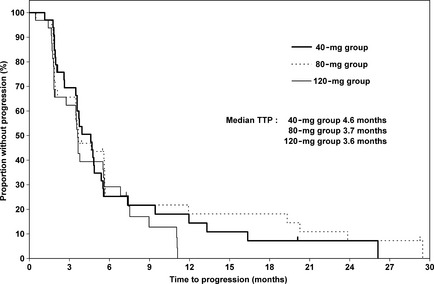
Kaplan–Meier estimates for time to progression (TTP) in postmenopausal Japanese women with breast cancer treated with three different doses of TAS‐108.
In the subgroup analysis of the patient population of tumor refractory to tamoxifen or AI, TAS‐108 treatment produced a CBR of 28.6% and 27.1%, respectively (Table 3).
Table 3.
Subgroup analysis for response to TAS‐108 within each dose group (tumor refractory to prior tamoxifen and aromatase inhibitor treatment)
| Tamoxifen refractory (n = 14) | Aromatase inhibitor refractory (n = 70) | |
|---|---|---|
| Clinical benefit rate (%) | 28.6 | 27.1 |
Safety
Most patients (72.2%) experienced drug‐related AEs (definite, probable, possible) including hot flushes, hyperhidrosis, and nausea as non‐hematological toxicities (Table 4). The most common was hot flush, reported by 22.7% of patients. A majority of these AEs observed were mild (grades 1–2) and there was no clear dose dependency regarding severity or frequency of AEs. Discontinuation due to TAS‐108‐related toxicity was rare (2/97; one patient with grade 3 hypoacusis and one patient with grade 3 dizziness in the 80‐mg group). One patient in the 80‐mg group had surgery for grade 3 cataracts in both eyes.
Table 4.
Drug‐related adverse events occurred in >10% of postmenopausal Japanese women with breast cancer treated with TAS‐108, in either dose group
| Eventa | Dose group | Total (n = 97) | ||||||
|---|---|---|---|---|---|---|---|---|
| 40 mg (n = 33) | 80 mg (n = 32) | 120 mg (n = 32) | ||||||
| All grades, n (%) | Grades 3–5, n (%) | All grades, n (%) | Grades 3–5, n (%) | All grades, n (%) | Grades 3–5, n (%) | All grades, n (%) | Grades 3–5, n (%) | |
| Hot flush | 7 (21.2) | 0 (0) | 8 (25.0) | 0 (0) | 7 (21.9) | 0 (0) | 22 (22.7) | 0 (0) |
| Hyperhidrosis | 2 (6.1) | 0 (0) | 3 (9.4) | 0 (0) | 6 (18.8) | 0 (0) | 11 (11.3) | 0 (0) |
| Nausea | 1 (3.0) | 0 (0) | 3 (9.4) | 0 (0) | 5 (15.6) | 0 (0) | 9 (9.3) | 0 (0) |
| Uterine leiomyoma | 0 (0) | 0 (0) | 4 (12.5) | 0 (0) | 1 (3.1) | 0 (0) | 5 (5.2) | 0 (0) |
| Blood cholesterol increased | 4 (12.1) | 0 (0) | 1 (3.1) | 0 (0) | 0 (0) | 0 (0) | 5 (5.2) | 0 (0) |
Patients could have had more than one event.
Exploratory analysis
Table 5 shows the results of analysis for each value. TAS‐108 did not cause significant endometrial thickening (median baseline ET, 3.3 mm; 24 weeks ET, 4.0 mm; n = 37). No change was observed in median BMD (−0.30%; n = 20), serum I‐CTP, and osteocalcin levels. The median triglyceride level decreased significantly from 104.0 to 86.0 mg/dL (P < 0.0001); there were no changes in other serum lipids. Increases in PRL, testosterone, and sex hormone‐binding globulin levels were observed.
Table 5.
Analysis of endometrial thickness (ET), bone mineral density (BMD), bone metabolism markers (BMMs), serum lipids, and endocrine hormones in postmenopausal Japanese women with breast cancer treated with TAS‐108
| Variable | n | Baseline (median) | 8a or 24b weeks (median) | Change or percentage changec from baseline to 8a or 24b weeks | ||
|---|---|---|---|---|---|---|
| Median | Range | P d | ||||
| ET, mm | 37 | 3.30 | 4.00 | 0.00 | −6.00–13.10 | 0.0850 |
| BMD, g/cm2 | 20 | 0.83 | 0.84 | −0.30 | −10.74–8.79 | 0.5220 |
| BMMs | ||||||
| Serum Osteocalcin, ng/mL | 50 | 9.50 | 9.20 | −0.62 | −38.75–85.42 | 0.8420 |
| Serum I‐CTP, ng/mL | 51 | 3.40 | 3.40 | −3.03 | −52.86–105.26 | 0.4120 |
| Serum lipids | ||||||
| Total‐cho, mg/dL | 93 | 208.00 | 212.00 | 0.00 | −78.00–94.00 | 0.4410 |
| HDL‐cho, mg/dL | 92 | 62.00 | 63.90 | 0.00 | −28.00–51.00 | 0.1960 |
| LDL‐cho, mg/dL | 91 | 121.00 | 127.00 | 3.00 | −57.00–81.00 | 0.0830 |
| Triglycerides, mg/dL | 93 | 104.00 | 86.00 | −13.00 | −217.00–301.00 | <0.0001 |
| APO‐A1, mg/dL | 93 | 147.00 | 151.00 | 4.00 | −45.00–59.00 | 0.1810 |
| APO‐B, mg/dL | 93 | 99.00 | 99.00 | 1.00 | −33.00–42.00 | 0.6680 |
| Endocrine hormones | ||||||
| E2, pg/mL | 93 | 10.00 | 11.00 | 0.00 | −11.00–33.00 | 0.1310 |
| FSH, mIU/mL | 93 | 44.49 | 44.80 | −2.60 | −24.27–26.88 | 0.3210 |
| Prolactin, ng/mL | 93 | 8.33 | 8.45 | 0.42 | −41.12–40.79 | 0.0280 |
| Testosterone, ng/dL | 93 | 0.21 | 0.22 | 0.02 | −0.23–0.31 | 0.0190 |
| TSH, μIU/mL | 93 | 2.58 | 2.51 | 0.00 | −5.90–94.30 | 0.6350 |
| Cortisol, μg/dL | 93 | 13.60 | 13.20 | 0.20 | −16.30–22.10 | 0.7540 |
| SHBG, nmol/L | 93 | 71.40 | 91.90 | 14.00 | −110.00–133.40 | <0.0001 |
Bone metabolism markers, serum lipids, and endocrine hormones were assessed at the 8‐week point in patients who received TAS‐108 for 8 weeks or more.
Endometrial thickness and BMD were assessed at the 24‐week point in patients who received TAS‐108 for 24 weeks or more.
Data are presented as change from baseline, except BMD and BMMs as percentage change from baseline.
P‐values based on the Wilcoxon signed‐rank test. APO‐A1, apolipoprotein A‐I; APO‐B, apolipoprotein B; HDL‐cho, high‐density lipoprotein cholesterol; I‐CTP, cross‐linked carboxy‐terminal telopeptide of type I collagen; LDL‐cho, low‐density lipoprotein cholesterol; SHBG, sex hormone‐binding globulin; Total‐cho, total cholesterol; TSH, thyroid‐stimulating hormone.
Discussion
This randomized phase II study was designed to evaluate the efficacy and safety of 40, 80, or 120 mg TAS‐108 given orally once daily in postmenopausal patients previously treated with one or two regimens of endocrine treatment (with a maximum of one regimen of chemotherapy), with HR‐positive MBC, particularly including prior AI‐ and/or tamoxifen‐resistant disease.
As the first step toward the best dose selection, we sought to find the “active” dose level(s) among the three dose groups based on the analysis of the primary end‐point. The tolerability at the active dose level(s) was subsequently assessed to achieve a relative balance between efficacy and toxicity of TAS‐108. In consequence, it is found that the lower dose of 40 mg showed, numerically, the highest CBR (30.3%) at 24 weeks and met the targeted expectations for clinical activity. This finding suggests that the two higher doses might have been beyond the plateau phase of the dose–response curve and therefore had a potential “reverse dose–response” effect. The safety parameters were similar between the three doses. In addition, secondary efficacy analyses supported the choice because TAS‐108 at a dose of 40 mg had similar but slightly higher antitumor activity than the two higher doses. The 40 mg dose of TAS‐108 was therefore recommended for further controlled studies against current therapeutic standards. The results observed in this study were largely similar to those reported by Buzdar et al.15
With the widespread use of AIs in the adjuvant setting, several drugs have recently been reported to be potentially effective in the treatment for breast cancer patients following the failure of AI treatment. Subgroup analysis revealed that there is biological evidence for a CBR of 28.6% (tamoxifen refractory) and 27.1% (AI refractory), and this finding supports the concept that there may be no major cross‐resistance between tamoxifen/AI and TAS‐108. These encouraging results suggest that this drug can expand the choice of endocrine therapy for MBC patients in that population.
The safety profile of TAS‐108 at all dose levels was favorable even when compared with the known safety profile of tamoxifen or other selective estrogen receptor modulators, which was similar to that in a phase I study by Saeki et al.13 The frequent drug‐related AEs were hot flush, hyperhidrosis, and nausea, which were of only mild severity (grade 1 or 2), and did not interfere with TAS‐108 treatment. The frequency and severity of AEs did not appear to be related to the dose of TAS‐108, which has been reported previously in a single‐dose study and repeated‐dose studies.10, 11, 12, 13 In the present study, grade 3 cataract was reported as a serious AE in one patient aged 63 years. Taking into account the report that tamoxifen can cause visual disorders, the relationship of the cataract to TAS‐108 was considered “possible”. No other clinical observations associated with the significant side‐effects of tamoxifen or AIs, such as thromboembolic events or bone fracture, have been reported. Therefore, the low toxicity profile of TAS‐108, coupled with the evidence of activity in MBC patients, justifies further clinical testing.
To date, there is no apparent clinical evidence of a stimulating effect of TAS‐108 on the endometrium in prior phase I studies involving Japanese and Caucasians patients,11, 13 and in the present exploratory analysis TAS‐108 did not cause significant endometrial thickening. In this study, no change was observed in BMD or BMMs, unlike with tamoxifen. This observation suggests that TAS‐108 may have few estrogenic effects on bone. Serum triglyceride was significantly decreased with no unfavorable changes in other cardiovascular risk factors tested in this study. TAS‐108 had no significant clinical effect on hormones. We acknowledge that because this was not an adjuvant study, there were several limitations to this exploratory analysis, such as a reduction in the number of MBC patients due to withdrawal from this study, and a relatively short length of drug exposure (particularly for analysis of the uterus and bone). Therefore, the effects of TAS‐108 on these values seemed tentative and need further investigation. However, the present analysis assessing the clinical potential impact of TAS‐108 suggests that this drug may not negatively affect the safety profile of postmenopausal patients.
In conclusion, TAS‐108 at the 40 mg dose level showed promising results regarding the primary end‐point of this study, and it was well tolerated at all dose levels in postmenopausal Japanese patients who had received one or two previous endocrine therapies. Based on these results, we determined the optimal dose of oral TAS‐108 to be 40 mg, once daily, for further clinical studies.
TAS‐108, a novel steroidal antiestrogen, may have the potential to develop into a clinically useful second‐ or third‐line endocrine therapy for HR‐positive breast cancer refractory to AI and/or tamoxifen.
Disclosure Statement
Hideo Inaji, Takahiro Nakayama, Naohito Yamamoto, Shigehira Saji, Toshiaki Saeki, and Shinzaburo Noguchi have received honoraria from Taiho Pharmaceuticals. Toshiaki Saeki and Shinzaburo Noguchi have received research funding from Taiho Pharmaceuticals. Tadashi Ikeda and Shinzaburo Noguchi have received consulting fees from Taiho Pharmaceuticals. The other authors have declared no conflicts of interest.
Abbreviations
- AE
adverse event
- AI
aromatase inhibitor
- BMD
bone mineral density
- BMM
bone metabolism marker
- CBR
clinical benefit rate
- CEC
Clinical Efficacy Committee
- CI
confidence interval
- CR
complete response
- E2
17β‐estradiol
- ER
estrogen receptor
- ET
endometrial thickness
- FSH
follicle‐stimulating hormone
- HR
hormone receptor
- I‐CTP
cross‐linked carboxy‐terminal telopeptide of type I collagen
- MBC
metastatic breast cancer
- ORR
objective tumor response rate
- PR
partial response
- PS
performance status
- SD
stable disease
- TAS‐108
(7α)‐21‐[4‐[(diethylamino)methyl]‐2‐methoxyphenoxy]‐7‐methyl‐19‐norpregna‐1,3,5(10)‐trien‐3‐ol 2‐hydroxy‐1,2,3‐propanetricarboxylate
- TTP
time to progression
Acknowledgments
This study was funded by Taiho Pharmaceuticals. We thank Prof. J. Patrick Barron of the Department of International Medical Communications of Tokyo Medical University (Tokyo, Japan), a remunerated consultant of Taiho Pharmaceuticals, for manuscript preparation. We thank all patients and investigators (physicians and staff) for their cooperation. We also thank Kunihiro Kishimoto and Kiyo Ryu for assistance in data management. The preliminary results of this study were presented in part at the 31st San Antonio Breast Cancer Symposium, San Antonio, TX, December 2008, and some other parts were presented at the 3rd Breast Cancer Symposium, San Francisco, CA, October 2009.
Appendix I.
The following institutions participated in this study: Hokkaido Cancer Center (Sapporo), Iwate Medical University (Morioka), Yamagata Prefectural Central Hospital (Yamagata), Tohoku University Hospital (Sendai), KKR Tohoku Kosai Hospital (Sendai), Tochigi Cancer Center (Utsunomiya), Saitama International Medical Center, Saitama Medical University (Hidaka), Saitama Red Cross Hospital (Saitama), Saitama Cancer Center (Ina), National Cancer Center Hospital (Tokyo), St. Luke's International Hospital (Tokyo), Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital (Tokyo), Chiba Cancer Center (Chiba), Tokai University School of Medicine (Isehara), Kanagawa Cancer Center (Yokohama), Yokohama Municipal Citizen's Hospital (Yokohama), Kitasato University School of Medicine (Sagamihara), Niigata Cancer Center Hospital (Niigata), Seirei Hamamatsu General Hospital (Hamamatsu), Aichi Cancer Center (Nagoya), Nagoya Medical Center (Nagoya), Nagoya City University Graduate School of Medical Sciences (Nagoya), Osaka Medical Center for Cancer and Cardiovascular Diseases (Osaka), Graduate School of Medicine, Osaka University (Suita), Osaka National Hospital (Osaka), Osaka Kouseinenkin Hospital (Osaka), Sakai Municipal Hospital (Sakai), Kansai Rosai Hospital (Amagasaki), Hyogo Cancer Center (Akashi), Shikoku Cancer Center (Matsuyama), Hiroshima University Hospital (Hiroshima), Kurashiki Central Hospital (Kurashiki), Kyushu Cancer Center (Fukuoka), and Kumamoto Municipal Hospital (Kumamoto).
(Cancer Sci, 2012; 103: 1708–1713)
References
- 1. Mouridsen H, Gershanovich M, Sun Y et al Superior efficacy of letrozole versus tamoxifen as first‐line therapy for postmenopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol 2001; 19: 2596–606. [DOI] [PubMed] [Google Scholar]
- 2. Nabholtz JM, Buzdar A, Pollak M et al Anastrozole is superior to tamoxifen as first‐line therapy for advanced breast carcinoma in postmenopausal women: results of a North American multicenter randomized trial. J Clin Oncol 2000; 18: 3758–67. [DOI] [PubMed] [Google Scholar]
- 3. Viale G, Giobbie‐Hurder A, Regan MM et al Prognostic and predictive value of centrally reviewed Ki‐67 labeling index in postmenopausal women with endocrine‐responsibe breast cancer: results from Breast International Group Trial 1‐98 comparing adjuvant tamoxifen with letrozole. J Clin Oncol 2008; 26: 5569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertelli G, Paridaens R. Optimal sequence of hormonotherapy in advanced breast cancer. Curr Opin Oncol 2006; 18: 572–7. [DOI] [PubMed] [Google Scholar]
- 5. Nayfield SG, Karp JE, Ford LG, Dorr FA, Kramer BS. Potential role of tamoxifen in prevention of breast cancer. J Natl Cancer Inst 1991; 83: 1450–9. [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto Y, Shibata J, Yonekura K et al TAS‐108, a novel oral steroidal antiestrogenic agent, is a pure antagonist on estrogen receptor α and a partial agonist on estrogen receptor β with low uterotrophic effect. Clin Cancer Res 2005; 11: 315–22. [PubMed] [Google Scholar]
- 7. Buzdar AU. TAS‐108: a novel steroidal antiestrogen. Clin Cancer Res 2005; 11: 906–8. [PubMed] [Google Scholar]
- 8. Yamamoto Y, Wada O, Takada I et al Both N‐ and C‐terminal transactivation functions of DNA‐bound ERα are blocked by a novel synthetic estrogen ligand. Biochem Biophys Res Commun 2003; 312: 656–62. [DOI] [PubMed] [Google Scholar]
- 9. Toko T, Shibata J, Sato K et al Antiestrogenic/estrogenic activities of TAS‐108 (SR16234), a new steroidal selective estrogen receptor modulator. Breast Cancer Res Treat 1999; 57: 52. [Google Scholar]
- 10. Yamaya H, Yoshida K, Kuritani J et al Safety, tolerability, and phrmacokinetics of TAS‐108 in normal healthy post‐menopausal female subjects: a phase I study on single oral dose. J Clin Pharm Ther 2005; 30: 459–70. [DOI] [PubMed] [Google Scholar]
- 11. Blakely LJ, Buzdar A, Chang HY et al A phase I and pharmacokinetic study of TAS‐108 in postmenopausal female patients with locally advanced, locally recurrent inoperable, or progressive metastatic breast cancer. Clin Cancer Res 2004; 10: 5425–31. [DOI] [PubMed] [Google Scholar]
- 12. Kumagai Y, Fujita T, Ozaki M et al Safety, tolerability and pharmacokinetics of TAS‐108, a novel anti‐oestrogen, in healthy post‐menopausal Japanese women: a phase I single oral dose study. Basic Clin Pharmacol Toxicol 2009; 104: 352–9. [DOI] [PubMed] [Google Scholar]
- 13. Saeki T, Noguchi S, Aogi K, Inaji H, Tabei T, Ikeda T. Evaluation of the safety and tolerability of oral TAS‐108 in postmenopausal patients with metastatic breast cancer. Ann Oncol 2009; 20: 868–73. [DOI] [PubMed] [Google Scholar]
- 14. Therasse P, Arbuck SG, Eisenhauer EA et al New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 15. Buzdar A, Vogel C, Schwartzberg L et al Randomized double‐blind phase 2 trial of 3 doses of TAS‐108 in patients with advanced or metastatic postmenopausal breast cancer. Cancer 2012; 118: 3244–53. [DOI] [PubMed] [Google Scholar]