

Abstract

Lipid phase separation in cellular membranes is thought to play an important role in many biological functions. This has prompted the development of synthetic membranes to study lipid–lipid interactions in vitro, alongside optical microscopy techniques aimed at directly visualizing phase partitioning. In this context, there is a need to overcome the limitations of fluorescence microscopy, where added fluorophores can significantly perturb lipid packing. Raman-based optical imaging is a promising analytical tool for label-free chemically specific microscopy of lipid bilayers. In this work, we demonstrate the application of hyperspectral coherent Raman scattering microscopy combined with a quantitative unsupervised data analysis methodology developed in-house to visualize lipid partitioning in single planar membrane bilayers exhibiting liquid-ordered and liquid-disordered domains. Two home-built instruments were utilized, featuring coherent anti-Stokes Raman scattering and stimulated Raman scattering modalities. Ternary mixtures of dioleoylphosphatidylcholine, sphingomyelin, and cholesterol were used to form phase-separated domains. We show that domains are consistently resolved, both chemically and spatially, in a completely label-free manner. Quantitative Raman susceptibility spectra of the domains are provided alongside their spatially resolved concentration maps.
Introduction
Lipid bilayers have been investigated for several decades to study lipid–lipid and lipid–protein interactions.1 They serve as model systems to aid our understanding of the heterogeneous organization of cellular membranes2 and in the investigation of many processes, including protein segregation in lipid domains, membrane protein function, drug–receptor interactions, and transmembrane transport. Notably, they are receiving increasing attention as building blocks in bottom-up synthetic biology approaches to creating artificial cells.3
A widely investigated physicochemical phenomenon of biomimetic membranes is the liquid–liquid phase coexistence occurring when saturated lipids and sterols condense to form a liquid-ordered (Lo) phase which separates from a liquid-disordered (Ld) phase rich in unsaturated lipids.4 This is an important phenomenon not only from a fundamental lipid-biophysics point of view but also in relation to the highly debated lipid raft hypothesis, which postulates that cholesterol (Chol)-rich ordered domains within the plasma membrane of cells serve as platforms for protein function and associated cell signaling and trafficking events.2 However, the quantitative characterization of these heterogeneous lipid domains in single bilayers is not trivial, and the presently utilized techniques have a number of drawbacks.
Atomic force microscopy (AFM) is a powerful tool to achieve nanometric topology resolution and has been utilized to distinguish the extremely small (∼1 nm) height differences between Lo and Ld lipid domains in bilayers.5,6 As a contact-force-based method, it must be utilized on supported bilayers adhering onto a substrate. Severe drawbacks of AFM are slow imaging speed and lack of chemical specificity.
Alternatively, optical phase-contrast techniques can provide information on the optical path length and, in turn, membrane thickness when combined with appropriate quantitative analysis. Especially, wide-field imaging techniques offer fast acquisition speeds and can interrogate supported bilayers7 as well as suspended bilayers such as giant unilamellar vesicles (GUVs).8 However, these methods are also not chemically sensitive.
Domain visualization in lipid membranes can be achieved using fluorescence microscopy with a range of fluorophore-labelled lipids and can also feature spatial resolution below the diffraction limit (<200 nm) when combined with super-resolution methods. The main limitations of this approach are photobleaching and perturbation to the native lipid behavior by the addition of the fluorescent moiety. For example, it has been shown that many raft-preferring lipids (e.g., sphingolipids and sterols), whose domain preference results from their molecular architecture, do not partition into ordered phases if fluorescently labelled, in contrast to their native counterparts.9 Incorporation of these labels into the bilayer structure may significantly perturb lipid packing,10 and labels have been reported to cause peroxidation of membrane lipids.11
Alternatively, two main types of label-free, chemically specific, high-resolution microscopy techniques have been used on lipid membranes: imaging mass spectrometry and vibrational optical microscopy. Imaging mass spectrometry6 offers very high chemical resolution, whereby biomolecular species can be distinguished and identified by accurate mass analysis. However, it is a destructive technique that works by desorbing and ionizing molecules from a sample surface. The spatial resolution is governed by the focusability of the ionizing beams and can be in the submicron scale using an energetic primary ion beam that ablates the sample surface and generates secondary ions, a process called secondary ion mass spectrometry. Nevertheless, the requirement for measurements to be performed under ultrahigh vacuum limits the application of imaging mass spectrometry to flash-frozen and freeze-dried lipid membranes, that is, membranes under nonphysiological conditions.
Vibrational optical microscopy is based on detecting the Raman-scattered light following the interaction of incident light with the intrinsic vibrational resonances of chemical bonds. It is therefore chemically specific without the need of labelling and also, in principle, noninvasive. However, spontaneous Raman scattering is a very weak process, with typical Raman scattering cross-sections of vibrating modes in organic molecules in the 10–29 cm2 range, resulting in very low photon fluxes of Raman-scattered light. It is therefore very challenging to detect single lipid membranes, which has led to various strategies to enhance the image contrast. One possibility is to exploit the local field enhancement effect in the vicinity of metallic tips12 (a process called tip-enhanced Raman scattering) and/or structured metallic substrates (surface-enhanced Raman scattering—SERS).13 This approach, however, requires additional sample preparation and the availability of reliable SERS substrates. Recently, Raman imaging was demonstrated on supported lipid monolayers prepared using the Langmuir–Blodgett method,14,15 which results in the lipid head groups being strongly attached to a glass substrate. Under these conditions, monolayers are extremely stable and can be imaged with high laser powers and long acquisition times. The use of Raman tags via deuterated lipids15 or lipids synthesized with a diyne moiety14 has provided additional chemical specificity. However, monolayers are not quite representative of lipid membranes, which exist as bilayers. Although lipid partitioning can be observed in lipid monolayers, the corresponding phases are different from the Ld and Lo phases of bilayers. Moreover, the presence of Raman tags might change lipid partitioning compared to unlabelled lipids.
Coherent Raman scattering (CRS) microscopy has emerged in the last decade as a chemically specific technique, which improves on the speed limit of spontaneous Raman microscopy, owing to the constructive interference of Raman-scattered light by identical vibrational modes coherently driven in the CRS excitation process.16,17 Briefly, CRS is a third-order nonlinearity (four-wave mixing) in which two laser fields of frequencies νp (pump) and νS (Stokes) coherently drive a molecular vibration in resonance at their frequency difference νvib = νp – νS. CRS can be detected as coherent anti-Stokes Raman scattering (CARS) or as stimulated Raman scattering (SRS). CARS is the anti-Stokes scattering of the pump field by the coherently driven vibration and occurs at the up-shifted frequency νp + νvib = 2νp – νS. SRS is the loss (or gain) at the pump (or Stokes) frequency from the homodyne interference of the coherent Raman-scattered field with the corresponding transmitted beam. Being a nonlinear process, CRS exhibits high spatial resolution beyond the one-photon diffraction limit and offers intrinsic optical sectioning. The coherent enhancement of CRS is particularly beneficial when imaging lipids, owing to the large number of identical CH bonds. Importantly, CRS is applicable to both supported and suspended lipid bilayers. Indeed, the visualization of spatially resolved lipid domains by CARS has been shown in GUVs and in supported lipid bilayers.18,19 In these works, chemical contrast was again achieved by inserting deuterated lipids and detecting them at the carbon deuterium (CD) vibrational resonance (∼2150 cm–1), which is significantly shifted compared to the nondeuterated CH stretch vibration (∼2850 cm–1). Deuterium labelling was implemented because CARS was excited/detected at a single vibrational resonance, which does not provide sufficient chemical specificity to distinguish lipids of different chemical compositions.
To achieve the full potential of CRS regarding its chemical sensitivity, a multiplex or hyperspectral approach must be applied,16,17 where for each spatial position, a wide spectrum of several vibrational resonances is acquired. This also enables a quantitative determination of the concentration of chemical components.20 Notably, while multiplex CARS spectroscopy was reported on supported planar lipid bilayers21 and on solutions containing unilamellar vesicles,22 spatially resolved quantitative imaging of lipid domains in single bilayers was not shown, possibly due to the slow acquisition speed of multiplex detection [utilizing a spectrometer and a charge-coupled device (CCD) camera] in these works.
We have recently developed rapid hyperspectral CARS and SRS imaging modalities based on spectral focussing of broadband femtosecond pulses,23,24 alongside a powerful quantitative data analysis method, to retrieve the spectra and concentration of chemical components.20,25,26 Here, we show the applicability of this approach to spatially resolve and chemically distinguish unlabelled Lo and Ld domains in single-membrane lipid bilayers.
Materials and Methods
Planar Lipid Bilayers
Samples were made using dioleoylphosphatidylcholine (DOPC), sphingomyelin (SM), and Chol in the molar ratios (DOPC/SM/Chol) of 1:0:0 (pure DOPC), 0:7:3 and 1:2:1 (enriched SM, forming a homogeneous Lo phase), and 2:2:1 and 2:1:1 (ternary mixtures, showing coexistence of Lo and Ld domains at room temperature). Samples 1:2:1 and 2:1:1 were prepared using porcine SM; for all other samples, chicken-egg SM was used. All lipids were purchased from Avanti Polar Lipids (Alabaster, USA). For fluorescence labelling, we used 0.5 mol % of the fluorescent lipid analogue 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DOPE) which partitions in the Ld phase. NBD-DOPE was from Sigma-Aldrich (Dorset, UK). Lipid stock solutions were handled and sealed under an inert atmosphere within a nitrogen glovebox (Cole-Parmer, UK). All lipids and fluorescent lipid analogues were suspended in 2:1 chloroform/methanol (volume/volume) or pure chloroform and stored at −20 °C under an inert atmosphere until used.
Planar lipid bilayers were formed via osmotically induced rupture of GUVs onto a hydrophilic glass surface (mixtures 1:0:0, 1:2:1, 2:1:1, and 2:2:1) or via spin-coating (mixtures 1:0:0 and 0:7:3). Glass coverslips were cleaned with acetone to remove inorganic contaminants. They were then submerged in 60 mL of sulfuric acid at 95 °C. 20 mL of hydrogen peroxide was added to the acid after several minutes. The mixture was allowed to react for 1 h, after which the coverslips were washed with distilled water and finally dried under a stream of nitrogen. The etching with acid and hydrogen peroxide served to remove leftover organic contaminants and increase the hydrophilicity of the coverslip surface. Etched coverslips were stored in nitrogen at around 5 °C in order to preserve the hydrophilicity.
GUVs were created using an electroformation protocol published previously.8 Briefly, 10 μL of lipid solution (1 g/L) was spread onto the surface of two tantalum electrodes, followed by removal of the solvent for 1 h under high vacuum. The electrodes were then suspended in 550 μL of deionized water at 60 °C and subjected to a 1.2 V peak-to-peak square waveform at 10 Hz for 1 h to induce GUV formation. Subsequently, the voltage was increased to 1.5 V peak-to-peak with a sinusoidal waveform, and the frequency was reduced to 5 Hz for 30 min, then 2 Hz for 15 min, and finally 1 Hz for 15 min to encourage the GUVs to go into solution. For GUVs forming lipid domains, the chamber was sealed, and cooling to room temperature was controlled in steps of 4 °C/h to control domain formation. A thin imaging spacer (120 μm thick with a 20 mm diameter opening, Grace Biolabs) was attached to the treated glass coverslip surface prior to deposition of and incubation with 8 μL of the aforementioned GUV solution for 15 min. Following incubation, 8 μL of 75 mM phosphate-buffered saline (PBS) was added to induce GUV rupture, and the chamber was immediately sealed with a clean glass slide. Preparation of the 2:2:1 sample was slightly different, with the last step involving deposition of 65 μL (rather than 8 μL) of GUV solution, which was topped up with another 65 μL after 5, 15, and 25 min; 65 μL of PBS was then added, and 65 μL of the medium was exchanged with 65 μL of PBS 10 times to remove any free-floating GUVs and other lipid debris; this was all done at 55 °C.
When using spin-coating, 150 μL of the mixture was spun on an etched coverslip at 3000 rpm with a 6 s constant acceleration and deceleration on either side of a 30 s period for a total of 42 s. The coverslip was then placed in a plastic centrifuge tube with a small piece of wet tissue. The tube was filled with nitrogen to prevent lipid peroxidation, sealed, and incubated in an oven at 37 °C for 1 h. An imaging spacer was then placed on the lipid-containing side of the coverslip to form a cylindrical well at the bottom of which the lipid sat. This was filled with degassed PBS. Multilamellar (multiple-bilayer) versions of the 0:7:3 and 1:0:0 samples were also made following the same procedure as above but using 20 times more lipid in the solution.
Optical Microspectroscopy
Epi-fluorescence and differential interference contrast (DIC) microscopy was performed on an Eclipse Ti-U Nikon microscope stand as described in our previous works.7,8 Here, we used a 60 × 1.27-NA water-immersion objective and a 1.34-NA oil-immersion condenser. DIC and fluorescence images were acquired using a CCD camera (Orca 285, Hamamatsu, Japan). DIC illumination was provided by a halogen tungsten lamp (V2-A LL 100W, Nikon) followed by a blue-green filter (BG40, Schott) to block near-infrared light, for which the DIC polarizers do not have sufficient extinction, and a green filter [GIF, transmission band (550 ± 20) nm; Nikon] to define the wavelength range for the quantitative differential interference contrast (qDIC) analysis. For the measurements shown on the DOPC/SM/Chol 2:1:1 sample, an exposure time of 0.1 s was used for each frame in the DIC images. The average over 512 frames was acquired for each angle of the polarizer (offset phase). For more details, see the Supporting Information. Fluorescence images were acquired with an exposure time of 2.5 s. Epi-fluorescence excitation was performed using a metal-halide lamp (Lumen L200/D, Prior Scientific, USA) at 10% power and an exciter/emitter/dichroic filter cube (GFP-A-Basic; Semrock, USA) suitable for the NBD dye.
Confocal Raman spectra of bulk lipids were taken using the Ti-U Nikon microscope stand equipped with a 20 × 0.75-NA objective. A 532 nm continuous-wave laser excitation was filtered with a Semrock laser line filter (LL01-532) and coupled into the microscope by a dichroic mirror (Semrock LPD01-532RS) with a power of 10 mW at the sample. Raman scattering was collected in the epi direction, filtered with a long-pass filter (Semrock BLP01-532R), dispersed by an imaging spectrometer (HORIBA iHR550) with a 600 lines/mm grating, and detected with a CCD camera (Andor Newton DU971N-BV) with a full width at half-maximum (fwhm) spectral resolution of about 2 cm–1.
CARS was measured using the setup and technique described in our previous work23 with a 60× 1.27-NA water-immersion objective and a 1.34-NA oil-immersion condenser (see the Supporting Information for more details). It consists of a home-built microscope with the same Ti-U Nikon microscope stand described above, using a single-broadband 5 fs pulsed Ti:Sa laser source (Venteon, Pulse:One PE) to provide the pump and Stokes beams for CARS, as well as a third beam for two-photon fluorescence (TPF). The pulses are centered at 685 or 689 nm (pump) and 806 nm (Stokes), with a bandwidth at 10% intensity of 65 and 200 nm, respectively. They are linearly chirped using glass blocks, and their delay time is tuned in order to drive vibrational resonances in the 2700–3100 cm–1 wave number range with a spectral resolution of about 10 cm–1, a technique known as spectral focussing.27 TPF excitation is centered at 940 nm, which is suitable to excite the NBD dye, with a Fourier-limited pulse duration of approximately 30 fs at the sample. CARS and TPF are collected in the forward direction by the condenser lens, separated using appropriate filters, and detected using photomultiplier tubes as described in our previous work.23 A pixel dwell time of 10 μs was used for all CARS and TPF measurements. Typical pump, Stokes, and TPF excitation powers at the sample were 40, 20, and 25 mW, respectively. Frame averaging was used as indicated in the figure captions. For correlative TPF/CARS imaging, TPF was conducted first; subsequently, the dye was fully photobleached using the CARS pump beam (typically, 10 rasterscans across the sample were sufficient to remove all residual fluorescence). In this way, CARS imaging was free from fluorescence artefacts. Backgrounds were measured under the same excitation and detection conditions with pump and Stokes pulses out of time overlap and were subtracted from the measured CARS intensities.
SRS imaging was performed using the setup described in detail in our previous work.24 Briefly, a pulsed Ti:Sa laser (Spectra-Physics Mai Tai) emitting 150 fs pulses centered at 820 nm is used as the pump beam for stimulated Raman loss (SRL). An optical parametric oscillator pumped by the second harmonic of the Ti:Sa laser (Radiantis Inspire) provides the Stokes beam. Here, its wavelength was set to 1070 nm, resulting in a central wave number of 2850 cm–1. A spectral range of 2700–3100 cm–1 was scanned by spectral focussing as described above, with a spectral resolution of about 30 cm–1. The sample was mounted on a Nikon Ti-U microscope with a 60× 1.27-NA water-immersion objective (Nikon MRD70650) and a 1.5× tube lens. For SRL detection, the Stokes beam was amplitude-modulated by an acousto-optic modulator using a 2.5 MHz square wave, and the resulting modulation of the pump beam in transmission, collected by a 1.34-NA condenser lens, was measured using a silicon photodiode and a lock-in amplifier. In this setup, forward CARS can be detected simultaneously with SRL using a photomultiplier.24 At the sample, the pump power was about 5 mW and the Stokes power was about 11 mW. A pixel dwell time of 1 ms was used without frame averaging.
All samples were imaged using DIC to identify the regions of interest and to check for photodamage, which was not observed under the indicated excitation and detection conditions. An example comparing DIC acquired before and after SRS imaging is shown in Supporting Information, Section 3.i. When performing long acquisitions for hyperspectral imaging, stability of the image focal plane and of the sample was monitored by repeating the in-plane image acquisition at 2850 cm–1 before and after the hyperspectral scan and verifying its reproducibility. Note that CARS hyperspectral datasets were acquired as averages over 10 frames as this repetition was observed to be within the stability and reproducibility limits on all investigated samples.
Results
CARS Microspectroscopy of a Single Lipid Bilayer
To
quantify the CARS signal strength and chemical contrast, we compare
the CARS spectrum measured on a single planar lipid bilayer consisting
of only DOPC with that of bulk DOPC in the CH stretch vibrational
range, as shown in Figure 1. For this experiment, we fabricated supported planar lipid
bilayers through osmotically induced rupture of GUVs onto a hydrophilic
glass surface (see Materials and Methods).
CARS microspectroscopy was performed with 10 cm–1 resolution via spectral focusing (see Materials
and Methods). xyz images were acquired to
locate the bilayer in the optimum focal plane, and a series of xy images at wave numbers of 2700–3100 cm–1 was recorded to resolve the lipid vibrational resonances. The resulting
hyperspectral CARS datasets were denoised and corrected from a spatially
varying background (using a polynomial fit) to account for slight
changes in the spatial overlap between pump and Stokes beams and/or
sample tilt over the scan range (for details, see the Supporting Information). The measured intensity
at the bilayer (spatially averaged) was then divided by the nonresonant
CARS intensity measured in the same image from a surrounding region
without the bilayer (also spatially averaged) under the same excitation
and detection conditions in order to correct for the varying temporal
overlap of the pump and Stokes beams and to derive a CARS intensity
ratio independent of excitation/detection parameters. The resulting
spectrum is shown in Figure 1 (center) and exhibits the characteristic dispersive line
shape from the interference between the resonant and nonresonant parts
of the CARS susceptibility, which is expected when the resonant material
fills only a small fraction of the focal volume, as is the case for
a lipid bilayer.21 The CARS intensity ratio
relative to the nonresonant CARS background measured in the glass
coverslip is also shown, with the dispersive lipid resonance being
superimposed onto the response from the surround aqueous buffer (PBS).
Note that the amplitude of the dispersive lipid resonance is only
a small fraction (∼3%) of the signal. The CARS intensity ratio
relative to glass, measured in bulk DOPC, is given in the top frame
for comparison and shows a much larger amplitude, as expected considering
the small thickness of the bilayer compared to the size of the axial
point-spread function (PSF). From the measured CARS ratio, the imaginary
part of the CARS susceptibility,  , is retrieved using a phase-corrected Kramers–Kronig
(PCKK) procedure,20 which recovers a resonant
Raman-like spectrum. The bottom panel of Figure 1 shows the
, is retrieved using a phase-corrected Kramers–Kronig
(PCKK) procedure,20 which recovers a resonant
Raman-like spectrum. The bottom panel of Figure 1 shows the 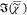 retrieved for bulk DOPC together with the
measured Raman spectrum. For the bilayer, we further applied a factorization
procedure20,28 (called FSC3) in order to remove
residual artefacts at the glass–PBS interface (see also the Supporting Information). The spatially averaged
spectrum in the bilayer region of the corresponding component is deduced.
This is shown for comparison in the bottom panel of Figure 1 as the mean spectrum for five
nominally identical bilayer samples. Notably, the retrieved
retrieved for bulk DOPC together with the
measured Raman spectrum. For the bilayer, we further applied a factorization
procedure20,28 (called FSC3) in order to remove
residual artefacts at the glass–PBS interface (see also the Supporting Information). The spatially averaged
spectrum in the bilayer region of the corresponding component is deduced.
This is shown for comparison in the bottom panel of Figure 1 as the mean spectrum for five
nominally identical bilayer samples. Notably, the retrieved 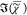 spectra of both bulk DOPC and the bilayer
greatly resemble the Raman spectrum of DOPC, but the amplitude of
spectra of both bulk DOPC and the bilayer
greatly resemble the Raman spectrum of DOPC, but the amplitude of 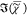 in bulk DOPC is higher than that in the
bilayer by a factor of 170. Since
in bulk DOPC is higher than that in the
bilayer by a factor of 170. Since 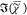 is linear in the number of chemical bonds
in the focal volume, this factor reflects the ratio between the bilayer
thickness and the effective axial extension of the PSF. Considering
a bilayer thickness of about 5 nm, this extension is estimated to
be 170 × 5 = 850 nm, consistent with the measured axial PSF of
0.9 μm fwhm for the CARS field in the setup used.29 It is also useful to estimate the number of
DOPC molecules generating the measured CARS for the bilayer, taking
into account the lateral PSF extension of 0.3 μm fwhm for the
CARS field29 and the area A per DOPC molecule in the bilayer. Using A = 70.15
Å2, as reported in the literature at room temperature,30 we estimate 2 × 105 DOPC molecules
contributing to the measured CARS signal. Note that, under the excitation
and detection conditions indicated in Figure 1, the signal-to-noise ratio observed in each
pixel of the CARS image of the bilayer (i.e., without spatial averaging)
was about 1 (see the Supporting Information S1). From this, we deduce a sensitivity limit of about 2000 DOPC molecules/
is linear in the number of chemical bonds
in the focal volume, this factor reflects the ratio between the bilayer
thickness and the effective axial extension of the PSF. Considering
a bilayer thickness of about 5 nm, this extension is estimated to
be 170 × 5 = 850 nm, consistent with the measured axial PSF of
0.9 μm fwhm for the CARS field in the setup used.29 It is also useful to estimate the number of
DOPC molecules generating the measured CARS for the bilayer, taking
into account the lateral PSF extension of 0.3 μm fwhm for the
CARS field29 and the area A per DOPC molecule in the bilayer. Using A = 70.15
Å2, as reported in the literature at room temperature,30 we estimate 2 × 105 DOPC molecules
contributing to the measured CARS signal. Note that, under the excitation
and detection conditions indicated in Figure 1, the signal-to-noise ratio observed in each
pixel of the CARS image of the bilayer (i.e., without spatial averaging)
was about 1 (see the Supporting Information S1). From this, we deduce a sensitivity limit of about 2000 DOPC molecules/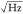 for the power and focussing conditions
used in Figure 1.
for the power and focussing conditions
used in Figure 1.
Figure 1.

Top: Spectrum
of CARS intensity ratio between bulk DOPC and the
glass coverslip support. Center: Spectrum of CARS intensity ratio
between a homogeneous DOPC lipid bilayer and either glass (black)
or the surrounding aqueous region (red). Bottom: Retrieved imaginary
part of the CARS susceptibility 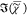 for bulk DOPC
and the bilayer (mean over
5 bilayer samples, see main text). The measured Raman spectrum of
bulk DOPC is shown for comparison (blue dotted curve). CARS spectra
in the lipid bilayer were obtained from spatially averaged hyperspectral
images of about 15 μm × 15 μm measured using a 60
× 1.27-NA water-immersion objective, a pixel dwell time of 0.01
ms, a pixel size of 0.1 μm, a 10-frame average, and a laser
power of 40 mW (pump) and 20 mW (Stokes) at the sample.
for bulk DOPC
and the bilayer (mean over
5 bilayer samples, see main text). The measured Raman spectrum of
bulk DOPC is shown for comparison (blue dotted curve). CARS spectra
in the lipid bilayer were obtained from spatially averaged hyperspectral
images of about 15 μm × 15 μm measured using a 60
× 1.27-NA water-immersion objective, a pixel dwell time of 0.01
ms, a pixel size of 0.1 μm, a 10-frame average, and a laser
power of 40 mW (pump) and 20 mW (Stokes) at the sample.
Fluorescence Microscopy and DIC of Lipid Domains
First, we demonstrated domain formation using conventional fluorescence microscopy. An example of a single-membrane planar lipid bilayer containing lipid domains is shown in Figure 2. We used a ternary mixture of DOPC, SM, and Chol in a 2:1:1 molar ratio, which is known to form Lo and Ld domains at room temperature.31 For fluorescence labelling, we used a small percentage (0.5 mol %) of the fluorescent lipid analogue NBD-DOPE, which partitions in the Ld phase.32 An epi-fluorescence image of the lipid bilayer exhibiting one bright Ld microdomain is shown in Figure 2c.
Figure 2.

(a) DIC differential phase (δ) image of a planar lipid bilayer with a ternary mixture of DOPC/SM/Chol in a 2:1:1 molar ratio exhibiting Lo and Ld domains. (b) Corresponding integrated phase (φ) image. (c) Epi-fluorescence image of the bilayer labelled with the fluorescent lipid analogue NBD-DOPE, which partitions in the Ld phase. (d) Thickness profile across the red line in (b), with transitions from the thinner Ld phase to the thicker Lo phase indicated by the arrows.
DIC microscopy measures the difference of the optical phase between two points in the sample plane spatially separated by an amount (the shear) typically comparable with the optical resolution. Based on this principle, we have developed a qDIC image acquisition and analysis procedure to measure the spatial distribution of the optical phase,8,33 which for lipid bilayers of known refractive index can in turn be used to calculate their thickness.7 Briefly, we start by acquiring a contrast image defined as
where I± are the transmitted intensities for opposite angles of the polarizer in a DIC setup with a de Sénarmont compensator.8 The differential phase is defined as δ = φ+ – φ–, where
is the optical phase accumulated in the sample for the beam passing through the point r ± s/2, s is the shear vector, and r is the DIC image coordinate on the sample plane. We calculate δ using the exact analytical solution of the relationship between Ic and δ (see the Supporting Information). The spatial distribution of the optical phase at the sample, φ(r), is then calculated from δ by performing a Wiener deconvolution procedure,8 further optimized to reduce integration artefacts as discussed in our recent work.7 In Figure 2a, we show the qDIC δ image of the bilayer. The corresponding integrated optical phase image is shown in Figure 2b. Notably, φ(r) correlates with the epi-fluorescence image as an inverted contrast, which is expected because Lo domains are thicker than Ld domains. The bilayer thickness t can be calculated taking into account that the optical phase introduced by the lipid bilayer is
where λ0 is the wavelength in vacuum, Δn is the refractive index change between the lipid bilayer and its surrounding aqueous medium, and t(r) is the thickness profile of the bilayer. Using Δn = 0.1159 under the experimental condition λ0 = 550 nm, we obtain the thickness profile shown in Figure 2d along the red line in Figure 2b. From this profile, we find a thickness of about 4 nm for the bilayer in the Ld domain, in good agreement with the thickness of a DOPC-only bilayer measured by others using ellipsometry,34 and an increase of about 1 nm going from the Ld phase to the Lo phase, also in agreement with findings from AFM studies35 and our recent qDIC work.7 Because of the measurement of the differential phase in DIC, thickness steps at the boundaries between domains can be reliably inferred, but absolute thicknesses over large distances carry increasing systematic errors. Even with these limitations, qDIC is a remarkably sensitive label-free optical method for distinguishing lipid domains based on their optical phase differences, as shown in our recent work where the accuracy and precision of the method are discussed in detail.7
Chemically Specific Hyperspectral CARS Imaging of Lipid Domains
We then performed hyperspectral CARS imaging
on the planar single
bilayer shown in Figure 2. Here, the entire hyperspectral image stack as a series of xy images at different frequencies in the 2700–3100
cm–1 range is analyzed using the PCKK phase retrieval
and FSC3 factorization methods described previously.20 The spatially resolved distribution of separated
chemical components is obtained together with the corresponding 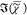 spectra. Importantly, the analysis is unsupervised,
that is, it does not require prior knowledge of the sample’s
chemical composition. We find that the method is able to distinguish
two main chemical components, as shown in Figure 3. The spatial distribution of the component
concentrations (Figure 3e,f) correlates very well with the lipid domains in Figure 2. It also correlates with the
intensity distribution of the single-frequency CARS image (as intensity
ratio to the surrounding) taken at the CH2 symmetric stretch
resonance, 2850 cm–1, shown in Figure 3b, which is expected on the
basis of the molecular packing density difference between Lo and Ld domains.36Figure 3c shows the phase-retrieved
CARS susceptibility at 2900 cm–1, where maximum
contrast of the domains in
spectra. Importantly, the analysis is unsupervised,
that is, it does not require prior knowledge of the sample’s
chemical composition. We find that the method is able to distinguish
two main chemical components, as shown in Figure 3. The spatial distribution of the component
concentrations (Figure 3e,f) correlates very well with the lipid domains in Figure 2. It also correlates with the
intensity distribution of the single-frequency CARS image (as intensity
ratio to the surrounding) taken at the CH2 symmetric stretch
resonance, 2850 cm–1, shown in Figure 3b, which is expected on the
basis of the molecular packing density difference between Lo and Ld domains.36Figure 3c shows the phase-retrieved
CARS susceptibility at 2900 cm–1, where maximum
contrast of the domains in  is obtained. Figure 3d is the TPF image
of the bilayer labelled
with the fluorescent lipid analogue NBD-DOPE, which partitions in
the Ld phase. The retrieved
is obtained. Figure 3d is the TPF image
of the bilayer labelled
with the fluorescent lipid analogue NBD-DOPE, which partitions in
the Ld phase. The retrieved  factorized spectra of the two chemical
components are shown in Figure 3a. They are compared with reference spectra measured on homogeneous
planar bilayers containing either only DOPC (the main component of
the Ld phase) or a SM-enriched DOPC/SM/Chol = 1:2:1 mixture
forming a homogeneous Lo phase. Reference spectra are shown
as spatial averages over the bilayer region (as discussed for Figure 1), and their amplitude
corresponds to having a spatially averaged concentration of 1. For
the spectra of the two chemical components, amplitudes are calculated
such that the spectral integral is equal to that of the corresponding
reference spectrum. Within the distribution from measurements repeated
on nominally identical samples, we find a very good correspondence
between the spectra retrieved from the unsupervised analysis in the
bilayers containing coexisting lipid domains (using the 2:1:1 mixture)
and the reference spectra from homogeneous bilayers, clearly showing
the ability of the technique to chemically resolve Lo and
Ld domains. We note the differences between the
factorized spectra of the two chemical
components are shown in Figure 3a. They are compared with reference spectra measured on homogeneous
planar bilayers containing either only DOPC (the main component of
the Ld phase) or a SM-enriched DOPC/SM/Chol = 1:2:1 mixture
forming a homogeneous Lo phase. Reference spectra are shown
as spatial averages over the bilayer region (as discussed for Figure 1), and their amplitude
corresponds to having a spatially averaged concentration of 1. For
the spectra of the two chemical components, amplitudes are calculated
such that the spectral integral is equal to that of the corresponding
reference spectrum. Within the distribution from measurements repeated
on nominally identical samples, we find a very good correspondence
between the spectra retrieved from the unsupervised analysis in the
bilayers containing coexisting lipid domains (using the 2:1:1 mixture)
and the reference spectra from homogeneous bilayers, clearly showing
the ability of the technique to chemically resolve Lo and
Ld domains. We note the differences between the 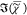 spectrum in the Lo phase, which
is narrower and more dominated by the CH2 symmetric stretch
vibration peak at 2850 cm–1, and the spectrum in
the Ld phase, which is broader with a pronounced shoulder
around 2930 cm–1 due to a combination of CH3 stretch vibrations and CH2 asymmetric stretch
enhanced by the broadening and shift of the CH deformations in the
liquid phase. These differences are consistent with previous knowledge
from Raman and CARS spectroscopy of saturated, more orderly packed
lipids versus unsaturated, disorderly packed lipids.21,37,38 Notably, the Raman spectrum of
pure SM reported in the literature39 (also
measured by us, see Supporting Information S16) exhibits a strong sharp peak near 2880 cm–1 not
observed in the
spectrum in the Lo phase, which
is narrower and more dominated by the CH2 symmetric stretch
vibration peak at 2850 cm–1, and the spectrum in
the Ld phase, which is broader with a pronounced shoulder
around 2930 cm–1 due to a combination of CH3 stretch vibrations and CH2 asymmetric stretch
enhanced by the broadening and shift of the CH deformations in the
liquid phase. These differences are consistent with previous knowledge
from Raman and CARS spectroscopy of saturated, more orderly packed
lipids versus unsaturated, disorderly packed lipids.21,37,38 Notably, the Raman spectrum of
pure SM reported in the literature39 (also
measured by us, see Supporting Information S16) exhibits a strong sharp peak near 2880 cm–1 not
observed in the 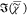 spectrum of
the Lo phase. This
is because pure SM at room temperature is in the gel (solid) phase.
Indeed, to obtain the Lo phase, SM has to be mixed with
Chol.31 On the other hand, the Raman spectrum
of pure Chol has a dominant peak near 2930 cm–1 from
the CH3 symmetric stretch bonds39 (see Supporting Information S16), while
in the
spectrum of
the Lo phase. This
is because pure SM at room temperature is in the gel (solid) phase.
Indeed, to obtain the Lo phase, SM has to be mixed with
Chol.31 On the other hand, the Raman spectrum
of pure Chol has a dominant peak near 2930 cm–1 from
the CH3 symmetric stretch bonds39 (see Supporting Information S16), while
in the 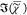 spectrum of the Lo phase, the
2930 cm–1 band appears as a shoulder of lower amplitude
compared to the dominant CH2 peak at 2850 cm–1. This can be understood considering that a Chol molecule has only
5 CH3 groups, compared to the long acyl chain with more
than 16 CH2 bonds in SM, and that the Lo phase
is enriched in SM with an equilibrium stoichiometry SM/Chol reported40 to be near 2:1.
spectrum of the Lo phase, the
2930 cm–1 band appears as a shoulder of lower amplitude
compared to the dominant CH2 peak at 2850 cm–1. This can be understood considering that a Chol molecule has only
5 CH3 groups, compared to the long acyl chain with more
than 16 CH2 bonds in SM, and that the Lo phase
is enriched in SM with an equilibrium stoichiometry SM/Chol reported40 to be near 2:1.
Figure 3.

Hyperspectral CARS imaging of the planar
bilayer in Figure 2 exhibiting Lo and
Ld domains. (a) Spectra obtained using an unsupervised
factorization into chemical components on nine nominally equal bilayer
patches exhibiting Lo and Ld domains as mean
(solid line) and standard deviation (bar). Red lines are spectra (mean
from five nominally equal samples) measured on homogeneous bilayers
containing a DOPC/SM/Chol = 1:2:1 mixture forming a homogeneous Lo phase (called SMe) or DOPC only. (b) CARS intensity relative
to the spatially averaged intensity in the region surrounding the
bilayer, imaged at 2850 cm–1. (c) Retrieved CARS
susceptibility 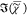 at 2900 cm–1. (d) TPF
image of the bilayer labelled with the fluorescent lipid analogue
NBD-DOPE partitioning in the Ld phase. (e,f) Spatial distribution
of the Lo and Ld concentration components. Gray
scales are from m to M. (g) Color overlay of Ld (blue)
and Lo (green). CARS measurements were performed using
a 0.01 ms pixel dwell time, 0.1 μm pixel size, 10-frame average,
and laser powers of 40 mW (pump) and 20 mW (Stokes) at the sample.
For TPF, the excitation power was 25 mW at the sample, and 20-frame
averaging was used.
at 2900 cm–1. (d) TPF
image of the bilayer labelled with the fluorescent lipid analogue
NBD-DOPE partitioning in the Ld phase. (e,f) Spatial distribution
of the Lo and Ld concentration components. Gray
scales are from m to M. (g) Color overlay of Ld (blue)
and Lo (green). CARS measurements were performed using
a 0.01 ms pixel dwell time, 0.1 μm pixel size, 10-frame average,
and laser powers of 40 mW (pump) and 20 mW (Stokes) at the sample.
For TPF, the excitation power was 25 mW at the sample, and 20-frame
averaging was used.
Having demonstrated that
hyperspectral CARS microscopy analyzed
with our unsupervised factorization procedure is able to separate
Lo and Ld domains spatially and spectrally in
single lipid bilayers, we then tested the method on label-free bilayers. Figure 4 shows the results
on eight nominally identical unlabelled bilayers exhibiting Lo and Ld domains. In this case, we performed an
unsupervised “global” factorization where we considered
spectral components common to all images rather than an individual
analysis for each sample separately, as was done to obtain the results
in Figure 3. A global
analysis was needed because the spatial separation between Lo and Ld domains was generally less marked in the unlabelled
bilayers, such that, in some cases, an individual unsupervised analysis
was unable to return separate domains. The retrieved  factorized global spectra of the two chemical
components corresponding to Lo and Ld are shown
in Figure 4a and compared
with the spectra of the labelled ternary bilayers from Figure 3a. Here, amplitudes are calculated
such that the spectral integral is equal to that of the corresponding
homogeneous bilayer mean spectrum shown in Figure 3a. Within errors, we observe a good agreement
between the spectral components of the labelled samples and those
of the unlabelled ones. An example of the corresponding spatial distributions
of the component concentrations is shown in Figure 4c,e and compares well with the spatial profile
measured in qDIC, as shown in Figure 4b,d. As mentioned, the spatial separation between Lo and Ld domains appeared less pronounced in the
unlabelled bilayers when compared to the labelled bilayers across
all investigated samples (see Supporting Information S6 and S7 for an overview). This suggests that the inclusion
of the dye, while not altering the spectra, influences the spatial
separation of the domains.
factorized global spectra of the two chemical
components corresponding to Lo and Ld are shown
in Figure 4a and compared
with the spectra of the labelled ternary bilayers from Figure 3a. Here, amplitudes are calculated
such that the spectral integral is equal to that of the corresponding
homogeneous bilayer mean spectrum shown in Figure 3a. Within errors, we observe a good agreement
between the spectral components of the labelled samples and those
of the unlabelled ones. An example of the corresponding spatial distributions
of the component concentrations is shown in Figure 4c,e and compares well with the spatial profile
measured in qDIC, as shown in Figure 4b,d. As mentioned, the spatial separation between Lo and Ld domains appeared less pronounced in the
unlabelled bilayers when compared to the labelled bilayers across
all investigated samples (see Supporting Information S6 and S7 for an overview). This suggests that the inclusion
of the dye, while not altering the spectra, influences the spatial
separation of the domains.
Figure 4.

CARS hyperspectral imaging on unlabelled lipid bilayers exhibiting Lo and Ld domains. (a) Unsupervised factorization into chemical components corresponding to the Lo and Ld domains. Spectra are obtained using global factorization on eight nominally identical unlabelled bilayers and compared to the spectra in the nine labelled samples. The spatial distribution of the concentrations is shown in (c) (Lo) and (e) (Ld). (b) DIC differential phase image of one of the lipid bilayers. (d) Optical phase image of the same bilayer. Scale bar: 5 μm. Gray scales are from m to M. CARS measurements were performed using a 0.01 ms pixel dwell time, 0.1 μ m pixel size, 10-frame average, and laser powers of 50 mW (pump) and 30 mW (Stokes) at the sample.
SRS Hyperspectral Imaging of Lipid Domains
After having
shown that CARS is able to image Lo and Ld domains
and determine their  spectra, we
investigated label-free single
lipid bilayers by SRS hyperspectral microscopy. We used the ternary
mixture 2:2:1 as this resulted in spatially well-separated Lo and Ld domains. SRS was measured in the form of SRL.
Our setup enabled simultaneous acquisition of forward-transmitted
CARS intensity (see Materials and Methods).
As was done for the CARS data sets, measured hyperspectral SRL data
sets were first denoised and corrected from a spatially varying background
(using a polynomial fit) to account for slight changes in the spatial
overlap between pump and Stokes beams and/or sample tilt over the
scan range (for details see the Supporting Information). The aforementioned unsupervised FSC3 procedure was
then applied to retrieve the spectra and concentration maps of the
Lo and Ld components. SRL spectra are not affected
by vibrationally nonresonant background and are thus Raman-like; therefore,
it is not necessary to apply the PCKK field-retrieval procedure which
is used for the CARS ratio.20
spectra, we
investigated label-free single
lipid bilayers by SRS hyperspectral microscopy. We used the ternary
mixture 2:2:1 as this resulted in spatially well-separated Lo and Ld domains. SRS was measured in the form of SRL.
Our setup enabled simultaneous acquisition of forward-transmitted
CARS intensity (see Materials and Methods).
As was done for the CARS data sets, measured hyperspectral SRL data
sets were first denoised and corrected from a spatially varying background
(using a polynomial fit) to account for slight changes in the spatial
overlap between pump and Stokes beams and/or sample tilt over the
scan range (for details see the Supporting Information). The aforementioned unsupervised FSC3 procedure was
then applied to retrieve the spectra and concentration maps of the
Lo and Ld components. SRL spectra are not affected
by vibrationally nonresonant background and are thus Raman-like; therefore,
it is not necessary to apply the PCKK field-retrieval procedure which
is used for the CARS ratio.20
In order to correct the SRL spectra for the varying pump-Stokes pulse overlap during spectral focusing, we used the CARS intensity, which is dominated by the nonresonant background and measured simultaneously to the SRL. We note that the CARS intensity is proportional to |Ep|4|Es|2, where Ep is the pump field and Es is the Stokes field, while the SRL intensity is proportional to |Ep|2|Es|. For the correction, we therefore divide the SRL signal by the square root of the spatially averaged CARS intensity, normalized to unity at the center wave number of the SRL hyperspectral scan range.
To represent the measured SRL
spectra as  , we then used the aqueous buffer PBS as
a reference value since the
, we then used the aqueous buffer PBS as
a reference value since the 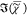 of PBS is known
(almost identical to that
of water).29 For this purpose, the SRL
signal at the center wave number, in the region outside the lipid
bilayer, was assigned to half the
of PBS is known
(almost identical to that
of water).29 For this purpose, the SRL
signal at the center wave number, in the region outside the lipid
bilayer, was assigned to half the  value of PBS since only one half of the
PSF contains the buffer, while the other half contains glass with
a negligible
value of PBS since only one half of the
PSF contains the buffer, while the other half contains glass with
a negligible 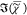 . This determined the factor to convert
SRL spectra from measured units (volts) into
. This determined the factor to convert
SRL spectra from measured units (volts) into 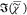 values, as discussed in more detail in
the Supporting Information (Section S4.ii).
values, as discussed in more detail in
the Supporting Information (Section S4.ii).
Figure 5a shows
the SRL spectra for the Lo and Ld domains obtained
on five different regions of the ternary 2:2:1 sample, where nominally
identical single lipid bilayers were found. SRL spectra were also
measured on control samples consisting of pure DOPC (a thick layer
formed by spin-coating) and a SMe homogeneous single bilayer (DOPC/SM/Chol
molar ratio 0:7:3) and are shown as red curves in Figure 5a (the SMe spectrum is the
mean over four nominally identical bilayers). For comparison to the
amplitude of the measured CARS spectra discussed in the previous sections,
the spectral integral of each SRL spectrum was set equal to that of
the corresponding  mean spectrum
of the homogeneous bilayer
measured by CARS (see Figure 3). We see that SRL spectra for Lo and Ld coincide with the reference spectra of the homogeneous samples within
errors. They are also consistent with the measured CARS spectra, shown
as green curves in Figure 5a (taken from the unlabelled ternary mixtures in Figure 4). The observed differences
between CARS and SRL spectra are attributed to the different spectral
resolutions and pulse temporal/spectral widths in the two measurements.
The CARS setup used offered a narrower spectral resolution and a broader
wave number range that can be accessed with a good signal-to-noise
ratio within the time overlap of the chirped pump and Stokes pulses
(see Materials and Methods). As a result,
SRS spectra exhibit a less-sharply-rising edge around 2850 cm–1 and have more noise in the spectral region above
2950 cm–1. Otherwise, spectra measured in CARS and
SRL have a comparable line shape, confirming the ability of our label-free
hyperspectral imaging and unsupervised analysis to consistently differentiate
between Lo and Ld domains. An example of the
spatial maps of the Lo and Ld components obtained
with SRL for one of the five investigated layers is shown in Figure 5 alongside the corresponding
DIC differential and integrated optical phase image. Also here, we
see excellent correlation between the spatial pattern of thicker domains
observed in DIC and the Lo domains found in SRL. More results
are shown in the Supporting Information (Section S5).
mean spectrum
of the homogeneous bilayer
measured by CARS (see Figure 3). We see that SRL spectra for Lo and Ld coincide with the reference spectra of the homogeneous samples within
errors. They are also consistent with the measured CARS spectra, shown
as green curves in Figure 5a (taken from the unlabelled ternary mixtures in Figure 4). The observed differences
between CARS and SRL spectra are attributed to the different spectral
resolutions and pulse temporal/spectral widths in the two measurements.
The CARS setup used offered a narrower spectral resolution and a broader
wave number range that can be accessed with a good signal-to-noise
ratio within the time overlap of the chirped pump and Stokes pulses
(see Materials and Methods). As a result,
SRS spectra exhibit a less-sharply-rising edge around 2850 cm–1 and have more noise in the spectral region above
2950 cm–1. Otherwise, spectra measured in CARS and
SRL have a comparable line shape, confirming the ability of our label-free
hyperspectral imaging and unsupervised analysis to consistently differentiate
between Lo and Ld domains. An example of the
spatial maps of the Lo and Ld components obtained
with SRL for one of the five investigated layers is shown in Figure 5 alongside the corresponding
DIC differential and integrated optical phase image. Also here, we
see excellent correlation between the spatial pattern of thicker domains
observed in DIC and the Lo domains found in SRL. More results
are shown in the Supporting Information (Section S5).
Figure 5.

SRL hyperspectral imaging on label-free planar lipid bilayers made of a ternary mixture of DOPC/SM/Chol in a 2:2:1 molar ratio exhibiting Lo and Ld domains. (a) Unsupervised factorization into chemical components corresponding to the Lo and Ld domains. Spectra are from five nominally identical unlabelled bilayers, shown as mean (solid curve) and standard deviation (bar). Red curves are reference SRL spectra measured on either an SMe bilayer or a pure DOPC homogeneous layer, as indicated. Green curves are the CARS spectra of the unlabelled domains from Figure 4. An example of the spatial distribution of the concentrations corresponding to the SRL spectra of the domains is shown in (c) (Lo) and (e) (Ld). (b) DIC differential phase image of one of the lipid bilayers. (d) Optical phase image of the same bilayer. Scale bar: 5 μm. Gray scales are from m to M. SRL measurements were performed using a pixel dwell time of 1 ms, a pixel size of 0.072 μm, and a laser power of 5 mW (pump) and 11 mW (Stokes) at the sample.
Conclusions
In summary, we have shown that hyperspectral CRS microscopy, combined with a quantitative unsupervised factorization of the measured data sets, can be used to resolve lipid partitioning and phase-separated domains in single lipid bilayers, both spatially and spectrally. Notably, to date, Raman microscopy of lipid domains in single membranes has largely exploited “Raman tags” to increase chemical specificity and has not yet taken full advantage of the inherently label-free capabilities of the technique.
By applying this analysis to lipid bilayers formed with ternary mixtures of DOPC, SM, and Chol, we have extracted the Raman spectra of Lo and Ld domains in susceptibility units relative to a nonresonant medium (glass or PBS) and correspondingly quantified the spatial distribution of the concentration of these components. We find that the spectra of the Ld components are equal within error to those of pure DOPC. Similarly, the spectra of the Lo domains are identical to those measured in homogeneous mixtures enriched in SM and Chol. A comparative study between bilayers formed using fluorescently labelled NBD-DOPE versus unlabelled DOPE in the ternary mixture suggests that the fluorescent lipid analogue is affecting the spatial distribution of the domains but not their spectra. It should be noted that the unsupervised factorization does not resolve the individual distribution of SM and Chol within the Lo domain. To that end, the use of Raman tags on a specific lipid species (e.g., SM) provides useful complementary information.
It is important to point out that imaging planar single bilayers pushes the detection sensitivity of CRS to its limit (as exemplified in Figure 1, comparing the signal from a bulk lipid to that of a single bilayer). This is because the coherent signal enhancement of the technique relies on having a large number of identical chemical bonds in the focal volume, and only a bidimensional layer of chemical bonds is available in a single bilayer. It is therefore remarkable that, despite the low signal levels, hyperspectral CRS imaging does encode sufficient information to retrieve the spectra and concentration of the domains via an unsupervised factorization algorithm with no prior knowledge.
In the future, this methodology could be applied to a variety of biomimetic membranes, including model systems closer to the complexity of cellular membranes (e.g., incorporating proteins). Notably, CRS is amenable to correlative fluorescence microscopy of tagged proteins in the same instrument, opening the prospect to non-invasively gain new insights into the relationship between lipid domains, their spatially resolved chemical composition, and lipid–protein interactions.
Acknowledgments
The microscope setup used here was developed with funding from the UK BBSRC (grant no. BB/H006575/1) and EPSRC (grant no. EP/M028313/1). D.R. acknowledges funding by an EPSRC DTA studentship (grant no. EP/M507842/1). P.B. and C.M. acknowledge the EPSRC for P.B.’s Leadership fellowship award (grant no. EP/I005072/1) which funded C.M.’s PhD studentship. W.L. acknowledges support by his Leverhulme Royal Society Research Fellowship (grant no. LT20085). P.B. acknowledges the Royal Society for her Wolfson Research Merit Award (grant no. WM140077). F.M. acknowledges the Ser Cymru II programme (Case ID 80762-CU-148) which is partly funded by Cardiff University and the European Regional Development Fund through the Welsh Government.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03179.
Quantitative DIC acquisition and analysis, CARS and SRS setups, and hyperspectral CARS and SRS analyses (PDF)
The authors declare no competing financial interest.
Notes
Information on the data presented here, including how to access them, can be found in the Cardiff University data archive http://doi.org/10.17035/d.2020.0112949840.
Supplementary Material
References
- Siontorou C.; Nikoleli G.-P.; Nikolelis D.; Karapetis S. Artificial Lipid Membranes: Past, Present, and Future. Membranes 2017, 7, 38. 10.3390/membranes7030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E.; Levental I.; Mayor S.; Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideau E.; Dimova R.; Schwille P.; Wurm F. R.; Landfester K. Liposomes and polymersomes: a comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. 10.1039/c8cs00162f. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandrini A.; Facci P. Phase transitions in supported lipid bilayers studied by AFM. Soft Matter 2014, 10, 7145–7164. 10.1039/c4sm01104j. [DOI] [PubMed] [Google Scholar]
- Mashaghi A.; Mashaghi S.; Reviakine I.; Heeren R. M. A.; Sandoghdar V.; Bonn M. Label-free characterization of biomembranes: from structure to dynamics. Chem. Soc. Rev. 2014, 43, 887–900. 10.1039/c3cs60243e. [DOI] [PubMed] [Google Scholar]
- Regan D.; Williams J.; Borri P.; Langbein W. Lipid Bilayer Thickness Measured by Quantitative DIC Reveals Phase Transitions and Effects of Substrate Hydrophilicity. Langmuir 2019, 35, 13805–13814. 10.1021/acs.langmuir.9b02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee C. I.; Zoriniants G.; Langbein W.; Borri P. Measuring the Lamellarity of Giant Lipid Vesicles with Differential Interference Contrast Microscopy. Biophys. J. 2013, 105, 1414–1420. 10.1016/j.bpj.2013.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E.; Levental I.; Grzybek M.; Schwarzmann G.; Mueller V.; Honigmann A.; Belov V. N.; Eggeling C.; Coskun Ü.; Simons K.; Schwille P. Partitioning, diffusion, and ligand binding of raft lipid analogs in model and cellular plasma membranes. Biochim. Biophys. Acta 2012, 1818, 1777–1784. 10.1016/j.bbamem.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Faller R. Molecular modeling of lipid probes and their influence on the membrane. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2353–2361. 10.1016/j.bbamem.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Ayuyan A. G.; Cohen F. S. Lipid Peroxides Promote Large Rafts: Effects of Excitation of Probes in Fluorescence Microscopy and Electrochemical Reactions during Vesicle Formation. Biophys. J. 2006, 91, 2172–2183. 10.1529/biophysj.106.087387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opilik L.; Bauer T.; Schmid T.; Stadler J.; Zenobi R. Nanoscale chemical imaging of segregated lipid domains usingtip-enhanced Raman spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 9978–9981. 10.1039/c0cp02832k. [DOI] [PubMed] [Google Scholar]
- Pahlow S.; März A.; Seise B.; Hartmann K.; Freitag I.; Kämmer E.; Böhme R.; Deckert V.; Weber K.; Cialla D.; Popp J. Bioanalytical application of surface- and tip-enhanced Raman spectroscopy. Eng. Life Sci. 2012, 12, 131–143. 10.1002/elsc.201100056. [DOI] [Google Scholar]
- Ando J.; Kinoshita M.; Cui J.; Yamakoshi H.; Fujita K.; Murata M.; Sodeoka M. Sphingomyelin distribution in lipid rafts of artificial monolayer membranes visualized by Raman microscopy. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 4558–4563. 10.1073/pnas.1418088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson S. H.; de Aguiar H. B. Molecular Imaging of Cholesterol and Lipid Distributions in Model Membranes. J. Phys. Chem. Lett. 2018, 9, 1528–1533. 10.1021/acs.jpclett.8b00235. [DOI] [PubMed] [Google Scholar]
- Zumbusch A.; Langbein W.; Borri P. Nonlinear vibrational microscopy applied to lipid biology. Prog. Lipid Res. 2013, 52, 615–632. 10.1016/j.plipres.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Camp C. H.; Cicerone M. T. Chemically sensitive bioimaging with coherent Raman scattering. Nat. Photon. 2015, 9, 295–305. 10.1038/NPHOTON.2015.60. [DOI] [Google Scholar]
- Li L.; Wang H.; Cheng J.-X. Quantitative Coherent Anti-Stokes Raman Scattering Imaging of Lipid Distribution in Coexisting Domains. Biophys. J. 2005, 89, 3480–3490. 10.1529/biophysj.105.065607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potma E. O.; Xie X. S. Direct Visualization of Lipid Phase Segregation in Single Lipid Bilayers with Coherent Anti-Stokes Raman Scattering Microscopy. ChemPhysChem 2005, 6, 77–79. 10.1002/cphc.200400390. [DOI] [PubMed] [Google Scholar]
- Masia F.; Glen A.; Stephens P.; Borri P.; Langbein W. Quantitative Chemical Imaging and Unsupervised Analysis Using Hyperspectral Coherent Anti-Stokes Raman Scattering Microscopy. Anal. Chem. 2013, 85, 10820–10828. 10.1021/ac402303g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurpel G. W. H.; Schins J. M.; Müller M. Direct Measurement of Chain Order in Single Phospholipid Mono- and Bilayers with Multiplex CARS. J. Phys. Chem. B 2004, 108, 3400–3403. 10.1021/jp037629j. [DOI] [Google Scholar]
- Rinia H. A.; Bonn M.; Müller M.; Vartiainen E. M. Quantitative CARS Spectroscopy Using the Maximum Entropy Method: The Main Lipid Phase Transition. ChemPhysChem 2007, 8, 279–287. 10.1002/cphc.200600481. [DOI] [PubMed] [Google Scholar]
- Pope I.; Langbein W.; Watson P.; Borri P. Simultaneous hyperspectral differential-CARS, TPF and SHG microscopy with a single 5 fs Ti:Sa laser. Opt. Express 2013, 21, 7096–7106. 10.1364/oe.21.007096. [DOI] [PubMed] [Google Scholar]
- Langbein W.; Regan D.; Pope I.; Borri P. Heterodyne dual-polarization epi-detected CARS microscopy for chemical and topographic imaging of interfaces. APL Photonics 2018, 3, 092402. 10.1063/1.5027256. [DOI] [Google Scholar]
- Di Napoli C.; Pope I.; Masia F.; Langbein W.; Watson P.; Borri P. Quantitative Spatiotemporal Chemical Profiling of Individual Lipid Droplets by Hyperspectral CARS Microscopy in Living Human Adipose-Derived Stem Cells. Anal. Chem. 2016, 88, 3677–3685. 10.1021/acs.analchem.5b04468. [DOI] [PubMed] [Google Scholar]
- Karuna A.; Masia F.; Wiltshire M.; Errington R.; Borri P.; Langbein W. Label-Free Volumetric Quantitative Imaging of the Human Somatic Cell Division by Hyperspectral Coherent Anti-Stokes Raman Scattering. Anal. Chem. 2019, 91, 2813–2821. 10.1021/acs.analchem.8b04706. [DOI] [PubMed] [Google Scholar]
- Rocha-Mendoza I.; Langbein W.; Borri P. Coherent Anti-Stokes Raman Microspectroscopy Using Spectral Focusing with Glass Dispersion. Appl. Phys. Lett. 2008, 93, 201103. 10.1063/1.3028346. [DOI] [Google Scholar]
- Masia F.; Karuna A.; Borri P.; Langbein W. Hyperspectral image analysis for CARS, SRS, and Raman data. J. Raman Spectrosc. 2015, 46, 727–734. 10.1002/jrs.4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuna A.; Masia F.; Borri P.; Langbein W. Hyperspectral volumetric coherent anti-Stokes Raman scattering microscopy: quantitative volume determination and NaCl as non-resonant standard. J. Raman Spectrosc. 2016, 47, 1167–1173. 10.1002/jrs.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Tristram-Nagle S.; Kučerka N.; Nagle J. F. Temperature Dependence of Structure, Bending Rigidity, and BilayerInteractions of Dioleoylphosphatidylcholine Bilayers. Biophys. J. 2008, 94, 117–124. 10.1529/biophysj.107.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett. 2005, 94, 148101. 10.1103/physrevlett.94.148101. [DOI] [PubMed] [Google Scholar]
- Baumgart T.; Hunt G.; Farkas E. R.; Webb W. W.; Feigenson G. W. Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim. Biophys. Acta 2007, 1768, 2182–2194. 10.1016/j.bbamem.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope I.; Payne L.; Zoriniants G.; Thomas E.; Williams O.; Watson P.; Langbein W.; Borri P. Coherent anti-Stokes Raman scattering microscopy of single nanodiamonds. Nat. Nanotechnol. 2014, 9, 940–946. 10.1038/nnano.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble S.; Patil S.; Kulkarni M.; Murthy A. V. R. Spectroscopic Ellipsometry of fluid and gel phase lipid bilayers in hydratedconditions. Colloids Surf., B 2019, 176, 55–61. 10.1016/j.colsurfb.2018.12.061. [DOI] [PubMed] [Google Scholar]
- Marquês J. T.; Viana A. S.; De Almeida R. F. M. Ethanol effects on binary and ternary supported lipid bilayers with gel/fluid domains and lipid rafts. Biochim. Biophys. Acta 2011, 1808, 405–414. 10.1016/j.bbamem.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Li L.; Cheng J.-X. Label-Free Coherent Anti-Stokes Raman Scattering Imaging of Coexisting Lipid Domains in Single Bilayers. J. Phys. Chem. B 2008, 112, 1576–1579. 10.1021/jp711527v. [DOI] [PubMed] [Google Scholar]
- Di Napoli C.; Masia F.; Pope I.; Otto C.; Langbein W.; Borri P. Chemically-specific dual/differential CARS micro-spectroscopy of saturated and unsaturated lipid droplets. J. Biophot. 2014, 7, 68–76. 10.1002/jbio.201200197. [DOI] [PubMed] [Google Scholar]
- Collard L.; Sinjab F.; Notingher I. Raman spectroscopy study of curvature-mediated lipid packing and sorting in single lipid vesicles. Biophys. J. 2019, 117, 1589–1598. 10.1016/j.bpj.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czamara K.; Majzner K.; Pacia M. Z.; Kochan K.; Kaczor A.; Baranska M. Raman spectroscopy of lipids: a review. J. Raman Spectrosc. 2014, 46, 4–20. 10.1002/jrs.4607. [DOI] [Google Scholar]
- Quinn P. J.; Wolf C. Egg-Sphingomyelin and Cholesterol Form a Stoichiometric Molecular Complex in Bilayersof Egg-Phosphatidylcholine. J. Phys. Chem. B 2010, 114, 15536–15545. 10.1021/jp107490a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.