

Abstract
Vascular endothelial growth factor (VEGF)‐targeted therapies show significant antitumor effects for advanced clear cell renal cell carcinomas (CC‐RCCs). Previous studies using VEGF inhibitors in mice models revealed that VEGF‐dependent capillaries were characterized by the existence of endothelial fenestrations (EFs). In this study, we revealed that capillaries with abundant EFs did exist, particularly in CC‐RCCs harboring VHL mutation. This finding was recapitulated in mice xenograft models, in which tumors from VHL null cells showed more abundant EFs compared to those from VHL wild‐type cells. Importantly, treatment with bevacizumab resulted in a significant decrease of tumor size established from VHL null cells. Additionally, a significant reduction of EFs and microvessel density was observed in VHL null tumors. Indeed, xenograft from 786‐O/mock (pRC3) cells developed four times more abundant EFs than that from 786‐O/VHL (WT8). However, introduction of the constitutively active form of hypoxia‐inducible factor (HIF)‐2α to WT8 cells failed to either augment the number of EFs or restore the sensitivity to bevacizumab in mice xenograft, irrespective of the equivalent production of VEGF to 786‐O/mock cells. These results indicated that HIF‐2α independent factors also play significant roles in the development of abundant EFs. In fact, several angiogenesis‐related genes including CCL2 were upregulated in 786‐O cells in a HIF‐2α independent manner. Treatment with CCL2 neutralizing antibody caused significant reduction of capillaries with EFs in 786‐O xenograft, indicating that they were also sensitive to CCL2 inhibition as well as VEGF. Collectively, these results strongly indicated that capillaries with distinctive phenotype developed in VHL null CC‐RCCs are potent targets for anti‐angiogenic therapy.
lt has been reported that the mutation of von Hippel‐Lindau tumor suppressor gene (VHL) is observed in approximately 60% of sporadic clear cell renal cell carcinomas (CC‐RCC).1, 2 It is well known that VHL loss of function mutations results in the upregulation of hypoxia‐inducible proteins such as vascular endothelial growth factor (VEGF),3 and transforming growth factor‐α,4 which contribute to RCC pathogenesis and tumor growth.5 It has been reported that VEGF plays important roles on angiogenesis6 and those inhibitors showed significant antitumor effects to advanced CC‐RCCs.7, 8, 9, 10, 11 As not all tumors respond to this therapy and a majority of responding tumors eventually become refractory to the treatment,12 further understanding of the characteristics of tumor microvasculature in CC‐RCCs is required to improve the clinical outcome.
We previously reported that capillary regression was observed in mice normal tissue vasculature such as pancreatic islets after the inhibition of VEGF signals and a consistent feature of these capillaries was the presence of endothelial fenestrations (EFs) approximately 60–70 nm in diameter.13 Abundant fenestrations were also observed in capillaries of pancreatic islet tumors developed in RIP‐Tag2 transgenic mice. Importantly, those tumor vessels showed high sensitivity to VEGF inhibition.14
Based on these backgrounds, we have examined the microvasculature of CC‐RCCs using electron microscopy. As expected, abundant fenestrations were observed in capillaries from CC‐RCC specimens and a significant correlation was observed between the abundance of fenestrations and the VHL status of specimens. Intriguingly, a mice xenograft model could recapitulate the characteristic phenotype of microvasculature in human VHL −/− CC‐RCC specimens and we showed that abundant EFs in VHL null CC‐RCCs disappeared after treatment with bevacizumab. Using xenograft models with different VHL status or hypoxia‐inducible factor (HIF) transactivity, we revealed that VEGF was not sufficient for the development EFs in VHL −/− CC‐RCC cells.
In fact, several angiogenesis‐related genes (i.e. CCL2, PGF, MMP2, and MMP9) were upregulated in a HIF‐independent manner in VHL −/− 786‐O cells. In these cells, the treatment of CCL2 neutralizing antibody decreased the number of fenestrations, indicating that CCL2 is required for the development of the characteristic microvasculature of VHL −/− CC‐RCC cells. Collectively, the present results strongly suggest that the endothelium with abundant fenestrations is a potent target of anti‐angiogenic therapy.
Material and Methods
Patients and RCC tissues
Tumor specimens were obtained from 24 consecutive patients with CC‐RCC who underwent surgery between March and October 2006 at Kyoto University (Kyoto, Japan) with appropriate informed consent. This study was approved by the institutional review board of Kyoto University (IRB approval number G52). Peripheral lesions of tumors without necrotic tissues were excised.
VHL genotyping and hypermethylation assays
Genomic DNA was extracted from the freshly frozen tumor specimens using QIAamp DNA mini kit (Qiagen, Hilden, Germany). We searched for mutation of VHL of all three exons.15 The methylation status in the CpG island of VHL was determined by methylation‐specific PCR as described previously.16
Cell culture
Cells (786‐O, A498, UMRC2, NC65, and Caki‐1) were cultured routinely in DMEM (Invitrogen, Carlsbad, CA, USA) containing 10% FBS supplemented with 1% penicillin/streptomycin. 786‐O subclones stably transfected with either pRc‐CMV (pRC3) or pRc‐CMV‐HA‐VHL (WT8)17 and UMRC2 with either pLenti6‐HA (UMRC2‐HA) or pLenti6‐HA‐LHVHL (UMRC2‐VHL) were a kind gift from Dr. William G. Kaelin (Dana‐Farber Cancer Institute, Boston, MA, USA). The cells were cultured as previously described.18 WT8 subclones stably transfected with either pIRES‐puro‐HA or pIRES‐puro‐HA‐HIF2α P531A19, 20 were cultured in the presence of 1.5 μg/mL puromycin in addition to G418. Lentiviral HIF1α shRNA vector (TRCN0000003810; target sequence, 5′‐GTGATGAAAGAATTACCGAAT‐3′) was purchased from Open Biosystems (Huntsville, AL, USA). Non‐silencing shRNAs (Open Biosystems) were used as negative control. The experimental procedures for shRNA transfection into UMRC2 were done as previously described.21 Following lentiviral infection, cells were maintained in the presence of 2 μg/mL puromycin.
Cell proliferation assays
Cells were seeded into 96‐well plates in triplicate in DMEM with 2.5% FBS and allowed to adhere overnight. The cultures were then washed and re‐fed with medium. For treatment with neutralizing antibodies, monoclonal anti‐VEGF antibody (bevacizumab; Roche Pharmaceuticals, Basel, Switzerland) or control saline was added to the medium. Proliferative activity was determined by the MTT assay using a microtiter plate reader at 540 nm.
Mouse xenograft assays
All experiments involving laboratory animals were done in accordance with the Guidelines for Animal Experiments of Kyoto University. A total of 1.0 × 107 cells were injected s.c. into both flanks of 6‐week‐old male BALB/cAJcl nude (nu/nu) mice (CLEA, Tokyo, Japan) and tumor volumes were measured once a week. After the drug trial, the animals were killed and xenograft tumors were excised. Peripheral lesions without necrotic tissues were used for sequential experiments. Experimental groups consisted of five mice per group, unless otherwise indicated.
Drugs and treatments
The dose of bevacizumab was determined based on previous studies.22 Treatment with bevacizumab (100 μg dissolved in 0.9% NaCl solution) or vehicle was started simultaneously in each cohort of xenografts when the average volume reached 100 mm3 at 3–4‐week intervals after tumor inoculation. Injections were done i.p. twice a week. Neutralizing antibodies to human CCL2 (MAB679; R&D Systems, Minneapolis, MN, USA) or control mouse IgG were given to mice with s.c. tumors from 786‐O cells by injecting i.p. twice a week at a dose of 10 μg/mouse.23
Microscopy
Samples were fixed with 1% glutaraldehyde and 1.44% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2). For scanning electron microscopy (SEM), after dehydration in a grade series of ethanol solutions, samples were freeze‐dried in a critical point apparatus, mounted onto metal stubs with carbon‐conductive paint, and coated with a thin layer of gold using a sputter coater. The tumor surfaces were observed at 10‐kV acceleration voltage (S‐4700; Hitachi, Tokyo, Japan). For transmission electron microscopy (TEM), the fixed and washed samples were post‐fixed with 1% OsO4 and 0.1 M sucrose in 0.1 M phosphate buffer for 2 h, then dehydrated with graded concentrations of ethanol and embedded in epoxy resin. Areas with abundant tumor capillaries were selected under a light microscope, ultrathin 80‐nm sections were cut on an ultramicrotome, placed on 150 mesh copper grids, stained with uranyl acetate followed by lead citrate, and examined using an electron microscope operated at 75‐kV acceleration voltage (H7000; Hitachi).
Quantitation of EFs
The EFs on the luminal surface of tumor capillaries were counted on SEM images. The EFs were identified as simple round openings of ~70‐nm diameter. We selected at random and captured 5–10 capillaries with 10–30‐μm diameter all over each tumor specimen. After three random segments with 4‐μm2 fields per vessel were captured and examined for each experimental variable (magnification, ×15 000), a total of 15–30 areas were counted. The number of EFs in each tumor and the mean number of fenestrations per 1‐μm2 endothelial surface area were calculated. For confirmation of diaphragmed fenestrations, observation was made on TEM images of thin, anuclear segments of endothelial profiles (magnification, ×35 000).
Protein extraction and immunoblot analysis
Whole‐cell proteins were isolated from snap‐frozen specimens or culture cells and followed by immunoblotting as previously described.18 Antibodies were HIF1α, HIF2α (both Novus Biologicals, Littleton, CO, USA), glucose transporter 1 (Glut1; Alpha Diagnostic International, San Antonio, TX, USA), HA (Covance, Berkeley, CA, USA), β‐tubulin, and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Enzyme‐linked immunosorbent assay
The VEGF protein levels in the supernatant and the tumor cytozol were quantified as previously described using a VEGF ELISA kit (R&D Systems).18, 24 Three independent wells were used for each set of experimental conditions. The VEGF concentration was normalized to total cell protein.
Immunohistochemistry and vascular density measurement
Immunohistochemical analysis was carried out on formalin‐fixed, paraffin‐embedded clinical tissues or OCT compound (Tissue Tek; Sakura Finetek, Torrance, CA, USA) fixed xenograft tissues. Primary antibodies were human CD31 (clone MEC 13.3; BD Pharmingen, San Diego, CA, USA), mouse CD31 (clone JC70A; Dako, Glostrup, Denmark), and CCL2 (R&D Systems). Tumor microvessel density (MVD) was evaluated using CD31 as the endothelial marker. The MVD was determined as suggested by Weidner.25
Reverse transcription and real time RT‐PCR analysis of angiogenesis‐ and invasion‐related genes
RT2 Profiler human angiogenesis PCR arrays (SA Biosciences, Frederick, MD, USA), containing 84 angiogenesis‐related genes, plus housekeeping genes and controls, were used according to the manufacturer's protocols.
Real‐time PCR
cDNA synthesis and real‐time PCR for CCL2, MMP2, MMP9, and GAPDH were carried out as described previously.26 The primer sequences were as follows: PGF, 5′‐GTTCAGCCCATCCTGTGTCT‐3′ (sense) and 5′‐CTTCATCTTCTCCCGCAGAG‐3′ (antisense); BNIP3, 5′‐CTCCTGGGTAGAACTGCACTTC‐3′ (sense) and 5′‐ACGCTCGTGTTCCTCATGCTG‐3′ (antisense); and HK2, 5′‐CAAAGTGACAGTGGGTGTGG‐3′ (sense) and 5′‐GCCAGGTCCTTCACTGTCTC‐3′ (antisense).
Oncomine database analysis
Oncomine data originated from Gumz et al.27 Expression levels of PGF, MMP‐2, MMP‐9, NRP2, EFNB2, THBS1, THBS2, VEGFC, and ID3 are compared between normal kidney and CC‐RCC.
Statistical analyses
Data are expressed as the mean ± SE. The significance of differences between means was assessed using Student's t‐test with Bonferroni's multiple comparison and χ2‐test. Inhibition of tumor growth was analyzed by two‐way repeated measures anova. A P‐value < 0.05 was considered statistically significant.
Results
Significant influence of VHL gene status on the development of EFs in CC‐RCC specimens
We first examined if fenestrations were observed on endothelial cells from 24 CC‐RCCs. VHL mutations were identified in 12 cases (50%) and none of them showed promoter methylation patterns (Table 1). Electron microscopy images revealed that the majority of capillaries from tumors with wild‐type (wt) VHL showed few fenestrations with thick endothelium (Fig. 1Aa,b). In contrast, those with mutant VHL showed attenuated endothelium with abundant fenestrations (Fig. 1Ac,d; arrows). Typically, these fenestrations were presented as the thinnest parts of capillary walls, consisting of long and thin cytoplasmic arms from the nucleus (N) in TEM images (Fig. 1Ad). The average number of fenestrations in wt and mutant groups was 2.5 ± 0.4 and 15.1 ± 1.7/μm2, respectively. Importantly, a significant difference was observed between them (Fig. 1B).
Table 1.
Patient characteristics, VHL alterations, and their relationship to clinicopathologic parameters in 24 sporadic clear cell renal cell carcinomas
| Parameter | VHL wild | VHL mutant[Link] | P‐value |
|---|---|---|---|
| Age (years) | |||
| Mean ± SD | 60.6 ± 12.0 | 64.5 ± 15.3 | 0.754 |
| Sex | |||
| Male | 9 | 7 | 0.386 |
| Female | 3 | 5 | |
| Stage[Link] | |||
| I | 7 | 7 | 0.788 |
| II | 2 | 1 | |
| III + IV | 3 | 4 | |
| Grade[Link] | |||
| 1 | 2 | 1 | 0.165 |
| 2 | 9 | 6 | |
| 3 | 1 | 5 | |
†No. with mutated VHL, 12/24 (50%). Location of VHL mutation: exon 1, 4; exon 2, 6; exon 3, 2. Type of VHL mutation: frameshift, 6; nonsense, 2; missense, 3; deletion, 1. No. with methylated VHL, 0/24 (0%). ‡Tumor stage and grade according to TNM (International Union Against Cancer, 6th edition, 2002). SD, standard deviation.
Figure 1.
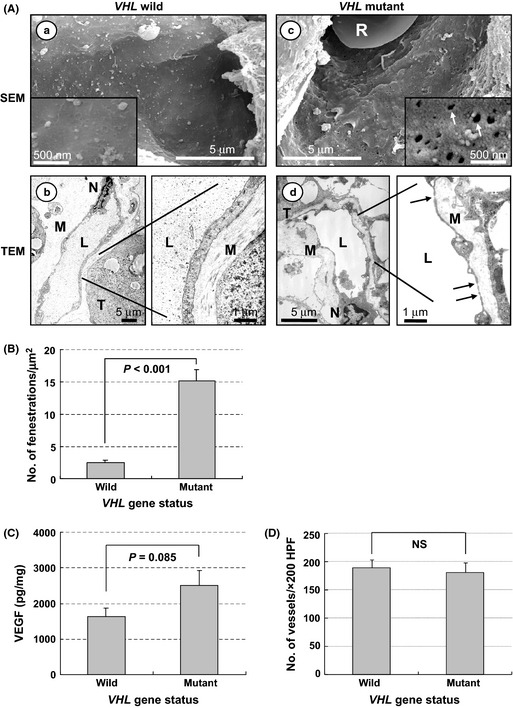
Phenotypes of tumor capillaries and VHL status in sporadic clear cell renal cell carcinomas (CC‐RCCs). (A) Electron micrographs of tumor capillaries in VHL wild‐type CC‐RCC (a,b) and VHL mutated CC‐RCC (c,d). Representative data are shown. Capillaries from tumors with wild‐type VHL show thick endothelium with few endothelial fenestrations (EFS); in contrast, those with mutant VHL show attenuated endothelium with abundant EFs (arrows). Inset shows a high magnification view (×50 000). L, capillary lumen; M, interstitial matrix; N, endothelial cell nucleus; R, red blood cell; SEM, scanning electron microscopy; T, tumor cell; TEM, transmission electron microscopy. (B) Quantitation of EFs in tumor capillaries with or without VHL mutation. Numbers of EFs were calculated per square micrometer. Columns, mean; bars, SE. (C) Amount of vascular endothelial growth factor (VEGF) in tumors with or without VHL mutation. Whole protein was extracted from tumors and analyzed by ELISA. Data are presented as pg VEGF protein/1 mg total protein. Columns, mean; bars, SE. (D) Microvessel density in tumors with or without VHL mutation. Columns, mean; bars, SE. HPF, high power field; NS, not significant.
We next examined the amount of VEGF in tumor tissues. Although tumors with mutated VHL produced a higher amount of VEGF compared to those without mutation, no statistically significant difference was observed between them (Fig. 1C). In fact, MVD was almost comparable in both groups (Fig. 1D). Collectively, these results indicate that the status of VHL significantly influenced the characteristics of endothelium of microvasculature of human CC‐RCC specimens, without affecting MVD.
Fenestration could be a potent predictive marker of the sensitivity of CC‐RCCs to VEGF inhibition
To further examine the effects of VHL on the endothelial cell morphology, a mouse xenograft model was established. As shown in Fig. 2A, administration of bevacizumab significantly inhibited tumor growth of xenografts from VHL −/− RCC cells, irrespective of H1H2 (UMRC2) or H2 (786‐O and A498) phenotype28, but not wt‐VHL RCC cells (NC65 and Caki−1). As tumors with abundant EFs were prone to VEGF inhibition,13, 14 we next assessed the effect of VEGF inhibition in these xenograft models. Capillaries from all VHL −/− cells presented abundant fenestrations at a similar level to those from VHL‐defective clinical specimens (Fig. 2B). In contrast, those from wt‐VHL cells showed less abundant fenestrations, indicating that the xenograft model recapitulated the VHL‐dependent formation of EFs in human CC‐RCC specimens. Importantly, SEM images revealed that the remaining capillaries after the treatment shows significantly less fenestration in the VHL null xenograft. In contrast, no significant differences were observed in those from wt‐VHL cells (Fig. 2B). Reduced MVD was also observed in the former cells (Fig. 2C), so these results strongly indicated that microvasculature with abundant EFs was sensitive to VEGF inhibition, and VHL status regulated the development of fenestrated tumor microvasculature.
Figure 2.
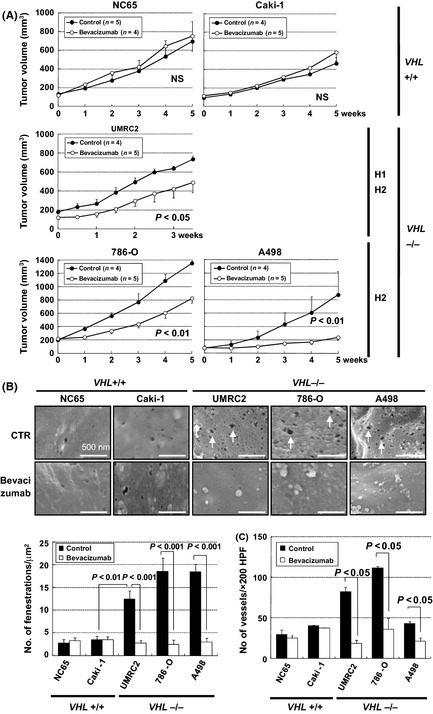
Effects of the bevacizumab treatment on ccRCC xenograft tumors. (A) Antitumor effects of bevacizumab in xenograft tumors from NC65 and Caki‐1 (wt VHL cells) or UMRC2, 786‐O, and A498 (VHL null cells). Each time point represents the mean ± SE of tumor volume in each group. The difference in tumor size between the treatment mice and controls was statistically significant in pVHL‐defective UMRC2, 786‐O, and A498 xenografts (P < 0.05, P < 0.01, P < 0.01, respectively, using two‐way repeated anova). H1H2, hypoxia‐inducible factor (HIF)‐1α and HIF‐2α; H2, HIF‐2α alone; NS, not significant. (B) Effects of bevacizumab treatment on endothelial fenestrations (EFs). Scanning electron micrographs of tumor capillaries treated with bevacizumab (bottom) or vehicle (CTR; top). Representative data are shown. Bars, 500 nm. Capillaries in xenograft tumors from UMRC2, 786‐O, and A498 cells developed abundant EFs (arrows). Reduction of the number of EFs in capillaries was detected only in VHL null xenografts treated with bevacizumab. Columns, mean; bars, SE. (C) Effects of bevacizumab treatment on microvessel density. CD31 staining of xenograft tumors with bevacizumab treatment showed significant reduction of microvessel density in UMRC2, 786‐O, and A498 xenografts. Columns, mean; bars, SE. HPF, high power field.
Abundance of EFs in microvasculature of CC‐RCCs and concomitant sensitivity to VEGF inhibition are regulated by pVHL in a HIF‐2α‐independent manner
To further clarify roles of VHL on tumor microvasculature, we next examined if HIF activity affected it. As it has been reported that HIF‐2α, rather than HIF‐1α, promotes pVHL‐defective renal carcinogenesis,21 we first assessed the effect of HIF‐2α on the morphology of the endothelium using a series of 786‐O subclones with different statuses of VHL or HIF transactivity. 786‐O subclone expressing HA‐tagged wt pVHL (WT8 cells) reduced the amount of HIF‐2α and canonical HIF target, Glut‐1, or VEGF. In contrast, both of them were up‐regulated by the introduction of constitutive active form of HIF‐2α (WT8 HIF2α P531A cells) (Figs 3A,S1).
Figure 3.
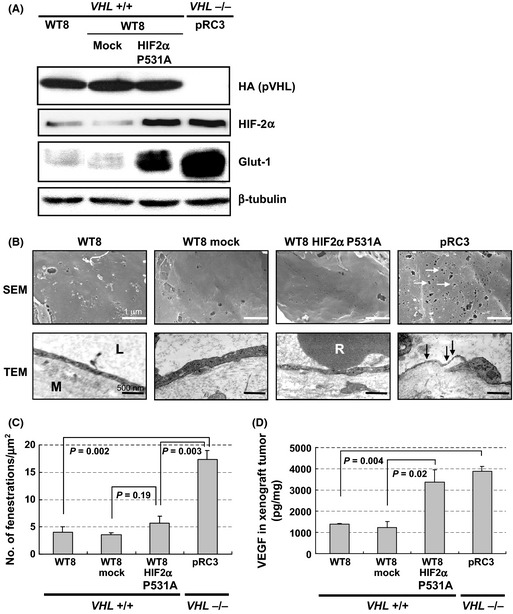
Characterization of xenografts from 786‐O subclones with different statuses of VHL and hypoxia‐inducible factor (HIF) activity. 786‐O VHL −/− human renal cell carcinoma cells stably transfected with plasmid encoding HA‐tagged wild‐type pVHL (WT8) or empty vector (pRC3) were used. WT8 cells transfected to produce HIF2α P531A (WT8/ H IF2α P531A) or empty vector (WT8/mock) were also examined. (A) Regulation of HIF target proteins by pVHL and HIF2α variant in the subclones. Immunoblot assays with the indicated antibodies. Glut1, glucose transporter 1. (B) Electron micrographs of tumor capillaries in xenografts from each cell line. Representative data are shown. Capillaries from VHL −/− pRC3 xenografts developed abundant EFs (arrows). L, capillary lumen; M, interstitial matrix; R, red blood cell. Scale bars = 1 μm (scanning electron microscopy [SEM], ×15 000) and 500 nm (transmission electron microscopy, ×25 000). (C) Quantitation of endothelial fenestrations in tumor capillaries from indicated xenografts. Columns, mean; bars, SE. (D) Amount of vascular endothelial growth factor (VEGF) in each xenograft. Whole protein was extracted from tumors and analyzed by ELISA. Data are presented as pg VEGF protein/1 mg total protein. Columns, mean; bars, SE.
Consistent with our previous report,20 WT8 HIF‐2α P531A cells restored their ability to form xenograft tumors in vivo, although introduction of HIF‐2α P531A to WT8 cells did not grossly affect the cell proliferation. None of them showed attenuated cell proliferation at up to 100 mg/mL bevacizumab in vitro (Figs S2,S3). Indeed, the mock transfected 786‐O subclone VHL −/− pRC3 xenograft developed four times more abundant EFs than that from WT8 cells (Fig. 3B,C). Unexpectedly, WT8 HIF‐2α P531A to cells failed to augment the number of EFs (Fig. 3C), although xenografts from these cells produced an almost comparable level of VEGF to those from pRC3 (Fig. 3D).
As for the sensitivity to VEGF inhibition, bevacizumab treatment significantly inhibited the tumor growth of xenografts from VHL −/− pRC3 but not wt‐VHL expressing WT8, WT8 mock, nor WT8 HIF‐2α P531A cells (Fig. 4A). Significant reduction of EFs was observed in pRC3 tumors but not others in accordance with its effect on MVD (Fig. 4B,C). The TEM images revealed that capillaries from pRC3 after bevacizumab treatment showed less fenestration with thick endothelium, the characteristics of WT8 HIF‐2α P531A, pVHL‐expressing subclones (Fig. 4C).
Figure 4.
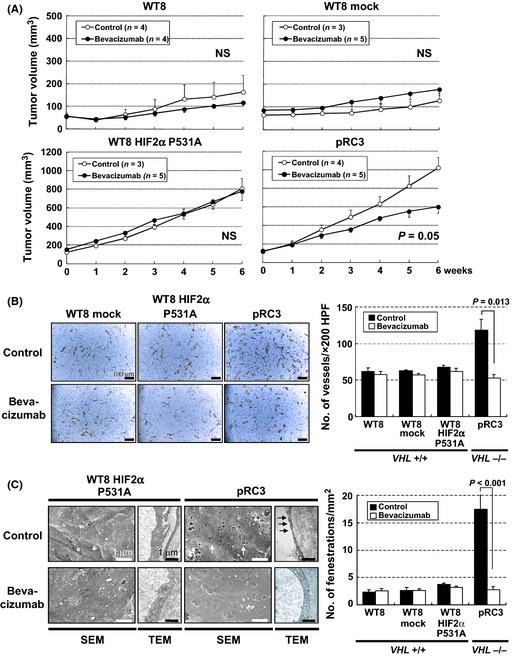
Effects of bevacizumab treatment in renal cell carcinoma xenografts from 786‐O subclones with different statuses of VHL and hypoxia‐inducible factor (HIF) activity. (A) Antitumor effects of bevacizumab in renal cell carcinoma tumors in nude mice. Each time point represents the mean ± SE of fold of tumor volume in each group. The difference in tumor size between the treatment mice and controls was statistically significant in pVHL‐defective pRC3 xenografts (P = 0.05, using two‐way repeated anova). (B) Effects of bevacizumab treatment on microvessel density. Left, CD31 staining of xenograft tumors treated with bevacizumab (bottom) or vehicle (top). Scale bars = 100 μm (×200). Right, significant reduction of microvessel density was observed only in pRC3 xenografts. Columns, mean; bars, SE. (C) Effects of bevacizumab treatment on endothelial fenestrations. Left, Scanning electron micrographs (SEM) and transmission electron micrographs (TEM) of tumor capillaries with bevacizumab treatment (bottom) or vehicle (top). Scale bars = 1 μm (SEM, ×15 000; TEM, ×20 000). Right, reduction in the number of endothelial fenestrations in capillaries was detected only in pRC3 xenograft treated with bevacizumab. Columns, mean; bars, SE.
Collectively, these results indicated that microvasculature with abundant EFs were sensitive to VEGF inhibition, however, other factors also played significant roles in the development of endothelium with distinctive phenotype.
Series of angiogenesis‐related genes regulated by VHL in a HIF‐independent manner
To explore the mechanisms by which EFs are regulated in VHL null CC‐RCCs, we compared the expression profile of a series of angiogenesis‐related genes in pRC3 and WT8 HIF‐2α P531A cells using PCR arrays. Those of WT8 and WT8 mock cells were also examined as a control. Although WT8 HIF‐2α P531A cells successfully upregulated VEGFA and VEGFC to almost comparable levels to pRC3 cells, the former cells failed to induce several genes such as CCL2 (Table 2). Quantitative PCR analysis with another set of primers confirmed the significant increase of CCL2 in pRC3 cells and xenografts from those cells showed abundant signals of this chemokine in immunohistochemical analysis (Fig. 5A,B). Similarly, CCL2 is also regulated by VHL in UMRC2 cells (Fig. 5C). We next examined the effect of HIF‐1α on the expression of this chemokine using shRNA‐mediated knockdown. As shown in Figure 5(D), this procedure did not significantly affect the abundance of CCL2 (P = 0.185), although the HIF‐1 target gene BNIP3 was effectively downregulated (P < 0.001).
Table 2.
mRNA changes between pRC3 versus WT8/HIF2aP531A
| Unigene | Symbol | Description | Fold change (pRC3/WT8) A | Fold change (HIF2aP531A/mock) B | Ratio A/B |
|---|---|---|---|---|---|
| Genes upregulated | |||||
| Hs.303649 | CCL2 | Chemokine (C‐C motif) ligand 2 | 88.91 | 0.60 | 149.19 |
| Hs.252820 | PGF | Placental growth factor | 30.75 | 2.82 | 10.91 |
| Hs.513617 | MMP2 | Matrix metallopeptidase 2 | 15.91 | 8.67 | 1.84 |
| Hs.297413 | MMP9 | Matrix metallopeptidase 9 | 8.05 | 3.19 | 2.52 |
| Hs.471200 | NRP2 | Neuropilin 2 | 7.60 | 1.12 | 6.76 |
| Hs.149239 | EFNB2 | Ephrin‐B2 | 4.00 | 1.26 | 3.17 |
| Hs.164226 | THBS1 | Thrombospondin 1 | 3.57 | 0.65 | 5.49 |
| Hs.371147 | THBS2 | Thrombospondin 2 | 3.42 | 0.41 | 8.32 |
| Hs.435215 | VEGFC | Vascular endothelial growth factor C | 3.38 | 2.35 | 1.44 |
| Hs.437008 | EPHB4 | EPH receptor B4 | 2.71 | 1.32 | 2.06 |
| Hs.133379 | TGFB2 | Transforming growth factor, beta 2 | 2.69 | 0.55 | 4.89 |
| Hs.76884 | ID3 | Inhibitor of DNA binding 3 | 2.64 | 1.23 | 2.14 |
| Hs.517356 | COL18A1 | Collagen, type XVIII, alpha 1 | 2.24 | 0.68 | 3.28 |
| Hs.73793 | VEGFA | Vascular endothelial growth factor A | 2.12 | 2.05 | 1.03 |
The RT2 Profiler human angiogenesis PCR Arrays (SA Biosciences) were carried out according to the manufacturer's protocols. Expression levels of genes involved in angiogenesis were compared between WT8 and pRC3, WT8/HIF2a P531A, and WT8/mock cells. Five housekeeping genes were used as controls for each gene expression calculation, and the extent of change in expression of each gene was calculated using the DCt method. Only genes whose expression was upregulated or downregulated at least 2‐fold are shown in Table 2. When DCt was over 12, and therefore the expression was thought to be extremely low, the gene was omitted from the analysis.
Figure 5.
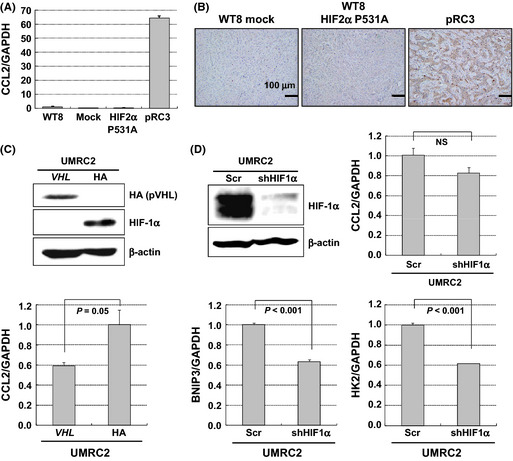
Regulation of CCL2 by VHL in a hypoxia‐inducible factor (HIF)‐independent manner. (A) Regulation of CCL2 by pVHL and HIF2α variant in the subclones. The expression of CCL2 was evaluated by real‐time PCR with a set of primers different from that of PCR arrays. Columns, mean; bars, SE. (B) Representative pictures of tumors from 786‐O subclones with CCL2 immunostaining. Scale bars = 100 μm. (C) Regulation of CCL2 by pVHL in HIF1α/HIF2α accumulated UMRC2 cells. UMRC2 cells stably transfected with plasmid encoding HA‐tagged wild‐type pVHL (VHL) or empty vector (HA) were used. Top, immunoblot assays with the indicated antibodies. Bottom, CCL2 expression in each cell type. Columns, mean; bars, SE. (D) HIF1α‐independent regulation of CCL2 in UMRC2 cells. UMRC2 cells transfected with lentivirus shRNA to knockdown HIF1α (shHIF1α) or non‐silencing shRNA (Scr) were examined. Top left, immunoblot assays with the indicated antibodies. Top right, CCL2 expression in each cell type. Bottom, BNIP3 and HK2 expression in each cell type. Columns, mean; bars, SE.
Previously, we and others reported that VHL regulated the expression of CCL2 in a HIF‐independent manner through JunB or inhibitory phosphorylation of CARD9, the nuclear factor‐κB agonist.26, 29 Importantly, the inhibition of CCL2 activity with a neutralizing antibody repressed xenograft tumor growth of VHL −/− CC‐RCC cells accompanied with decreased MVD.26 As shown in Figure 6(A), SEM images revealed that the remaining endothelium after treatment showed significantly less fenestration compared to controls, indicating that microvasculature with abundant EFs were also sensitive to CCL2 inhibition. Importantly, this chemokine is significantly highly expressed in tumor specimens with mutant VHL (Fig. 6B). Thus, these results strongly suggested that CCL2 is also involved in the development of fenestrated capillaries of VHL −/− CC‐RCC specimens.
Figure 6.
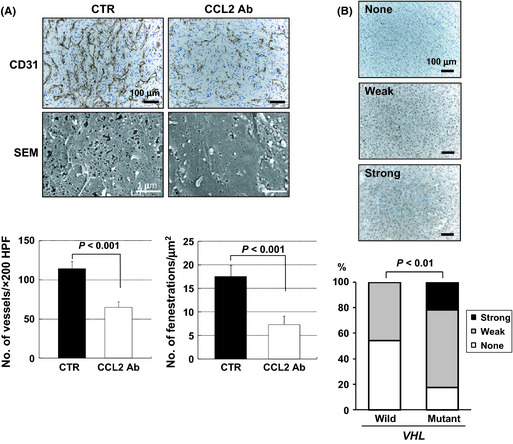
Involvement of CCL2 in the development of fenestrated capillaries of VHL −/− clear cell renal cell carcinomas. (A) Effects of CCL2 neutralizing antibody (CCL2 Ab) treatment on microvasculature. Representative data are shown. CD31, scale bars = 100 μm; scanning electron micrographs (SEM), scale bars = 1 μm. Bottom left, 786‐O xenograft tumors in CCL2 Ab treatment group showed significant reduction in microvessel density compared to those in vehicle treatment group (CTR). Bottom right, quantitation of endothelial fenestrations in capillaries after treatment with CCL2 Ab. Columns, mean; bars, SE. (B) Expression of CCL2 in tumors with or without VHL mutation. Top, representative pictures of immunohistochemistry sections of tumors showing none, weak, or strong staining for CCL2. Bottom, ratio of CCL2 expression pattern and correlation of VHL status.
In addition to CCL2, quantitative PCR analysis using 786‐O subclones confirmed that PGF, MMP2, and MMP9 were also regulated by VHL, mainly in a HIF‐independent manner (Fig. S4). Although the exact roles of each gene in the development of EFs should be further clarified, results in the present study suggest that VHL regulates the development of characteristic microvasculature through the regulation of angiogenesis‐related genes and determine the sensitivity to anti‐angiogenic therapy.
Discussion
It has been shown that VEGF‐targeted therapy had a significant clinical benefit for patients with metastatic RCC.10 However, some patients are inherently resistant to these therapies and most patients acquire resistance.12 Therefore, the precise mechanisms of the antitumor effects of this therapy need to be clarified.
Previously, we and others showed that capillaries with abundant EFs disappeared after the inhibition of VEGF signals in mice normal tissue or tumor microvasculature.13, 14 In the present study, we revealed that abundant EFs were observed in capillaries from human CC‐RCCs harboring VHL mutation and this finding was recapitulated in mice xenograft models. Importantly, VHL −/− CC‐RCC xenografts did show significantly better response to bevacizumab, irrespective of H1H2 or H2 phenotype. It was also revealed that the remaining capillaries after treatment showed significantly less fenestration.
In terms of the clinical implications of this study, Flaherty et al. examined the relationship between the tumor vascular permeability of RCC and clinical outcomes of sorafenib using dynamic contrast‐enhanced MRI. They reported that decreased tumor vascular permeability by the treatment was associated with improved clinical outcome.30 As EFs are related to increased vascular permeability,31 it will be assumed that decreased permeability in affected tumors was caused by reduction of fenestrations in the tumor microvasculature.
Additionally, retrospective analyses of VEGF‐targeted therapy for CC‐RCC indicated that patients with loss of functional VHL had a higher response rate or prolonged time to tumor progression compared to those with wt‐VHL.32, 33 Choueiri et al. reported that no responses (0 of 21) were seen in patients with wt VHL treated with bevacizumab or sorafenib, whereas six of 19 patients with mutated VHL responded to them.32 When the results in the present study were also concerned, it would be suggested that bevacizumab and sorafenib exhibit their anti‐tumor effects preferentially against CC‐RCCs with VHL mutation, at least in part, through the regression of microvasculature with abundant fenestrations.
As for the mechanistic insights into the development of EFs, it is well known that VEGF induces fenestrations.34, 35, 36 However, the precise roles of VEGF in inducing EFs are still undefined.37 We first evaluated whether HIF target proteins, including VEGF, play major roles in the development of EFs, as HIF escapes destruction and is free to activate its target genes in VHL −/− CC‐RCC cells.5 Unexpectedly, WT8 HIF2 α P531A cells failed to increase the comparable number of EFs or elicit a comparable response to bevacizumab to that of VHL −/− CC‐RCC cells. Previously, it had been reported that activation of HIF could not lead to increased MVD of VHL −/− tumor microvasculature.20, 38 Recently, Verheul et al.39 reported that higher VEGF expression was not associated with higher MVD in clinical CC‐RCC samples. Indeed, no significant correlation was observed between the levels of VEGF expression and VHL gene status in tumor specimens in our analysis. These findings suggested that VEGF was not sufficient for the development of characteristic microvasculature in VHL −/− CC‐RCC cells, and that other factors regulated by VHL enhanced the induction of EFs. In fact, leptin has been shown to act synergistically with VEGF to induce fenestrations.40
In the present study, we showed the possibility that CCL2 augmented the formation of EFs in pVHL‐defective CC‐RCC. It was revealed that CCL2 was regulated by VHL, and HIF‐1α and HIF‐2α scarcely affected its expression. This chemokine was significantly abundant in tumor specimens with mutant VHL compared to those without, and treatment with CCL2 neutralizing antibody decreased the number of fenestrations in the VHL −/− CC‐RCC xenograft model.
In addition to CCL2, it was shown that a number of angiogenesis‐related genes were regulated by VHL. Intriguingly, gene expression data on the Oncomine website indicated that CC‐RCC specimens showed significantly higher expression of PGF, MMP2, MMP9, NRP2, EFNB2, THBS1, THBS2, VEGFC, and ID3 compared to those from normal kidney (Fig. S5). Of these, we reported that MMP2 and MMP9 were regulated by VHL mainly through JunB.26 It is still unclear how VHL regulates the remaining genes, but it is assumed that these genes, including CCL2, act synergistically with VEGF to induce fenestrations in VHL−/− CC‐RCC.
In addition to RCC, rectal tumors also showed partial regression with bevacizumab monotherapy.41 When we think of the clinical implications of the present study, we need to evaluate whether EFs could be observed in other types of tumor and whether EFs could act as biomarkers to predict the clinical efficacy of bevacizumab in cancer.42
In conclusion, we have identified capillaries with abundant fenestrated endothelium in clinical samples from VHL −/− CC‐RCC. It was revealed that those characteristic capillaries recapitulated by xenograft models were sensitive to the inhibition of CCL2 as well as VEGF. Collectively, these results strongly indicated that capillaries with distinctive phenotype developed in VHL null CC‐RCCs are potent targets for anti‐angiogenic therapy. At present, it is still unclear why microvasculature with endothelial fenestrations can be inhibited by bevacizumab and this question must be clarified in future studies.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Characterization of 786‐O subclones with different status of VHL and hypoxia‐inducible factor (HIF) reactivity.
Fig. S2. Xenograft formation of 786‐O subclones.
Fig. S3. Effect of bevacizumab treatment on the cell proliferation of 786‐O subclones in vitro.
Fig. S4. Quantitative PCR analysis of several angiogenesis‐related genes in 786‐O subclones with different statuses of VHL and hypoxia‐inducible factor (HIF) reactivity.
Fig. S5. Oncomine database analysis of a series of angiogenesis‐related genes regulated by VHL in normal kidney versus clear cell renal cell carcinomas.
Acknowledgments
We thank Dr. William G. Kaelin (Dana‐Farber Cancer Institute, Boston, MA, USA) for providing us with the different RCC cell lines. We thank Makio Fujioka for technical support with electron microscopy, Tomoko Matsushita, Chie Hagihara and Megumi Kuraguchi for their technical assistance, and members of the Osamu Ogawa's laboratory and cancer course at Kyoto University Graduate School of Medicine for helpful discussions. This work was supported by the following grants: Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (No. 18591751), Kobayashi Institute for Innovative Cancer Chemotherapy, and Takeda Science Foundation, Japan (E. Nakamura).
(Cancer Sci 2012; 103: 2027–2037)
References
- 1. Cohen HT, McGovern FJ. Renal‐cell carcinoma. N Engl J Med 2005; 353: 2477–90. [DOI] [PubMed] [Google Scholar]
- 2. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991–5004. [DOI] [PubMed] [Google Scholar]
- 3. Gnarra JR, Zhou S, Merrill MJ et al Post‐transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Nat Acad Sci USA 1996; 93: 10589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gunaratnam L, Morley M, Franovic A et al Hypoxia inducible factor activates the transforming growth factor‐alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(−/−) renal cell carcinoma cells. J Biol Chem 2003; 278: 44966–74. [DOI] [PubMed] [Google Scholar]
- 5. Kaelin WG Jr. The von Hippel‐Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 2008; 8: 865–73. [DOI] [PubMed] [Google Scholar]
- 6. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9: 669–76. [DOI] [PubMed] [Google Scholar]
- 7. Motzer RJ, Hutson TE, Tomczak P et al Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. N Engl J Med 2007; 356: 115–24. [DOI] [PubMed] [Google Scholar]
- 8. Escudier B, Pluzanska A, Koralewski P et al Bevacizumab plus interferon alfa‐2a for treatment of metastatic renal cell carcinoma: a randomised, double‐blind phase III trial. Lancet 2007; 370: 2103–11. [DOI] [PubMed] [Google Scholar]
- 9. Escudier B, Eisen T, Stadler WM et al Sorafenib in advanced clear‐cell renal‐cell carcinoma. N Engl J Med 2007; 356: 125–34. [DOI] [PubMed] [Google Scholar]
- 10. Brugarolas J. Renal‐cell carcinoma–molecular pathways and therapies. N Engl J Med 2007; 356: 185–7. [DOI] [PubMed] [Google Scholar]
- 11. Yang JC, Haworth L, Sherry RM et al A randomized trial of bevacizumab, an anti‐vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003; 349: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rini BI, Atkins MB. Resistance to targeted therapy in renal‐cell carcinoma. Lancet Oncol 2009; 10: 992–1000. [DOI] [PubMed] [Google Scholar]
- 13. Kamba T, Tam BY, Hashizume H et al VEGF‐dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol 2006; 290: H560–76. [DOI] [PubMed] [Google Scholar]
- 14. Inai T, Mancuso M, Hashizume H et al Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 2004; 165: 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamano K, Esumi M, Igarashi H et al Biallelic inactivation of the von Hippel‐Lindau tumor suppressor gene in sporadic renal cell carcinoma. Journal Urol 2002; 167: 713–7. [DOI] [PubMed] [Google Scholar]
- 16. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Nat Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr, Goldberg MA. Negative regulation of hypoxia‐inducible genes by the von Hippel‐Lindau protein. Proc Nat Acad Sci USA 1996; 93: 10595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakamura E, Abreu‐e‐Lima P, Awakura Y et al Clusterin is a secreted marker for a hypoxia‐inducible factor‐independent function of the von Hippel‐Lindau tumor suppressor protein. Am J Pathol 2006; 168: 574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL‐defective tumor growth. PLoS Biol 2003; 1: E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel‐Lindau protein. Cancer Cell 2002; 1: 237–46. [DOI] [PubMed] [Google Scholar]
- 21. Shen C, Beroukhim R, Schumacher SE et al Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov 2011; 1: 222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res 2005; 65: 671–80. [PubMed] [Google Scholar]
- 23. Lu X, Kang Y. Chemokine (C‐C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 2009; 284: 29087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Turner KJ, Moore JW, Jones A et al Expression of hypoxia‐inducible factors in human renal cancer: relationship to angiogenesis and to the von Hippel‐Lindau gene mutation. Cancer Res 2002; 62: 2957–61. [PubMed] [Google Scholar]
- 25. Weidner N. Intratumor microvessel density as a prognostic factor in cancer. Am J Pathol 1995; 147: 9–19. [PMC free article] [PubMed] [Google Scholar]
- 26. Kanno T, Kamba T, Yamasaki T et al JunB promotes cell invasion and angiogenesis in VHL‐defective renal cell carcinoma. Oncogene 2012; 31: 3098–110. [DOI] [PubMed] [Google Scholar]
- 27. Gumz ML, Zou H, Kreinest PA et al Secreted frizzled‐related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res 2007; 13: 4740–9. [DOI] [PubMed] [Google Scholar]
- 28. Gordan JD, Lal P, Dondeti VR et al HIF‐alpha effects on c‐Myc distinguish two subtypes of sporadic VHL‐deficient clear cell renal carcinoma. Cancer Cell 2008; 14: 435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang H, Minamishima YA, Yan Q et al pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF‐kappaB agonist Card9 by CK2. Mol Cell 2007; 28: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flaherty KT, Rosen MA, Heitjan DF et al Pilot study of DCE‐MRI to predict progression‐free survival with sorafenib therapy in renal cell carcinoma. Cancer Biol Ther 2008; 7: 496–501. [DOI] [PubMed] [Google Scholar]
- 31. Eriksson A, Cao R, Roy J et al Small GTP‐binding protein Rac is an essential mediator of vascular endothelial growth factor‐induced endothelial fenestrations and vascular permeability. Circulation 2003; 107: 1532–8. [DOI] [PubMed] [Google Scholar]
- 32. Choueiri TK, Vaziri SA, Jaeger E et al von Hippel‐Lindau gene status and response to vascular endothelial growth factor targeted therapy for metastatic clear cell renal cell carcinoma. Journal Urol 2008; 180: 860–5; discussion 5–6. [DOI] [PubMed] [Google Scholar]
- 33. Rini BI, Jaeger E, Weinberg V et al Clinical response to therapy targeted at vascular endothelial growth factor in metastatic renal cell carcinoma: impact of patient characteristics and Von Hippel‐Lindau gene status. BJU Int 2006; 98: 756–62. [DOI] [PubMed] [Google Scholar]
- 34. Cao R, Eriksson A, Kubo H, Alitalo K, Cao Y, Thyberg J. Comparative evaluation of FGF‐2‐, VEGF‐A‐, and VEGF‐C‐induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ Res 2004; 94: 664–70. [DOI] [PubMed] [Google Scholar]
- 35. DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol 2004; 287: G757–63. [DOI] [PubMed] [Google Scholar]
- 36. Yokomori H, Oda M, Yoshimura K et al Vascular endothelial growth factor increases fenestral permeability in hepatic sinusoidal endothelial cells. Liver Int 2003; 23: 467–75. [DOI] [PubMed] [Google Scholar]
- 37. Maharaj AS, D'Amore PA. Roles for VEGF in the adult. Microvasc Res 2007; 74: 100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurban G, Hudon V, Duplan E, Ohh M, Pause A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res 2006; 66: 1313–9. [DOI] [PubMed] [Google Scholar]
- 39. Verheul HM, van Erp K, Homs MY et al The relationship of vascular endothelial growth factor and coagulation factor (fibrin and fibrinogen) expression in clear cell renal cell carcinoma. Urology 2010; 75: 608–14. [DOI] [PubMed] [Google Scholar]
- 40. Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF‐2 and VEGF. Proc Nat Acad Sci USA 2001; 98: 6390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Willett CG, Boucher Y, di Tomaso E et al Direct evidence that the VEGF‐specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 2004; 10: 145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol 2010; 11: 1172–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of 786‐O subclones with different status of VHL and hypoxia‐inducible factor (HIF) reactivity.
Fig. S2. Xenograft formation of 786‐O subclones.
Fig. S3. Effect of bevacizumab treatment on the cell proliferation of 786‐O subclones in vitro.
Fig. S4. Quantitative PCR analysis of several angiogenesis‐related genes in 786‐O subclones with different statuses of VHL and hypoxia‐inducible factor (HIF) reactivity.
Fig. S5. Oncomine database analysis of a series of angiogenesis‐related genes regulated by VHL in normal kidney versus clear cell renal cell carcinomas.