

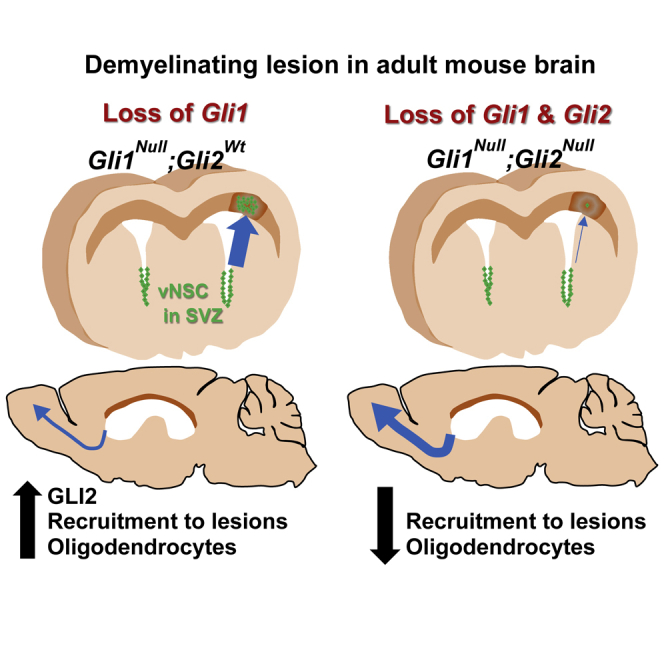
Summary
Enhancing repair of myelin is an important therapeutic goal in many neurological disorders characterized by demyelination. In the healthy adult brain, ventral neural stem cells (vNSCs) in the subventricular zone, marked by GLI1 expression, do not generate oligodendrocytes. However, in response to demyelination, their progeny are recruited to lesions where they differentiate into oligodendrocytes and ablation of GLI1 further enhances remyelination. GLI1 and GLI2 are closely related transcriptional activators but the role of GLI2 in remyelination by vNSCs is not clear. Here, we show that genetic ablation of Gli1 in vNSCs increases GLI2 expression and combined loss of both transcription factors decreases the recruitment and differentiation of their progeny in demyelinated lesions. These results indicate that GLI1 and GLI2 have distinct, non-redundant functions in vNSCs and their relative levels play an essential role in the response to demyelination.
Keywords: neural stem cell, GLI1, GLI2, remyelination, Gli1 inhibition, Gli2 inhibition, adult subventricular zone
Graphical Abstract
Highlights
-
•
Loss of Gli1 increases GLI2 expression in SVZ neural stem cells upon demyelination
-
•
Loss of Gli1 and Gli2 inhibits recruitment of neural stem cell progeny to the lesion
-
•
Loss of Gli1 and Gli2 decreases their differentiation into oligodendrocytes
In this article, Samanta and colleagues show that GLI2 is necessary for enhanced remyelination of the mouse brain mediated by loss of Gli1 in adult neural stem cells.
Introduction
Neural stem cells in the subventricular zone (SVZ) of adult mammalian brains are a heterogeneous population (Chaker et al., 2016), which include cells expressing GLI1 residing in the ventral SVZ (Ahn and Joyner, 2005). These ventral neural stem cells (vNSCs) do not generate oligodendrocyte progenitor cells (OPCs) or mature oligodendrocytes (OLs) and their progeny are excluded from all white matter tracts in the adult mouse brain. Instead, the vNSCs generate interneurons and astrocytes in the olfactory bulb (OB) and astrocytes in the gray matter of the adult brain (Garcia et al., 2010; Ihrie et al., 2011). Remarkably, GLI1 vNSCs respond to loss of OLs by recruitment of their progeny and by differentiating into OLs in demyelinating lesions (Samanta et al., 2015; Sanchez and Armstrong, 2018). The number of vNSC-derived cells recruited to the lesions and the proportion of these cells generating remyelinating OLs increases significantly when GLI1 is inactivated genetically or pharmacologically (Samanta et al., 2015).
GLI1 and GLI2 act as major transcriptional activators of the Sonic hedgehog (Shh) pathway, but have different expression patterns in the SVZ (Petrova et al., 2013). While genetic ablation of Gli1 does not affect development, knocking out either Gli2 alone (Mo et al., 1997) or both Gli1 and Gli2 (Park et al., 2000) is embryonic lethal, indicating their distinct functions. In this study, we found an increase in GLI2 expression in the SVZ of Gli1 null mice in response to demyelination leading us to examine whether GLI2 plays a role in the enhanced remyelination observed by GLI1 inhibition. Our results show that the combined ablation of Gli1 and Gli2 in vNSCs not only impairs the recruitment of their progeny into demyelinated lesions, but also their differentiation into OLs. In addition, the in vivo loss of both transcription factors substantially directs the migration of cells derived from vNSCs away from the lesions, thus indicating that the physiological migration of vNSC-derived cells to the OB versus recruitment to lesions are mechanistically distinct. These results highlight the non-overlapping functions of GLI1 and GLI2 in response to a demyelinating injury.
Results
Gli2 Is Upregulated in vNSCs following Demyelination
GLI2 is broadly expressed in the NSCs along the entire adult SVZ (Petrova et al., 2013) in contrast to GLI1, which is limited ventrally in healthy mice. We examined GLI2 expression in the SVZ of Gli1-LacZ knockin mice after inducing demyelination with cuprizone, a toxin that causes oligodendroglial cell death (Matsushima and Morell, 2001). We observed a significant (2.3 ± 0.37-fold) increase in the levels of Gli2 mRNA in the Gli1LacZ/LacZ SVZ lacking GLI1 expression at 6 weeks of cuprizone diet as compared with the SVZ of healthy mice on a regular diet (Figures 1A and 1B). More importantly, there was a significant increase in the proportion of GLI1 vNSCs co-expressing GLI2 in both the Gli1LacZ/LacZ SVZ (48.9% ± 4.4% on a cuprizone diet versus 8.6% ± 0.7% on a regular diet) and the Gli1LacZ/WT SVZ (42.6% ± 6.7% on a cuprizone diet versus 16.3% ± 2.6% on a regular diet) at peak demyelination (Figures 1C and 1D). Thus, Gli2 is upregulated in the vNSCs in response to demyelination, suggesting a role in remyelination.
Figure 1.

Gli2 Expression Increases in the Gli1NULL SVZ on Demyelination
(A) Schematic for tissue harvested for qRT-PCR in (B) and immunofluorescence in (C and D).
(B) qRT-PCR showing Gli2 mRNA expression in the SVZ on demyelination induced with 6 weeks of cuprizone diet.
(C) Immunofluorescence for co-localization of Gli2 (green) and LacZ (magenta) in the ventral SVZ of mice on 6 weeks of regular or cuprizone diets. Scale bar, 50 μm. Hoechst, nuclei.
(D) Quantification of the Gli1-LacZ NSCs co-expressing Gli2 in (C).
One-way ANOVAs with Tukey's post-hoc t tests; data presented as mean ± SEM; n = 3 mice/group. SVZ, subventricular zone; CUP, cuprizone diet; REG, regular diet.
Combined Loss of Gli1 and Gli2 Decreases the Recruitment of vNSC-Derived Cells to Demyelinated Lesions
To determine if GLI2 expression is required for recruitment of vNSCs-derived cells to the demyelinated lesion, we examined the effects of conditional ablation of Gli2 specifically in adult GLI1 vNSCs using Gli1CreER;Gli2FX mice. After confirming that Gli2 is ablated from 84.7% ± 0.4% of the GLI1 vNSCs (Figures S1A and S1B), we analyzed the fate of the GLI1 vNSC progeny in the corpus callosum (CC) at 2 weeks of recovery from a cuprizone diet (Figures 2A and 2B). As expected from previous studies, Gli1 fate-mapped cells were observed in the CC only upon demyelination and significantly more Gli1 fate-mapped cells were found in the Gli1NULL CC compared with the Gli1HET CC when GLI2 expression was intact (Figure 2B) (Samanta et al., 2015). The number of infiltrating Gli1 fate-mapped cells in Gli1HET CC did not change upon ablation of Gli2 (Figures 2A and 2B). In contrast, loss of one copy of Gli2 in the Gli1NULL mice (Gli1NULL;Gli2HET) reduced the number of Gli1 fate-mapped cells in the CC by 81.5% ± 5.6%, and complete ablation of Gli2 (Gli1NULL;Gli2NULL) reduced the number of fate-mapped cells by 88.7% ± 4.3% compared with the Gli1NULL mice with intact Gli2 expression (Gli1NULL;Gli2WT) (Figures 2A and 2B) without altering the extent of demyelination (Figures S2A and S2B). Thus, haploinsufficiency of Gli2 in the Gli1NULL vNSCs is sufficient to reverse the enhanced recruitment of their progeny into the CC upon demyelination.
Figure 2.

Ablation of Gli2 Decreases Recruitment of Gli1NULL vNSC Progeny to the White Matter Lesion.
(A) Immunofluorescence for GFP + fate-mapped Gli1 cells in the CC of mice at 2 weeks of recovery from a cuprizone diet. Scale bar, 200 μm, n = 5 mice/group.
(B) Quantification of the total number of fate-mapped Gli1HET (white) and Gli1NULL (green) vNSCs in (A). Two-way ANOVAs with Tukey's post-hoc t tests within Gli1HET and Gli1NULL groups; mean ± SEM. n = 5 mice/genotype.
(C) Quantification of the proportion of GFP + fate-mapped Gli1 vNSCs co-expressing Nestin in the SVZ at 6 weeks of cuprizone diet. n = 3 mice/genotype.
(D) Immunofluorescence for co-expression of GFAP (magenta) and GFP (green) in fate-mapped Gli1HET (left) or Gli1NULL (right) vNSCs in the SVZ at 2 weeks of recovery from a cuprizone diet. Gli1 + GFAP + NSCs (arrows); Gli1 + GFAP- cells (arrowheads). Inset shows enlarged boxed area. n = 3 mice/genotype, Scale bars, 50 μm.
(E) Quantification of the proportion of fate-mapped Gli1 vNSCs co-expressing GFAP (D) in Gli1HET and Gli1NULL mice. n = 3 mice/genotype.
(F) Immunofluorescence for EdU incorporation (green) in RFP + fate-mapped Gli1 vNSCs (red) from Gli1HET and Gli1NULL mice in vitro. n = 3 replicates, Scale bar, 50 μm.
(G) Quantification of the proportion of RFP + fate-mapped Gli1 vNSCs labeled by EdU in (F).
For (C), (E), and (G): one-way ANOVA within Gli1 groups followed by t tests comparing groups with controls. Data presented as mean ± SEM. CC, corpus callosum; SVZ, subventricular zone; CUP, cuprizone diet; REG, regular diet; Gli2-OE, Gli2 overexpression.
Although loss of Gli2 does not alter neurogenesis from vNSCs in the healthy SVZ (Petrova et al., 2013), its role in maintaining neurogenesis along with quiescence and activation of these cells upon demyelination is unknown. We quantified the Gli1 fate-mapped cells in the SVZ that co-express NESTIN, a marker of activated NSCs and GFAP, a marker of quiescent NSCs at peak demyelination (Codega et al., 2014). Loss of one or both copies of Gli2 did not change the number of Gli1 fate-mapped cells co-expressing NESTIN at peak demyelination or GFAP at 2 weeks of recovery from demyelination in the Gli1HET or Gli1NULL SVZ (Figures 2C–2E). To further examine the effect of Gli2 on proliferation of Gli1 fate-mapped vNSCs from adult Gli1HET and Gli1NULL SVZ, we quantified the rate of EdU incorporation in vNSCs overexpressing Gli2 (Roessler et al., 2005) and in cells treated with GANT58 inhibitor to reduce Gli2 expression in vitro (Lauth et al., 2007). GANT58 treatment did not change the proportion of Gli1 fate-mapped vNSCs derived from the Gli1HET or Gli1NULL SVZ (Figure S2C) but reduced the Gli2 mRNA expression by ~50% in the NSCs, comparable with that in Gli2 heterozygous mice (data not shown). Conversely, overexpression of Gli2 increased its expression about 2-fold in NSCs (data not shown) without affecting the proportion of Gli1 fate-mapped cells (Figure S2C). Overexpression or inhibition of Gli2 in the Gli1HET vNSCs did not alter EdU incorporation (Figures 2F and 2G). However, decreasing Gli2 in Gli1NULL vNSCs increased EdU incorporation (0.6% ± 0.1% cells in control versus 9.9% ± 1.8% cells with GANT58 treatment) in fate-mapped vNSCs, while overexpression of Gli2 had no effect (Figures 2F and 2G), indicating that inhibition of Gli2 in combination with loss of Gli1 increases the proliferation of vNSCs in vitro. The unchanged proliferation rate in Gli1NULL vNSCs with Gli2 overexpression is consistent with the lack of alteration in number of activated NSCs in Gli1NULL vNSCs at peak demyelination. Thus, the decrease in recruitment of Gli1 fate-mapped cells to the CC following demyelination on combined loss of Gli1 and Gli2 may not be due to a reduction in proliferation of vNSCs.
Taken together, these results indicate that Gli2 is essential for recruitment of Gli1NULL vNSC-derived cells to the site of demyelinating injury but does not affect their quiescence or activation following demyelination.
Combined Loss of Gli1 and Gli2 Increases Migration of vNSC-Derived Cells to the Olfactory Bulb on Demyelination
Our results raise the possibility that loss of Gli2 could alter the physiological migration of vNSCs into the OB via the rostral migratory stream (RMS) in the demyelinated brain. Loss of Gli2 in the healthy SVZ does not affect the deeper layer granule interneurons in the OB produced by vNSCs, suggesting that GLI2 is not essential for vNSC migration to the OB (Ihrie et al., 2011; Petrova et al., 2013; Young et al., 2007). However, the role of GLI2 in the migration of vNSCs-derived cells lacking GLI1 to the OB and upon demyelination is not clear. We quantified the number of fate-mapped Gli1 cells in the OB of Gli1HET and Gli1NULL brains with varying levels of Gli2 expression at 2 weeks of recovery from demyelination. Loss of Gli1 did not alter the number of Gli1 fate-mapped cells in the OB of healthy mice on regular diet (Figures 3A and 3B). However, following recovery from demyelination there was a significant increase in the number of fate-mapped Gli1NULL vNSCs in the OB (2.6- ± 0.13-fold in Gli1NULL;Gli2HET and 2.7- ± 0.41-fold in Gli1NULL;Gli2NULL) compared with the Gli1HET vNSCs (4,670 ± 1,040 cells/mm3 in Gli1HET;Gli2WT versus 11,777 ± 1,898 cells/mm3 in Gli1NULL;Gli2HET and 11,142 ± 1,168 cells/mm3 in Gli1NULL;Gli2NULL) (Figures 3A and 3B). However, loss of Gli2 did not affect the position and morphology of fate-mapped vNSCs within the OB following demyelination. Cells with neuronal morphology were located in the deeper granule cell layers and those with an astrocytic morphology were predominantly in the external plexiform layer of the OB (Figures 3A and 3A’).
Figure 3.

Ablation of Gli2 Increases Recruitment of Gli1NULL vNSC Progeny to the Olfactory Bulb
(A) Immunofluorescence for GFP + fate-mapped vNSCs (green) in Gli1HET (top) and Gli1NULL (bottom) OB on regular diet or at 2 weeks of recovery from a cuprizone diet. (A’) Enlarged portion of the OB (dotted rectangle) showing (1) granule and (2) external plexiform layers.
(B) Quantification of the fate-mapped Gli1HET and Gli1NULL vNSCs in (A). One-way ANOVAs followed by multiple t tests within Gli1 groups and comparing all groups with Gli1HET;Gli2WT controls.
(C) Quantification of the combined number of fate-mapped Gli1HET and Gli1NULL cells in the CC and OB. One-way ANOVAs followed by multiple t tests within Gli1 groups.
Data presented as mean ± SEM; n = 3 mice/genotype. Scale bar, 50 μm. OB, olfactory bulb; CC, corpus callosum; SVZ, subventricular zone; CUP, cuprizone diet; REG, regular diet.
To determine the total number of vNSC-derived cells that had migrated out of the SVZ, we combined the fate-mapped cells recruited to the CC and the OB. Consistent with an increase in proliferation in Gli1NULL SVZ on demyelination, we found a 2.16- ± 0.03-fold increase in the total Gli1NULL NSC-derived cells compared with those from Gli1HET NSCs (Figure 3C). This increase in total number of recruited cells in Gli1NULL CC and OB remained constant irrespective of the Gli2 gene dosage. In addition, loss of Gli2 did not alter the percentage of Gli1 fate-mapped vNSCs in the SVZ co-expressing the neuroblast marker doublecortin (DCX) at peak demyelination and at 2 weeks of recovery from demyelination, indicating that there was no reduction in generation of neuroblasts destined for the OB (Figures S3A–S3C). Since the total number of cells recruited out of the SVZ remained constant with loss of Gli2 but the location of the recruited cells was altered with respect to the CC versus OB, these results also indicate that loss of Gli2 redirects Gli1NULL vNSC-derived cells from the site of demyelination to the RMS for migration to the OB. Together, these data show that, while inhibition of Gli1 with simultaneous increase in Gli2 promotes migration of vNSC-derived cells to demyelinated lesions, loss of both Gli1 and Gli2 prevents their recruitment toward the demyelinated lesion.
Gli2 Promotes the Differentiation of Gli1-null vNSC-Derived Cells into OLs
In the healthy brain, GLI2 is expressed in all NSCs in the SVZ and in parenchymal astrocytes, but not in cells of the OL lineage, including OPCs and mature OLs (Garcia et al., 2010). However, its role in cell fate decisions in the demyelinated brain remains unknown. We examined if loss of GLI2 altered the differentiation of the few vNSC-derived cells recruited to the CC in response to demyelination. Ablation of Gli2 significantly reduced the number of fate-mapped mature OLs expressing CC1 in the Gli1NULL callosum (81.2% ± 2.2% in Gli1NULL;Gli2WT versus 24.9% ± 11.2% in Gli1NULL;Gli2HET and 33.2% ± 12.3% in Gli1NULL;Gli2NULL) but did not change the OLs in the Gli1HET callosum at 2 weeks of recovery from a cuprizone diet (Figures 4A and 4B). Interestingly, loss of Gli2 did not alter the generation of PDGFRα-expressing OPCs in the Gli1HET and Gli1NULL CC (Figures 4A and 4B) suggesting that GLI2 is required for terminal differentiation of OLs in the absence of GLI1. To further examine the effect of GLI2 on differentiation, we overexpressed and pharmacologically inhibited Gli2 in the Gli1 fate-mapped vNSCs from adult Gli1HET and Gli1NULL SVZ in vitro. Neither overexpression nor inhibition of Gli2 expression in Gli1HET- or Gli1NULL-derived vNSCs changed the number of Gli1 fate-mapped OPCs expressing NG2 (Figures 4C and 4D). However, overexpression of Gli2 significantly increased the differentiation of Gli1NULL vNSCs into mature OLs expressing MBP and this effect was abrogated by inhibition of Gli2 (0.05% ± 0.03% in control cells versus 1.52% ± 0.35% on Gli2 overexpression versus 0.03% ± 0.03% with GANT58 treatment) (Figures 4E and 4F). These results indicate that overexpression of Gli2 is necessary, in the absence of Gli1, for terminal differentiation to mature OLs.
Figure 4.

Gli2 Promotes Oligodendrocyte Differentiation from vNSCs
(A) Immunofluorescence for fate-mapped Gli1NULL vNSCs (green) co-expressing PDGFRα (OPC) or CC1 (OL) (magenta). n = 5 mice/genotype, GFP + cell (arrows) and marker + GFP co-expressing cells (arrowheads).
(B) Quantification of the proportion of GFP + fate-mapped Gli1HET (white) and Gli1NULL (green) vNSCs that co-express markers of OPC (PDGFRα) and OL (CC1) in (B).
(C) Immunofluorescence for OPCs (NG2, green) and RFP + Gli1 fate-mapped vNSCs (red) following overexpression (Gli2-OE) or inhibition (GANT58 treatment) of Gli2 in vitro. n = 3 replicates.
(D) Quantification of Gli2 overexpression or inhibition in (C).
(E) Immunofluorescence for MBP + mature OLs (green) and RFP + fate-mapped Gli1 vNSCs (red) following overexpression (Gli2-OE) or inhibition (GANT58 treatment) of Gli2 in vitro. n = 3 replicates.
(F) Quantification of Gli2 overexpression (Gli2-OE) or inhibition (GANT58 treatment) in (E).
One-way ANOVA with Tukey's post-hoc t tests within Gli1 genotypes; all data presented as mean ± SEM. Scale bars, 50 μm. CC, corpus callosum; OPC, oligodendrocyte progenitor cells; OL, oligodendrocyte; CUP, cuprizone diet; Gli2-OE, Gli2-overexpression.
To examine the role of GLI2 in astrocyte generation from vNSCs, we quantified the number of GFAP-expressing astrocytes and Nestin-expressing astrocytes/progenitor cells in the white matter following 2 weeks of recovery from a cuprizone diet in vivo. Interestingly, loss of Gli2 reduced the proportion of Gli1 fate-mapped cells expressing GFAP modestly in the Gli1HET CC (15.5% ± 2.4% in Gli1HET;Gli2WT versus 0.4% ± 0.4% in Gli1HET;Gli2HET and 1.7% ± 1.0% in Gli1HET;Gli2NULL), but had no effect on number of NESTIN-expressing cells (Figures S4A–S4C). However, the proportion of Gli1 fate-mapped cells co-expressing GFAP and NESTIN were unaltered in the Gli1NULL CC irrespective of the Gli2 gene dosage (Figures S4A–S4C). We further examined this effect on astrocyte differentiation in vitro, but did not detect any effect with overexpression or inhibition of Gli2 in Gli1HET- or Gli1NULL-derived vNSCs (Figures S4D and S4E), suggesting that the in vivo effect on number of GFAP-expressing cells in Gli1HET CC is probably not a direct effect of loss of Gli2. Taken together, these results indicate that GLI2 is required for generation of mature OLs in the absence of GLI1, further highlighting the importance of relative expression levels of the two transcription factors following demyelination.
Discussion
This study shows that GLI2 plays an essential role in differentiation and recruitment of vNSC-derived cells to demyelinated lesions in the absence of GLI1. We observed an increase in GLI2 expression following demyelination in the SVZ and when we ablated Gli2 in the Gli1-deficient vNSCs, the resulting progeny were preferentially recruited to the OB. This altered recruitment pattern suggests that GLI2 expression may normally be required to divert Gli1NULL vNSC-derived cells toward the lesion. In addition, any cell that is recruited to the lesion may fail to differentiate into mature OLs in the absence of GLI2.
Gli1 knockout mice are viable and loss of GLI1 does not seem to alter the expression of Shh target genes (Lipinski et al., 2006). On the contrary, Gli2 knockout mice as well as Gli1/Gli2 double homozygous mutant mice show embryonic lethality (Bai et al., 2002; Park et al., 2000). Notably, global loss of Gli1 combined with heterozygous ablation of Gli2 also results in death soon after birth, indicating a haploinsufficient effect of Gli2 (Park et al., 2000). While Gli2HET mice are considered normal, they show developmental defects following teratogen exposure, further underscoring the functional effects of Gli2 haploinsufficiency (Heyne et al., 2016; Kietzman et al., 2014). Gli2 has also been shown to promote migration of cranial neural crest and glioma cells in the brain (Huang et al., 2018; Timberlake et al., 2019). Similarly, our results showing reduction of recruitment of vNSC-derived cells to the demyelinated CC in Gli1NULL; Gli2HET mice point to the role of Gli2 haploinsufficiency.
Our data further show that overexpression of GLI2 is sufficient to increase differentiation into mature OLs by Gli1NULL but not by Gli1HET vNSCs, further indicating the distinct functions of relative levels of GLI1 and GLI2. This is consistent with studies showing different genes being activated by GLI1 alone or GLI2 alone or combined actions of both transcription factors (Tolosa et al., 2020). Although ablation of Gli2 reduced astrocyte differentiation from Gli1HET vNSCs progeny recruited to the demyelinated CC, in vitro overexpression and inhibition of Gli2 in these vNSCs did not alter their differentiation into astrocytes. In addition, loss of Gli2 in Gli1NULL vNSCs did not alter astrocyte differentiation upon demyelination, similar to that observed in healthy brains where the loss of both GLI1 and GLI2 activity produces an activated phenotype without a change in the number of astrocytes (Garcia et al., 2010). These results suggest that GLI2 does not play a direct role in astrocyte differentiation. Alternatively, these data may reflect differences in the microenvironment upon demyelination. Indeed, demyelination changes the expression of several growth factors, including fibroblast growth factor and transforming growth factor β1 (Huang and Dreyfus, 2016), which can regulate Gli2 expression (Brewster et al., 2000; Javelaud et al., 2011) and astrocyte generation (Nataf, 2020; Savchenko et al., 2019).
Taken together, our results indicate that GLI2 is necessary in the absence of GLI1 for enhanced migration of vNSC progeny and oligodendrogenesis in demyelinated lesions. These data highlight that GLI1 and GLI2 have distinct functions in response to demyelination, consistent with their distinct transcriptional targets (Ali et al., 2019; Tolosa et al., 2020). They also suggest that therapeutic strategies that target the transcriptional effectors of Shh (Lauth et al., 2007) could have very different outcomes depending on the relative specificity for GLI1 versus GLI2. In conclusion, our results indicate that specific GLI1 inhibitors that do not target GLI2 may be required for enhancing remyelination.
Experimental Procedures
Fate Mapping and Demyelination
All animals were maintained according to the SVM IACUC protocols at University of Wisconsin-Madison. Mouse lines obtained from Jackson Laboratory: Gli1CreERT2 (Jax no. 007913), Gli1nLacZ (Jax no. 008211), Gli2Flox (Jax no. 007926), Rosa-CAG-EGFP (RCE) (Jax no. 032037) for in vivo experiments and Rosa-CAG-TdTomato (Ai9) (Jax no. 007909) for in vitro experiments. The genotypes used: Gli1HET;Gli2WT (Gli1CreER/WT;RCEFX/FX), Gli1HET;Gli2HET (Gli1CreER/WT;Gli2FX/WT;RCEFX/FX), Gli1HET;Gli2NULL (Gli1CreER/WT;Gli2FX/FX;RCEFX/FX), Gli1NULL;Gli2WT (Gli1CreER/CreER;RCEFX/FX), Gli1NULL;Gli2HET (Gli1CreER/CreER;Gli2FX/WT;RCEFX/FX), and Gli1NULL;Gli2NULL (Gli1CreER/CreER;Gli2FX/FX;RCEFX/FX) for in vivo analysis; and for in vitro analysis: Gli1HET (Gli1CreER/WT;Ai9FX/WT) and Gli1NULL(Gli1CreERT2/CreERT2;Ai9FX/WT). All mice were maintained on C57Bl/6 background. For fate mapping, 10-week-old mice were administered 5 mg/day tamoxifen (no. 13258, Cayman Chemical, Ann Arbor, MI) in corn oil for four intraperitoneal injections on alternate days. No labeling was seen in the absence of tamoxifen administration. Demyelination was induced 1 week after tamoxifen administration by feeding 0.2% cuprizone in the chow for 5–6 weeks.
In Vivo Immunofluorescence
Cryosections were processed for immunofluorescence with rabbit or chicken anti-GFP antibody and these antibodies: rat anti-PDGFRα; mouse anti-CC1, anti-GFAP, anti-MBP, and anti-LacZ, guinea pig anti-GLI2 (1:50, kind gift from Jonathan Eggenschwiler, University of Georgia) (Cho et al., 2008), and anti-donkey or anti-goat secondary antibodies conjugated with Alexa Fluor (Table S1). Nuclei were counterstained with Hoechst 33258 (1:5,000, Invitrogen, no. H3570).
Primary NSC Culture
Seven- to 12-week-old Gli1CreER/WT;Ai9FX/WT or Gli1CreER/CreER;Ai9FX/WT mice were administered 5 mg of tamoxifen to fate-map Gli1-expressing cells 3 days before euthanasia. The SVZ from two brains were pooled for each NSC culture as described in the Supplemental Experimental Procedures.
In Vitro Immunofluorescence
NSCs plated on Matrigel-coated coverslips were fixed in ice-cold methanol and labeled with the primary antibodies, including: rabbit anti-NG2 and rat anti-RFP to label TdTomato (Table S1). Images were analyzed as described in the Supplemental Experimental Procedures.
Statistical Analysis
All experiments were replicated at least three times. For in vivo experiments, at least five sections per mouse were analyzed and data from three to five mice were combined. All data are expressed as mean ± standard error of mean. Statistical analysis was performed using Student's t test, one-way ANOVA, or two-way ANOVA with Tukey's post-hoc t test. Differences were considered statistically significant at p < 0.05.
Author Contributions
D.Z.R., J.S., and H.M.M. performed the research, analyzed and interpreted the results, and edited the manuscript. S.K.B., E.D.C., M.M.T., and C.A.O. performed imaging and in vitro cell counts. J.R.H.-S. performed preliminary cell counts. J.S. designed the experiments and wrote the manuscript. J.L.S. edited the manuscript.
Conflicts of Interests
A patent on the method of targeting GLI1 as a strategy to promote remyelination has been awarded, with J. L.S., J.S., and G.F. listed as co-inventors. J.L.S. is a consultant for and has ownership interests in Glixogen Therapeutics.
Acknowledgments
This research was supported by grants to J.L.S. from NIH NS0100867 and NY State DOH01-STEM5-2016-00305 and to J.S. from the Nancy and Jean-Pierre Boespflug Foundation for Myopathic Research.
Published: October 29, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.10.003.
Supplemental Information
References
- Ahn S., Joyner A.L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Ali S.A., Niu B., Cheah K.S.E., Alman B. Unique and overlapping GLI1 and GLI2 transcriptional targets in neoplastic chondrocytes. PLoS One. 2019;14:e0211333. doi: 10.1371/journal.pone.0211333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C.B., Auerbach W., Lee J.S., Stephen D., Joyner A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Brewster R., Mullor J.L., Ruiz i Altaba A. Gli2 functions in FGF signaling during antero-posterior patterning. Development. 2000;127:4395–4405. doi: 10.1242/dev.127.20.4395. [DOI] [PubMed] [Google Scholar]
- Chaker Z., Codega P., Doetsch F. A mosaic world: puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip. Rev. Dev. Biol. 2016;5:640–658. doi: 10.1002/wdev.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho A., Ko H.W., Eggenschwiler J.T. FKBP8 cell-autonomously controls neural tube patterning through a Gli2- and Kif3a-dependent mechanism. Dev. Biol. 2008;321:27–39. doi: 10.1016/j.ydbio.2008.05.558. [DOI] [PubMed] [Google Scholar]
- Codega P., Silva-Vargas V., Paul A., Maldonado-Soto A.R., Deleo A.M., Pastrana E., Doetsch F. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 2014;82:545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A.D., Petrova R., Eng L., Joyner A.L. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J. Neurosci. 2010;30:13597–13608. doi: 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyne G.W., Everson J.L., Ansen-Wilson L.J., Melberg C.G., Fink D.M., Parins K.F., Doroodchi P., Ulschmid C.M., Lipinski R.J. Gli2 gene-environment interactions contribute to the etiological complexity of holoprosencephaly: evidence from a mouse model. Dis. Models Mech. 2016;9:1307–1315. doi: 10.1242/dmm.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D., Wang Y., Xu L., Chen L., Cheng M., Shi W., Xiong H., Zalli D., Luo S. GLI2 promotes cell proliferation and migration through transcriptional activation of ARHGEF16 in human glioma cells. J. Exp. Clin. Cancer Res. 2018;37:247. doi: 10.1186/s13046-018-0917-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Dreyfus C.F. The role of growth factors as a therapeutic approach to demyelinating disease. Exp. Neurol. 2016;283:531–540. doi: 10.1016/j.expneurol.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie R.A., Shah J.K., Harwell C.C., Levine J.H., Guinto C.D., Lezameta M., Kriegstein A.R., Alvarez-Buylla A. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron. 2011;71:250–262. doi: 10.1016/j.neuron.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javelaud D., Alexaki V.I., Dennler S., Mohammad K.S., Guise T.A., Mauviel A. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71:5606–5610. doi: 10.1158/0008-5472.CAN-11-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzman H.W., Everson J.L., Sulik K.K., Lipinski R.J. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS One. 2014;9:e89448. doi: 10.1371/journal.pone.0089448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M., Bergstrom A., Shimokawa T., Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U S A. 2007;104:8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R.J., Gipp J.J., Zhang J., Doles J.D., Bushman W. Unique and complimentary activities of the Gli transcription factors in Hedgehog signaling. Exp. Cell Res. 2006;312:1925–1938. doi: 10.1016/j.yexcr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Matsushima G.K., Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo R., Freer A.M., Zinyk D.L., Crackower M.A., Michaud J., Heng H.H., Chik K.W., Shi X.M., Tsui L.C., Cheng S.H. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- Nataf S. The demonstration of an Aqp4/Tgf-beta 1 pathway in murine astrocytes holds implications for both neuromyelitis optica and progressive multiple sclerosis. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21031035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.L., Bai C., Platt K.A., Matise M.P., Beeghly A., Hui C.C., Nakashima M., Joyner A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Petrova R., Garcia A.D., Joyner A.L. Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J. Neurosci. 2013;33:17490–17505. doi: 10.1523/JNEUROSCI.2042-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E., Ermilov A.N., Grange D.K., Wang A., Grachtchouk M., Dlugosz A.A., Muenke M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 2005;14:2181–2188. doi: 10.1093/hmg/ddi222. [DOI] [PubMed] [Google Scholar]
- Samanta J., Grund E.M., Silva H.M., Lafaille J.J., Fishell G., Salzer J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature. 2015;526:448–452. doi: 10.1038/nature14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M.A., Armstrong R.C. Postnatal Sonic hedgehog (Shh) responsive cells give rise to oligodendrocyte lineage cells during myelination and in adulthood contribute to remyelination. Exp. Neurol. 2018;299:122–136. doi: 10.1016/j.expneurol.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Savchenko E., Teku G.N., Boza-Serrano A., Russ K., Berns M., Deierborg T., Lamas N.J., Wichterle H., Rothstein J., Henderson C.E. FGF family members differentially regulate maturation and proliferation of stem cell-derived astrocytes. Sci. Rep. 2019;9:9610. doi: 10.1038/s41598-019-46110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timberlake A.T., Jin S.C., Nelson-Williams C., Wu R., Furey C.G., Islam B., Haider S., Loring E., Galm A., Yale Center for Genome A. Mutations in TFAP2B and previously unimplicated genes of the BMP, Wnt, and Hedgehog pathways in syndromic craniosynostosis. Proc. Natl. Acad. Sci. U S A. 2019;116:15116–15121. doi: 10.1073/pnas.1902041116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E.J., Fernandez-Barrena M.G., Iguchi E., McCleary-Wheeler A.L., Carr R.M., Almada L.L., Flores L.F., Vera R.E., Alfonse G.W., Marks D.L. GLI1/GLI2 functional interplay is required to control Hedgehog/GLI targets gene expression. Biochem. J. 2020;477:3131–3145. doi: 10.1042/BCJ20200335. [DOI] [PubMed] [Google Scholar]
- Young K.M., Fogarty M., Kessaris N., Richardson W.D. Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J. Neurosci. 2007;27:8286–8296. doi: 10.1523/JNEUROSCI.0476-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.