

Abstract
European canker, caused by the necrotrophic fungal phytopathogen Neonectria ditissima, is one of the most damaging apple diseases worldwide. An understanding of the molecular basis of N. ditissima virulence is currently lacking. Identification of genes with an up-regulation of expression during infection, which are therefore probably involved in virulence, is a first step towards this understanding. Reverse transcription quantitative real-time PCR (RT-qPCR) can be used to identify these candidate virulence genes, but relies on the use of reference genes for relative gene expression data normalisation. However, no report that addresses selecting appropriate fungal reference genes for use in the N. ditissima-apple pathosystem has been published to date. In this study, eight N. ditissima genes were selected as candidate RT-qPCR reference genes for gene expression analysis. A subset of the primers (six) designed to amplify regions from these genes were specific for N. ditissima, failing to amplify PCR products with template from other fungal pathogens present in the apple orchard. The efficiency of amplification of these six primer sets was satisfactory, ranging from 81.8 to 107.53%. Analysis of expression stability when a highly pathogenic N. ditissima isolate was cultured under 10 regimes, using the statistical algorithms geNorm, NormFinder and BestKeeper, indicated that actin and myo-inositol-1-phosphate synthase (mips), or their combination, could be utilised as the most suitable reference genes for normalisation of N. ditissima gene expression. As a test case, these reference genes were used to study expression of three candidate virulence genes during a time course of infection. All three, which shared traits with fungal effector genes, had up-regulated expression in planta compared to in vitro with expression peaking between five and six weeks post inoculation (wpi). Thus, these three genes may well be involved in N. ditissima pathogenicity and are priority candidates for further functional characterization.
Introduction
The filamentous fungus Neonectria ditissima, (Tul. & C. Tul.) Samuels & Rossman is the causal agent of European canker (EC) in apple. Although able to infect a wide range of hardwood trees species [1] the disease in apple can be especially destructive with significant economic ramifications in wet, moderate climates mainly due to twig dieback [2]. EC has been recorded in the apple growing regions of North and South America, Europe, Asia and New Zealand resulting in tree loss. In some countries, fruit loss challenges the profitability of production [3–6]. EC early symptoms of infection are reddish-brown lesions around a wound, such as a leaf scar, spur or pruning wound. Over time a canker develops that can ultimately girdle the trunk or branch, causing the death of any distal shoots [7]. This disease occurs predominantly during wet seasons when dispersal of ascospores and conidia, and infection in orchards is facilitated [8]. Ascospores, produced in red perithecia, can be observed within a year after initial canker formation. These two-celled spores can be expelled from the perithecium and wind-dispersed, or exuded as a white-cream sticky mass and splash dispersed, during high humidity periods [9]. In the early stages of canker, conidia are released from white-cream sporodochia and splash dispersed between adjacent trees. Thus, compared to ascospores, conidia can only be locally spread. However, regardless of spore type, spore dispersal, germination and infection are highly facilitated by rainfall.
Control measures for EC focus on inoculum removal through pruning and fungicide application, which work as a temporary protection barrier against fungal ingress. Even when using the most stringent fungicide programmes combined with pruning, the incidence of canker still increases but at a rate that is slower than if no control measures were adopted [4]. A promising direction to realise sustainable control comes from identification of Malus x domestica germplasm that varies in susceptibility to N. ditissima [10,11]. Genetic mapping has identified an N. ditissima resistance locus, Rnd1, from the cultivar (cv) ‘Robusta 5’ [12]. SNP markers have been developed for this locus, hinting at the development of an EC-resistant apple cultivar using marker-assisted selection (MAS, [12]).
Although deployment of resistance in germplasm can be effective for disease control, resistance can often be overcome by the pathogen. An understanding of underlying molecular mechanisms and evolutionary forces at play are needed to develop a long-term management strategy to effectively control the disease. Knowledge of the molecular basis of the interaction between N. ditissima and apple is very limited. No specific molecular resistance mechanisms have yet been reported and very little is known about the interaction that precedes either symptom expression or successful host defence. Indeed, there is a dearth of knowledge regarding N. ditissima virulence mechanisms. Analysing the gene expression of candidate virulence genes in N. ditissima during plant infection is the first step towards filling this knowledge gap, with an up-regulation of gene expression during growth in planta compared to that in vitro suggesting an involvement in virulence.
Although the price of RNAseq experiments is continuously falling, providing a means of accurately analysing gene expression, reverse transcription quantitative real-time PCR (RT-qPCR) remains a valid methodology, especially where detailed time courses of infection are investigated including multiple time points, the scale of which could still render analysis by RNAseq too costly. Indeed, significant progress has been made in understanding plant-fungal interactions since the development of RT-qPCR, due to its sensitivity and ease of use, although it does require rigorous standardisation to accurately interpret the data and generate reliable results [13]. Quantification errors in RT-qPCR data can occur due to variations in RNA concentration, RNA quality, efficiency of cDNA synthesis and PCR amplification. Moreover, in order to allow comparison of expression levels in a disease time course, reference genes are required to account for differences in fungal biomass between samples and potential differences in total RNA extraction efficiencies.
Typically ‘housekeeping’ genes (genes required for basic cellular functions) have been used as reference genes for data normalisation with expression independent of the experimental condition [14,15]. However, such genes are still regulated to some extent, reinforcing the opinion that there is no universal reference gene with expression levels that remain constant across all conditions [16,17]. Since even small variations of an internal control can lead to inaccuracies in expression data, it is critical to validate stable expression of reference genes prior to their use for normalisation in RT-qPCR analysis. When validating reference genes, there is a “circular problem” of evaluating the expression stability of a candidate gene when there is no reliable measure available to normalise the candidate. However, statistical algorithms such as geNorm [18], NormFinder [19], and BestKeeper [20] permit a careful selection of a set of genes that display minimal variation across different biological conditions.
To date, a study dedicated to the selection and validation of suitable reference genes in N. ditissima has not been reported. Therefore, the purpose of this study was to identify a robust set of reference genes to be used for gene expression profiling in the fungal pathogen N. ditissima. A set of housekeeping genes was selected as potential reference genes to be tested for their stability across different growth conditions. As a test case, the most stable reference genes were then applied to quantify expression of candidate virulence genes in N. ditissima, during a time course of infection in planta.
Methods
Fungal material, growth conditions and sampling
The N. ditissima pathogenic isolate 23606 from the International Collection of Microorganisms from Plants (ICMP; Manaaki Whenua—Landcare Research, New Zealand; previously referred to as RS324p), collected from a 12-year-old M. x domestica cv. ‘Golden Delicious’ tree, New Plymouth, Taranaki, 2009, was used in this study [21]. ICMP 23606 was cultured on a modified version of Matsushima’s medium (MM, Matsushima 1961) as adjusted by Dubin and English [22] under near ultraviolet (NUV) light at 20°C to encourage conidial production. Conidia for either seeding liquid cultures or for use in pathogenicity assessments were collected by washing 4-week-old MM cultures with sterile water (SW, Milli-Q Integral Water Purification System for Ultrapure water, Merck KGaA, MA, USA), then filtered through glass wool to remove mycelial debris. When required, spore concentrations were adjusted with the aid of a haemocytometer.
ICMP 23606 was sub-cultured under 10 different liquid culture conditions for gene expression stability assessment. For a control of vegetative growth under nutrient-rich conditions, the isolate was cultured in Potato Dextrose Broth (PDB, DifcoTM, NJ, USA). Starvation conditions were realised by culturing in MM which also encouraged spore production. Additives to impose stress were added to PDB and adjusted to pH 6.5. For osmotic stress: 1 M sorbitol or 3 M sodium chloride (NaCl). For cell wall stress: 1% (w/v) aqueous Congo Red to a final concentration of 100 μg/mL or 0.03% (w/v) Calcofluor White, both filter-sterilised through a 0.22 μM filter (Ahlstrom-Munksjö, Helsinki, Finland) prior to adding to PDB. For oxidative stress: 30 mM hydrogen peroxide (H2O2). For toxic stress: 0.22 μM filter-sterilised caffeine to a final concentration of 2.5 mM. Cold and heat stress were induced by growing N. ditissima in PDB at 4°C and 37°C, respectively. Liquid cultures that were not exposed to cold and heat stress were incubated at 20°C. All liquid cultures were incubated for 24 hours at 90 rpm and then filtered through Miracloth (Merck KGaA, MA, USA), by rinsing with SW, to collect the mycelium, which was snap-frozen in liquid nitrogen prior to RNA extraction.
Botryosphaeria stevensii RS3 (from the Plant & Food Research Culture Collection, Auckland, New Zealand), Cladosporium sp. ICMP 15697, Colletotrichum acutatum ICMP 13946, Colletotrichum gloeosporioides ICMP 10112, Neofabraea alba CBS 518 (from the Centraalbureau voor Schimmelcultures–Westerdijk Fungal Biodiversity Institute, CBS-KNAW, Netherlands), Neofabraea malicortis CBS 102863, Neofabraea perennans CBS 102869, Venturia inaequalis isolate ICMP 1639 [23], V. inaequalis isolate MNH120 (ICMP 13258, [24]) and V. inaequalis isolate EU-B04 [23] were cultured on Potato Dextrose Agar (PDA, DifcoTM, NJ, USA) at 20°C, under NUV light, for 7 days.
Plant material and inoculation
One-year-old dormant Malus x domestica cv. ‘Royal Gala’ trees on rootstock ‘M793’ were inoculated with N. ditissima, isolate ICMP 23606, in September 2017. Forty-eight potted 1-year-old trees were arranged in a glasshouse, in a randomised block design, with four replicates, each comprising 12 trees. Of these 12 trees, six were inoculated (five inoculation sites per tree) and six mock-inoculated (three sites per tree), with sampling at six time points. Bud scars were used as inoculation sites and were made by breaking off the buds. Only buds on the main leader were used [12]. The scars were at least 25 cm apart and were inoculated within 2 hrs of being made with 10 μl of 1x105 spores/mL conidial suspension or 10 μL of SW. After inoculation, the relative humidity in the glasshouse was increased to 100% for three days, and then kept at 75%. The average temperature in the glasshouse was 19°C, with lows of 8.5°C and a high of 32°C, over the duration of the experiment. During the inoculation period, temperatures ranged from 22.5°C to 23.5°C, and 10.5°C to 26.5°C during the period of 100% humidity. For future standardisation of sampling times, thermal time (cumulative daily mean air temperature above a base temperature, units = °C days; [25]) were calculated for each sampling date, assuming a threshold base temperature of 0°C, since the fungus still grows at 1°C [21], and any temperature above 16°C was capped at 16°C, as this threshold was identified as the temperature above which the disease does not increase faster [2,26,27].
Branch samples were taken at three (262°C days), four (359°C days), five (455°C days), six (552°C days), eight (753°C days) and 14 weeks post inoculation (wpi; 1347°C days), with inoculated and control samples taken for each time point. Although four biological replicates (consisting of individual trees) were inoculated with pathogen or water for sampling at each time point, single inoculation sites were randomly selected from only three of the biological replicates for RNA isolation. Tissue samples, approximately 1 cm in length including the bud scars, were halved longitudinally with one half being snap-frozen in liquid nitrogen for RNA extraction and the other fixed for light microscopy. For light microscopy, in planta samples from six and 14 wpi were sectioned in 1 μm-thick sections of resin-embedded material and stained in a 0.05% solution of toluidine blue in benzoate buffer (pH 4.4), washed in distilled water, dried, mounted in Shurmount (Triangle Biomedical Sciences, St Louis, MO), and observed using an Olympus Vanox AHBT3 microscope (Olympus Optical, Tokyo).
RNA extraction
Snap-frozen in vitro and in planta samples were ground with a pestle and mortar, under liquid nitrogen, to a fine powder and stored at -80°C prior to RNA extraction. RNA was extracted from the in vitro samples using the SpectrumTM Plant Total RNA kit (Sigma-Aldrich, MO, USA) according to the manufacturers’ instructions. RNA from in planta samples was extracted following a modified version of the rapid CTAB extraction procedure proposed by Gambino et al. [28]. Briefly, ground samples (approximately 100mg) were transferred to 900 μl of Extraction Buffer (2% (w/v) hexadecyl(trimethyl)ammonium bromide (CTAB), 2.5% (w/v) polyvinylpyrrolidne (PVP)-40, 2 M sodium chloride, 100 mM TRIS hydrochloride (Tris-HCl) pH 8.0 and 25 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0) with 18 μL of β-mercaptoethanol, pre-warmed to 65°C, then briefly vortexed. Samples were incubated at 65°C for 10 min, then extracted with an equal volume of chloroform:isoamyl alcohol (IAA; 24:1 v/v). Samples were vortexed briefly prior to centrifugation at 11,000 g for 10 min at 4°C. This chloroform:IAA extraction was repeated prior to precipitating the RNA by the addition of lithium chloride (12 M) to a final concentration of 3M. Samples were incubated on ice for at least 30 min. The RNA was then collected by centrifugation at 21,000 g for 20 min at 4°C, then re-suspended in 500 μL SSTE buffer (1 M sodium chloride, 0.5% sodium dodecyl sulphate, 10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0), which was pre warmed to 65°C. The RNA was then extracted using an equal volume of chloroform:IAA as described above. RNA was precipitated from the retrieved aqueous phase by the addition of 0.7 volumes of cold (4°C) isopropanol. Centrifugation was carried out at 21,000 g for 15 min at 4°C. The remaining pellet was washed in 500 μL of 70% (v/v) ethanol followed by centrifugation at 21,000 g for 10 min at 4°C.
The RNA was dried for 10 min in a laminar flow hood, and then re-suspended in 30 μL of UltraPureTM DNase/RNase-Free Distilled Water (InvitrogenTM, Thermo Fisher Scientific, MA, USA). RNA was treated with DNase I (InvitrogenTM, Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions to remove any contaminating gDNA. Absence of gDNA in the samples was confirmed by end-point PCR (see below) using primers specific for N. ditissima myo-inositol-1-phosphate synthase (mips, Table 1) and Malus x domestica glyceraldehyde-3-phosphate dehydrogenase (gapdh, [29]).
Table 1. Candidate reference genes of Neonectria ditissima, primers and their efficiencies when used in qRT-PCR.
| Gene name | Gene ID and accession number | Abbreviation | Forward Primer | Reverse Primer | Size (bp) | Efficiency (E) | R2 value* |
|---|---|---|---|---|---|---|---|
| Actin | Ndactin MT040710 | actin | CTCTGTTCCAGCCCTCAGTC | TCGGACATCGACATCACACT | 92 | 1.97 | 0.9940 |
| Beta-tubulin | Ndbtub MT040711 | Btub | TGGAAGTCAAGCACGATGAG | ATGTGCCCCACATCTCTTTC | 90 | 2.07 | 0.9959 |
| Myo-inositol-1-phosphate synthase | Ndmips MT040712 | mips | TGTTCAACATCTGCGAGGAC | GCCTTCCACTGGATACGAGA | 94 | 1.96 | 0.9895 |
| Elongation factor thermo unstable | NdEfTu MT040713 | EfTu | GATGCCAGTGGATCTTCACC | TGAGGCTTTGTCGAGTGTTG | 82 | 1.84 | 0.9813 |
| 18s ribosomal RNA adenine methylase transferase | Nd18sAMT MT040714 | 18sAMT | TCCGCAAGAACAAGACACTG | ACCATCCTCGATGTCCATGT | 167 | 2.01 | 0.9883 |
| 40S ribosomal protein subunit S8 | NdS8 MT040715 | S8 | CTCTTACCACCCCTCGAACA | TTCTTCACGTCCTCCTCGAC | 183 | 1.91 | 0.9879 |
| 40S ribosomal protein subunit S27a | NdS27a MT040716 | S27a | TCGACAACGTCAAGTCCAAG | CTTCTTGGGGGTGGTGTAGA | 203 | Not tested | Not tested |
| Ubiquitin-conjugating enzyme | NdE2 MT040717 | E2 | CTCCGACATGGAGAGGAGAG | GAGAGGCCCAGATACCCTTC | 235 | Not tested | Not tested |
* Coefficient of correlation.
RNA sample concentration and purity was assessed using the DeNovix DS-11 spectrophotometer (DeNovix Inc., Wilmington, DE, USA) and concentration and integrity were assessed using the Agilent RNA 6000 Nano Kit (Agilent Technologies, Waldbronn, Germany) in conjunction with the Agilent 2100 Bioanalyzer software according to the manufacturer’s instructions. RNA samples of sufficient integrity, with a RIN value of 7 or more, were used for cDNA synthesis.
cDNA synthesis
cDNA was synthesised from RNA using the High Capacity cDNA Reverse Transcription kit (InvitrogenTM, Thermo Fisher Scientific, MA, USA) following the manufacturer’s instructions for ‘cDNA synthesis without RNase inhibitor’. 10 μL of reaction mix was combined with 10 μL (67 ng/ μL) of DNase-treated RNA and cDNA synthesised using the following cycling conditions; annealing at 25°C for 10 min; extension at 37°C for 120 min and denaturation at 85°C for 5 min. Two reactions were carried out; one with the reverse transcriptase enzyme and one without it. Subsequently, an end-point PCR (see below) was carried out to confirm absence of gDNA contamination and successful synthesis of cDNA, using primers specific for N. ditissima mips (Table 1) and Malus x domestica gapdh [29]; duplicate reverse transcriptase reactions were pooled if judged successful by these criteria.
Candidate reference and virulence gene selection, and primer design
Candidate reference genes were selected based on their use for normalisation in other fungal pathosystems: actin, β-tubulin (Btub), mips, thermo-unstable elongation factor (EfTu), 18s ribosomal RNA adenine methylase transferase (18sAMT), 40S ribosomal protein subunit S8 (S8), 40S ribosomal protein subunit S27a (S27a), ubiquitin conjugating enzyme 2 (E2) [15]. For these genes, putative N. ditissima ICMP 23606 orthologues were selected utilising BLASTn [30] of the N. ditissima ICMP 23606 genome [31] against annotated genes from the N. ditissima R09/05 genome [32]. Putative identification of orthologues from N. ditissima ICMP 23606 was confirmed by utilising BLASTn and BLASTp against the databases of the reference RNA sequences (refseq_rna) and reference protein sequences (refseq_protein) respectively, in NCBI. (accessed November, 2019; S1 Table).
To maximise the chance of designing specific primer sets for the candidate reference genes, the predicted N. ditissima amplification product sequence from every gene was compared against sequences from 10 apple pathogens (that are associated with Malus tissue and/or expected to be found in the apple-growing orchard) in the Reference RNA sequences (refseq_rna) database held at the NCBI (accessed November, 2019) using BLASTn 2.9.0 [30] with a statistically significant expect value of e-10 (p = 0.005) as a cut-off. The apple pathogens included were: B. stevensii RS3, Cladosporium sp. ICMP 15697, C. acutatum ICMP 13946, C. gloeosporioides ICMP 10112, N. alba CBS 518, N. malicortis CBS 102863, N. perennans CBS 102869, V. inaequalis isolate ICMP 1639, V. inaequalis isolate MNH120 (ICMP 13258) and V. inaequalis isolate EU-B04. Primers were designed to bind to sequences that were dissimilar in N. ditissima and the 10 apple pathogens. Primers had melting temperatures between 56.4 and 60.3°C, lengths between 19 to 21 bp, GC content of 50 to 60%, and amplicon sizes from 82 to 235 bp (Table 1).
Candidate virulence genes were selected following screening of the predicted proteome of N. ditissima ICMP 23606, using SignalP 4.1 [33], to identify putatively secreted proteins and EffectorP 2.0 [34], to identify putative effectors. Predicted protein products were also analysed with InterProScan 5 [35] to identify protein domains. The Pathogen-Host Interaction database (PHI-base [36]) was screened with the candidate genes to identify any similar genes/protein products involved in virulence in other pathogens. Primers were designed using Primer3 software [37] to be specific to either gDNA or cDNA.
End-point PCR
End-point PCR was carried out in a 20 μL reaction volume using a final concentration of 0.2 μM primers (forward and reverse), 0.2 mM dNTPs, 10X PCR buffer (200mM Tris-HCl), 2 mM magnesium chloride (MgCl2), one unit of PlatinumTM Taq DNA polymerase enzyme (InvitrogenTM, Thermo Fisher Scientific, MA, USA) and 20 ng of DNA template. The PCR cycling programme consisted of an initial denaturation at 95°C for 2 min, followed by 40 cycles of 95°C for 10 sec, 55°C to 60°C, depending on primer set, for 30 sec and 72°C for one min per kb; and a final extension period at 72°C for 10 min. PCR amplification products were visualised following gel electrophoresis in a 2% (w/v) agarose gel in 1x Tris Acetate-EDTA (TAE) buffer with RedSafeTM (Intron Biotechnology, SEL, Korea), at the manufacturer’s recommended concentration, and the ChemiDocTM XRS+ system (Bio-Rad, CA, USA). Estimation of amplification product sizes was made by comparison with the 1kb Plus DNA Ladder (InvitrogenTM, Thermo Fisher Scientific, MA, USA).
RT-qPCR
RT-qPCR analysis was carried out using in vitro samples to test gene primer efficiency and gene expression stability of candidate reference genes. RT-qPCR master mix was based on a 10μL reaction volume using a final primer concentration of 0.25 μM (forward and reverse), 5 μL of LightCycler® 480 SYBR Green I Master Mix (Hoffmann-La Roche, BSL, Switzerland), 1.5 μL of PCR-grade water and 2.5 μL of cDNA (concentration ranged from 50 pg/μL to 500 ng/μL). RT-qPCR master mix was aliquoted into a 384-well plate manually, with three technical replicates per biological replicate. RT-qPCR analysis was carried out on a LightCycler® 480 instrument (Hoffmann-La Roche, BSL, Switzerland) using the SYBR Green detection system. The RT-qPCR cycling conditions for all analysis consisted of a pre-incubation period of 95°C for 5 min followed by 40 amplification cycles of 95°C for 10 sec, 60°C for 10 sec and 72°C for 15 sec. This was followed by a melting curve analysis cycle of 95°C for five sec and 65°C for 1 min, with cooling at 40°C for 10 sec. The quantification cycle (Cq) values and associated melting curves from all analyses were recorded using the LightCycler® 480 software 1.5.0.39 (Hoffmann-La Roche, BSL, Switzerland).
RT-qPCR analysis was carried out using in planta samples for gene expression analysis during an infection time course. RT-qPCR master mix was based on a 28 μL reaction volume using a final forward and reverse primer concentration of 0.5 μM, 14 μL of LightCycler® 480 SYBR Green I Master Mix (Hoffmannn-La Roche, BSL, Switzerland) and 7 μL of cDNA. A Biomek 2000 workstation (Beckman Coulter, CA, USA) was used to aliquot the RT-qPCR master mix into a 384-well plate, with four technical replicates per biological replicate. The RT-qPCR programme was the same used for the in vitro samples. The Cq values and associated melting curves from all analyses were recorded using the LightCycler® 480 software 1.5.0.39 (Hoffmannn-La Roche, BSL, Switzerland).
Primer specificity test
To confirm specificity, end-point PCR was conducted with the primer sets for each gene using genomic DNA (gDNA) from the 10 apple pathogens (previously mentioned) used in the design of the primers as template with gDNA of N. ditissima as a positive control. gDNA was extracted from PDA cultures using the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) following the manufacturers protocol. End-point PCR was carried out as described previously.
Primer efficiency test
For the primer efficiency test, N. ditissima cDNA, from in vitro samples grown in PDB liquid culture, was used as template. Five samples from a 10-fold dilution series of the cDNA (from 500 ng to 50 pg) were used as separate templates. Three technical replicates were used for each dilution including three non-template control replicates. RT-qPCR analysis was carried out as described previously. Primer efficiency was calculated from the Cq values obtained from the average of the technical replicates. A slope was generated of the regression between the average Cq values and the log values of each sample dilution. Primer efficiency was calculated for the N. ditissima candidate reference and virulence genes.
Expression stability of candidate reference genes
cDNA samples from N. ditissima isolate ICMP 23606 cultured under the 10 different growth treatments were used as template. The RT-qPCR analysis for each candidate reference gene consisted of three biological replicates per growth treatment with three non-template control replicates as a negative control. Three technical replicates were carried out for each sample. RT-qPCR analysis was carried out as described previously. The raw Cq data were used as input in three gene expression stability analysis software programs; geNorm [18], NormFinder [19] and BestKeeper [20]. Moreover, geNorm, NormFinder and BestKeeper algorithms were used to generate relative stability values (RSVs) and comprehensive stability values (CSVs). The RSVs were calculated based on the stability values (SV) of each algorithm [38]. The CSVs, allowing the ranking of the candidate reference genes combining the outputs from the three algorithms, were computed from the geometrical mean (GM) of the RSVs of each candidate reference gene.
Gene expression analysis
For gene expression analysis, cDNA samples were used to assess the expression of three genes of interest from N. ditissima using two stably-expressed reference genes identified in this study. The cDNA samples included those from six time points (week 3, 4, 5, 6, 8, and 14) post inoculation, with water (‘mock-inoculated’ samples) and N. ditissima (‘inoculated’ samples) having three biological replicates per treatment for each time point. Similarly, cDNA of three in vitro samples was also included for expression comparisons of N. ditissima candidate virulence genes. The cDNA samples from 6 and 8 weeks post-inoculation were also analysed using the two least stably-expressed reference genes identified in this study. RT-qPCR analysis was carried out as described previously. The raw Cq data were normalised using the delta delta Cq method [39].
Statistical analysis
One-way analysis of variance (ANOVA) was used to determine overall statistically significant differences in gene expression among all the groups (wpi) with a significance level of p < 0.05. For determination of differences in gene expression occurring between specific sets of groups (wpi), a post-hoc Tukey-Honest Significant Difference (HSD) test was used with a significance level of p < 0.01. A post-hoc Tukey-HSD test was only run if ANOVA analysis gave a significant p value. Analyses were carried out using the Genstat software 18th Edition (VSN International, 2017).
Results
Candidate reference genes
In this study eight N. ditissima genes were selected and assessed for suitability as RT-qPCR reference genes (Table 1 and S1–S3 Tables) by screening them across a set of 10 different growth conditions, 9 of which exerted abiotic stress.
The majority of primers designed for RT-qPCR are specific for N. ditissima
None of the predicted amplification products from N. ditissima were identical to the sequences of the eight candidate reference genes from the other apple pathogens present in the NCBI Reference RNA sequences (refseq_rna) database (S2 Table). Conventional end-point PCR demonstrated that the primer sets for amplification of the actin, mips, S8 and 18sAMT sequences were specific and generated amplification products of the expected size only when template from N. ditissima was used (Fig 1 and S1 Fig). The use of the Btub and EfTu primer sets resulted in amplification products of the expected size when template from N. ditissima was used (Fig 1), but also resulted in amplified products of different sizes from 200 bp to 3 kb. The E2 and S27a primer sets showed evidence of cross-reactivity with the template from at least one of the other apple pathogens giving an amplification product of a similar size to that obtained when template was derived from N. ditissima (Fig 1) and, thus, were excluded from further evaluation. Additionally, a single peak was observed in melt curve analysis of all primer pairs indicating a single PCR product when used in RT-qPCR (S2 Fig).
Fig 1. End-point PCR to test specificity of eight Neonectria ditissima candidate reference gene primer sets.
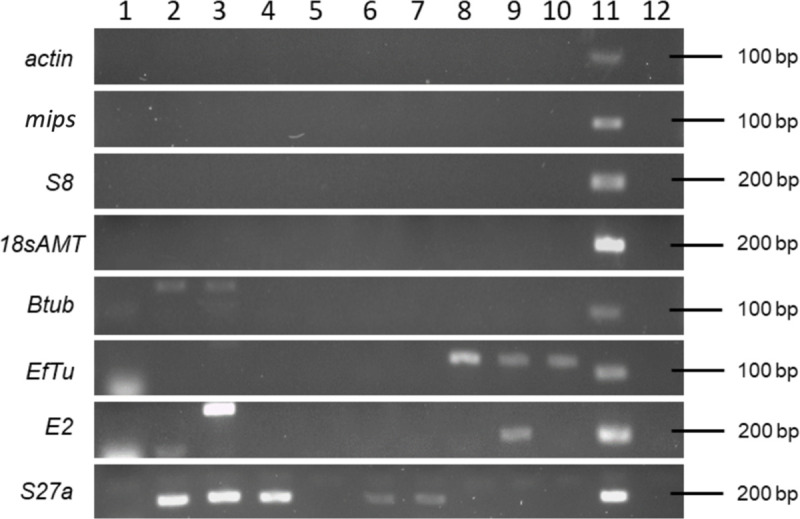
Amplification using gDNA template from (1) Botryosphaeria stevensii RS3, (2) Cladosporium sp. ICMP 15697, (3) Colletotrichum acutatum ICMP 13946, (4) Colletotrichum gloeosporioides ICMP 10112, (5) Neofabraea alba CBS 518, (6) Neofabraea malicortis CBS 102863, (7) Neofabraea perennans CBS 102869, (8) Venturia inaequalis 1639, (9) Venturia inaequalis MNH120 ICMP 13258, (10) Venturia inaequalis EU-B04, (11) N. ditissima ICMP 23606 as a positive control and (12) non-template negative control.
The efficiency of all primer sets is suitable for RT-qPCR analysis
When evaluating primer efficiency (E) and the coefficient of correlation (R2) using the standard curve analysis, the average Cq values ranged from 18.64 (for actin) to 23.64 (for mips, S3 Table) when using undiluted cDNA from N. ditissima as template. Primer efficiency data are shown in Table 1. Standard curve details for each primer set are detailed in S5 Table. All had a satisfactory linear relationship (R2 >0.9813). The actin, mips and 18sAMT primer sets had efficiencies (E) closer to 2, with values of 1.96, 1.97 and 2.01, respectively. The efficiency for the Btub primer set displayed a higher value (2.07). The primer sets for S8 and EfTu had lower efficiency values but above 1.8, thus, six candidate reference genes were carried into the gene stability analysis.
Actin and mips are the most stably expressed genes
The expression of six N. ditissima candidate reference genes was analysed by RT-qPCR following growth under 10 different regimes, including 9 stress conditions. The results showed that the Cq values among all genes ranged from 15.1 to 21.9 (S3 Fig). EfTu showed the highest expression level, whereas S8 exhibited the lowest (S3 Fig). Diverse results were obtained following analysis of gene expression stability using geNorm, NormFinder and BestKeeper algorithms (Fig 2).
Fig 2. Stability values of candidate reference genes from Neonectria ditissima.

Values based on the three algorithms geNorm (orange), NormFinder (yellow), and BestKeeper (white): Ribosomal protein S8 (S8), β-tubulin (Btub), 18s ribosomal RNA adenine methylase transferase (18sAMT), Elongation factor thermos-unstable (EfTu), myo-inositol-1-phosphate synthase (mips), actin.
Overall, all candidate reference target genes displayed high stability according to geNorm parameters (M ≤ 0.5). geNorm analysis ranked actin and mips as genes with the highest expression stability based on their small stability values (M = 0.211, 0.212, respectively; Fig 2 and Table 2). geNorm also indicated a low pairwise variation when combining these two genes (V2/3 = 0.088), which is lower than the suggested cut-off threshold of 0.15 that indicates that the geometric mean of these two genes can be used as the optimal normalisation factor in a RT-qPCR analysis (Fig 3).
Table 2. Ranking summary of six Neonectria ditissima candidate reference genes using geNorm, NormFinder and BestKeeper.
| geNorm | NormFinder | BestKeeper | ||||
|---|---|---|---|---|---|---|
| Gene | M value | Gene | S value | Gene | SD | |
| 1 | actin | 0.211 | actin | 0.132 | actin | 0.171 |
| 2 | mips | 0.212 | mips | 0.138 | 18sAMT | 0.201 |
| 3 | EfTu | 0.292 | 18sAMT | 0.245 | mips | 0.231 |
| 4 | 18sAMT | 0.345 | EfTu | 0.362 | EfTu | 0.319 |
| 5 | Btub | 0.405 | S8 | 0.414 | Btub | 0.468 |
| 6 | S8 | 0.437 | Btub | 0.531 | S8 | 0.521 |
| actin & mips a | mips & EfTu b | actin & 18sAMT c | ||||
a Optimum pair of reference genes based on the average pairwise variation V (V2/3 = 0.088).
b Optimum pair of reference genes based on stability value (S = 0.06).
c Optimum pair of reference genes based on highest correlation (r = 0.991, p < 0.001).
Fig 3. Determination of the optimal number of reference genes by geNorm software.

V2/3 represents the pairwise variation between the two most stably expressed genes according to geNorm, actin and mips. V3/4 represents the variation adding third and fourth place, EfTu and 18sAMT. V4/5, includes 18sAMT and Btub. V5/6, includes Btub and S8.
Based on the NormFinder stability value (S), actin and mips were ranked as the most stably expressed genes with S values below 0.2 (Fig 2). 18sAMT, EfTu, S8 and Btub showed less stable expression with S values larger than 0.2 but below 0.6 (Fig 2). NormFinder selected mips and EfTu as the best combination of two reference genes with the lowest S value (0.060, Table 2). BestKeeper designated all genes as having satisfactory stability, with no standard deviation (SD) values greater than 1.0 (i.e. two-fold change). actin and 18sAMT were ranked according to BestKeeper as the most stably expressed genes with SD values of 0.171 and 0.201, respectively (Fig 2), and the best combination of genes to use based on their high correlation (r = 0.991, p < 0.001, Table 2). EfTu and mips were ranked second with SD values less than 0.4 and Btub and S8 had the lowest rank with SD values higher than 0.4 but lower than 0.6 (Fig 2).
Due to each software program suggesting a different combination of genes to be used as references in a RT-qPCR analysis (Table 2), the output data from the three programs were used to generate comprehensive stability values (CSVs) based on individual stability values (SVs) from each algorithm. The comprehensive rank based on the CSV values indicated that actin and mips are the most stable genes in N. ditissima displaying relatively stable expression patterns under all conditions assessed (Table 3).
Table 3. The relative (RSV) and comprehensive (CSV) stability values of six Neonectria ditissima candidate reference genes.
| geNorm | NormFinder | BestKeeper | Comp. Rank2 | |||||
|---|---|---|---|---|---|---|---|---|
| Gene | RSV1 | Gene | RSV | Gene | RSV | Gene | CSV3 | |
| 1 | actin | 1.000 | actin | 1.000 | actin | 1.000 | actin | 1.0000 |
| 2 | mips | 1.004 | mips | 1.045 | 18sAMT | 1.175 | mips | 1.1233 |
| 3 | EfTu | 1.039 | 18sAMT | 1.856 | mips | 1.351 | 18sAMT | 1.4164 |
| 4 | 18sAMT | 1.302 | EfTu | 2.742 | S8 | 1.865 | EfTu | 1.7453 |
| 5 | Btub | 1.441 | S8 | 3.136 | EfTu | 2.737 | S8 | 2.4586 |
| 6 | S8 | 1.555 | Btub | 4.023 | Btub | 3.047 | Btub | 2.5129 |
1RSV: Relative stability value
2Comp. Rank: Comprehensive rank
3CSV: Comprehensive stability value.
Candidate virulence genes
The optimised reference genes were used in a test case of in planta gene expression of three candidate virulence genes (Table 4 and S4 Table).
Table 4. Primer sequences of Neonectria ditissima candidate virulence genes for qRT-PCR analysis.
| Gene name | Gene ID and accession number | Abbreviation | Forward Primer | Reverse Primer | Size (bp) | Efficiency (E) | R2 value* |
|---|---|---|---|---|---|---|---|
| Candidate effector gene g4542 | Nd_g4542 MT040718 | g4542 | GCGGCTTTGTGTGACTATGG | AGATATTGCCTCCCCAAGCT | 148 | 2.02 | 0.9954 |
| Candidate effector gene g5809 | Nd_g5809 MT040719 | g5809 | CTCGGTATTGGCCAGACTCA | AGCCAGACCATCTCCCAAC | 127 | 1.92 | 0.9948 |
| Candidate effector gene g7123 | Nd_g7123 MT040720 | g7123 | GAATGGTGAGGGTTGGGAGT | AGTTGATAGACCCGGTGCAA | 143 | 1.94 | 0.9951 |
* Coefficient of correlation.
The candidate virulence genes (g4542, g5809 and g7123), selected are single copy genes, predicted to encode effectors, in that the protein products are small, predicted to be canonically secreted (with the presence of a signal peptide as identified by SignalP 4.1), and are predicted by the algorithm EffectorP [34] to be an effector with probability values of 0.873, 0.827, 0.856, respectively (S3 and S4 Tables). No domains, in addition to the signal peptide, were identified when the predicted protein products were analysed by Interproscan (S3 Table). No genes or proteins similar to g4542 and its predicted encoded product were identified in the public domain. However, a gene encoding a hypothetical protein most similar to g5809 was found in Phialemoniopsis curvata and a protein most similar to the putative product of g5809 was found in Scedosporium apiospermum (both belonging to the Class Sordariomycetes [40,41]). Moreover, a hypothetical gene and protein most similar to g7123 were identified in Fusarium oxysporum. None of the genes were similar to characterised virulence determinants in PHI-base, thus they could be considered N. ditissima-specific gene sequences with a potential virulence function, sharing characteristics with many fungal effectors.
Infection progress across the time course
When assessing in planta phenotype after inoculation, the first external canker symptoms were observed at inoculation sites six wpi. Obvious canker symptoms were visible seven wpi. Internal browning of tissue was apparent at six wpi. Following sectioning, at six wpi, fungal hyphae were visible externally at the inoculation point and extended approximately 50 μm in to the tissue, with some initial disruption of the plant cell wall. Later in the infection time course (at 14 wpi) fungal hyphae were clearly visible extending from the wound, accompanied by extensive plant tissue degradation (Fig 4).
Fig 4. Symptoms caused by Neonectria ditissima on apple (cv. ‘Royal Gala’) twigs.
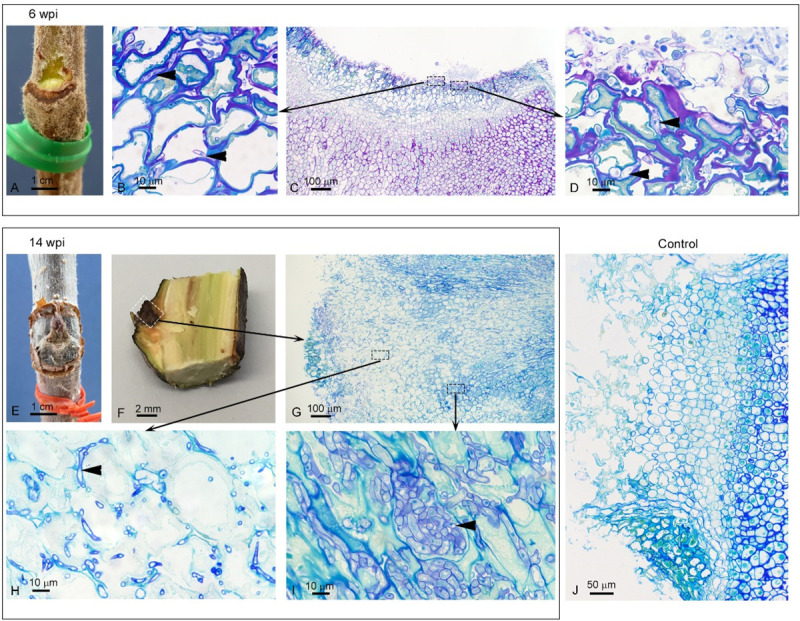
Observation of one representative biological replicate from six (A-D) and 14 (E-I) weeks post inoculation (wpi). Six wpi—A: External canker symptoms. C: 1 μm-thick section of resin-embedded material at the entrance point of infection. Black-dashed rectangles indicate close-ups in B and D. B and D: Fungal hyphae (arrow heads: purple staining) penetrating through the tissue. 14 wpi—E: External canker symptoms. F: Longitudinal section through apple twig at inoculation point (white dashed rectangle). G: 1 μm-thick section of resin-embedded material at the entrance point of infection (close-up of area enclosed by white dashed rectangle in F). Black-dashed rectangles indicate close-ups in H and I. H: Plant cell wall break down (pale green staining) as fungal hyphae (arrow heads: purple staining) ramify through the tissue, associated with the apoplast. I: Significant fungal growth (arrow heads: purple staining). J: Control (14 wpi) with no evidence of fungal infection.
Candidate effector genes expression is upregulated in planta versus in vitro
Expression of the three N. ditissima candidate virulence genes (g4542, g5809, g7123) in planta at all the time points post-inoculation was significantly different to their expression in vitro (p < 0.001, Fig 5).
Fig 5. The relative expression of three Neonectria ditissima candidate virulence genes during in planta infection.

g4542, g5809 and g7123 relative expression in vitro versus in planta during a time course of infection. wpi: weeks post-inoculation. Error bars represent standard error of the mean (SEM) among biological replicates. For each gene, letters indicate significant differences between time points at p < 0.01. Relative expression was measured through data normalisation using actin and mips as the reference gene combination.
Expression of g4542 was up-regulated at three, four, five and six wpi (57-, 63-, 68- and 66-fold, respectively) and was significantly different from its in vitro expression (3-fold, p < 0.001, Fig 5). A significant decrease at eight and 14 wpi (22- and 14-fold, respectively, p = 0.0091) was observed, however this low expression during the late stages of infection was still significantly greater than in vitro expression (p = 0.0058). Expression of g5809 was up-regulated at three wpi (42-fold), with increases in the following weeks (four wpi, 86-fold and five wpi, 83-fold), reaching a significant peak of expression by six wpi (271-fold, p < 0.001). By eight wpi (75-fold), and 14 wpi (39-fold), g5809 showed a decrease in expression significantly different from six wpi (p < 0.001), but similar to that recorded during the initial stages of infection (p = 0.88). At all time points, g5809 expression in planta was significantly greater that its expression in vitro (5-fold, p = 0.0076, Fig 5). A similar pattern was observed for g7123, where gene expression was upregulated at all time points of infection and this was significantly different from its in vitro expression (p = 0.0083, Fig 5). Up-regulation was noticed at three wpi (38-fold), four wpi (72-fold) with a significant increase by five wpi (111-fold, p = 0.0068). g7123 expression started decreasing by six wpi (77-fold), with a similar level to that recorded at four wpi (p = 0.65). Significant g7123 expression reduction was observed late in the infection time course at eight wpi (19-fold, p = 0.0084) and 14 wpi (14-fold, p = 0.0097), however it remained significantly different from expression in vitro (3-fold, p = 0.0082). There was no amplification of fungal sequences during RT-qPCR analysis when mock-inoculated cDNA was used as template throughout the time course.
Overall, the patterns of expression of the three candidate virulence genes were very similar when either the combination of the most stable reference genes actin and mips, or the two least stably expressed genes, S8 and Btub, were used for normalisation. However, the relative expression values were greater when actin and mips were used for normalisation. Thus, when S8 and Btub were used, differences in expression of the candidate virulence genes at different time points were deemed to be insignificant (p = 0.1231 to 0.6586, S4 Fig), where with actin and mips differences were significant (p < 0.001, S4 Fig). Moreover, expression in planta versus in vitro was not significantly different in g5809 (p = 0.5477) and g7123 (p = 0.5980) when using S8 and Btub, whereas a significant difference can be observed when using actin and mips (p < 0.001, S4 Fig).
Discussion
Suitable reference genes for gene expression analysis in N. ditissima have been identified. The first step in the validation of reference genes for N. ditissima expression profiling by RT-qPCR required analysis of specificity. A specificity test may not be required when analysing, for example, tissue-cultured plants inoculated with a specific fungus, which have no contaminants. However, replication of natural canker infections, for analysis of pathogenicity, requires the use of mature host material, typically derived from an orchard or glasshouse, hence the requirement for highly specific primer sets. Indeed, in the test case of in planta gene expression, samples for the infection time course came from a glasshouse experiment, with the apple material originating from an open hardstand which had the potential of carrying contaminating organisms that are commonly found in orchards. Two of the eight primer sets for the candidate genes (S27a and E2) lacked specificity, even after annealing temperature adjustments in end-point PCR, showing cross-reactivity with the DNA of other apple pathogens. These primer sets were excluded from further analysis. Moreover, the use of Btub and EfTu primer sets resulted in non-specific amplification in other organisms, however these primer sets were not initially discarded from further analysis since the non-specific amplified products were not the same size as the amplicon for N. ditissima, and thus, during RT-qPCR analysis could be distinguished if amplified. Indeed, during the RT-qPCR assays only single amplification products were observed during the melt-curve analysis (S1 Fig).
When defining the optimal reference genes for a RT-qPCR assay, gene expression stability is a decisive factor. Therefore, the software packages geNorm, NormFinder and BestKeeper were used to provide a reliable measure of gene expression stability. These algorithms have been broadly utilised for validation of reference genes in plant-fungal interactions; such as in cereal-Fusarium graminearum [42], rice-Magnaporthe oryzae [43], oil palm-Ganoderma [44], wheat-rust [16] and sugarcane-Sporisorium scitamineum [38] interactions. geNorm software ranked actin and mips as the most stable genes with the smallest M values, but even genes such as S8 and Btub that had the lowest ranking, had M values below the 0.5 cut-off suggested by the software (Fig 2). This threshold is somewhat arbitrary. Indeed, putative reference genes of cucumber with M values less than 1.5 were considered stably expressed [45]. Similarly, a study carried out in M. oryzae defined M ≥ 1 as the cut-off for a gene to be defined as unstable [43]. Therefore, according to geNorm software, all six candidate genes assessed are suitable as reference genes.
The ranking given by NormFinder was very similar to geNorm, with actin and mips ranked as the most stable genes. The S values obtained in this study (0.132 to 0.531) are similar to the S values obtained when F. graminearum reference genes were analysed under stress conditions (0.144 to 0.616, [42], unsurprising given that Neonectria and Fusarium are closely related. mips and EfTu were selected as the best combination of two reference genes using NormFinder, excluding the most stable gene actin. This result can be explained when observing the Cq data where the values for mips and EfTu were similar (18 to 20) but different to the actin values (16 to 17, S2 Fig).
BestKeeper identified actin and 18sAMT as the two most stably expressed genes with the lowest variation (SD values of 0.171 and 0.201, respectively) based on the pair-wise correlation analysis of all pairs of candidate reference genes. Pfaffl et al. [20] considered the expression of any gene with an SD higher than one to be inconsistent which, similar to geNorm, indicated that all six candidate reference genes chosen in this study are stably expressed. However, the outputs from BestKeeper differed to those from the other two software programs. This is a pattern that has been observed in previous studies [15,38,46]. The best combination of two genes to use as references differed among the analyses conducted using the three programs, thus, reinforcing that a comprehensive rank is needed for a clearer consensus. Thus, a comprehensive rank, based on the software output values converted to relative stability values, indicated actin and mips as the most stably expressed genes in N. ditissima.
Previously, actin has been studied extensively for its use as a reference gene for data normalisation in many species [15,42–44] due to its conserved function in cytoskeleton assembly. Even though its expression can vary across different fungal (and plant) species, and experimental conditions, it has proven to be a stable gene to be used for gene expression analysis in N. ditissima. Mips is an enzyme known to play a crucial role in cellular structure serving to synthesise a precursor of inositol which is a key component of cellular membranes [47,48]. Its study has been mainly focused on its contribution to resistance towards abiotic and biotic stress and regulation of growth in plants [49–51], oxidative stress in bacteria [52] and regulation of cell growth, structure and intracellular signalling in yeast and fungi [53,54]; however, in this study, mips has shown stable expression when N. ditissima is grown under stress conditions making it an ideal reference gene for data normalisation. This study has identified, for the first time, a set of reference genes in N. ditissima, where actin, mips, or their combination can be recommended as a reliable tool for normalisation of the expression of genes of interest, facilitating gene expression analysis in the apple–N. ditissima interaction.
Three N. ditissima genes (g4542, g5809, g7123) were selected in this study for their potential role in N. ditissima virulence. g4542, g5809 and g7123 candidate virulence genes showed evidence of upregulation peaks, compared to expression in vitro, at different time points of infection. N. ditissima progresses slowly over weeks rather than hours or days, even when in a conducive environment [55], as used during the time course experiment. g4542 was upregulated during the early stages (three to six wpi) and g5809 and g7123 were mainly upregulated at six wpi and five wpi, respectively. Upregulation of expression of g4542 and g7123 compared to in vitro ceased at six wpi and g5809, at eight wpi. The roles of these predicted virulence genes are currently unknown, although the predicted encoded products of all three share traits with fungal effectors in that they are small, secreted proteins, and have been predicted to be an effector by the algorithm EffectorP. Effector genes often have tailored expression profiles, altering expression depending on their function, the stage of infection and their targets in the plant [56,57]. In the late stages of infection, the expression of the three candidate effector genes was low. However, it would be inaccurate to consider that at 14 wpi, the expression of these genes is down-regulated. In contrast, at 14 wpi, the three candidate effector genes still show relative expression values, compared with expression in vitro, from 14-fold to 39-fold induction.
When expression levels of the candidate virulence genes at two time points were analysed using either the two least-stably expressed genes, S8 and Btub, or actin and mips, the patterns of gene expression were similar, although relative expression levels were lower when S8 and Btub were used. This is unsurprising since although they were the least-stably expressed, S8 and Btub were classified as suitable to be used as reference genes by all the software packages. This phenomenon has also been observed in other studies with a decrease in expression when lower stability genes are used for normalisation [58,59]. However, more dramatic differences in expression levels can also be observed with significant alterations of relativities depending on the reference genes used [60]. Although patterns of expression were retained, statistical analysis of the differences between the relative expression levels was affected, with a decrease in the significance of the differences between expressions at different time points in planta compared with expression in vitro. This indicates that use of the sub-optimal genes for normalisation may lead to nuanced differences in gene expression being overlooked.
The RT-qPCR data demonstrate that g4542, g5809 and g7123 expression is likely to be required when N. ditissima is infecting apple woody stem tissue, but not during growth in vitro, and that they appear to be important during the pre-symptomatic through to the symptomatic phase of the infection. This result suggests that, in common with other studies, the traditional way of looking at necrotrophs as pathogens that do not have an intimate interaction with their host but instead only release toxic molecules and lytic enzymes to decompose plant tissue for nutrition is too simplistic [61], and that N. ditissima effectors may well be involved in a nuanced interaction with apple [62]. Indeed, necrotrophic pathogens can rely on effectors that require an interaction with a host susceptibility protein. When this susceptibility protein is the product of a plant resistance (R) gene [63], recognition of the effector by the susceptibility protein initiates a cell death response that, rather than restricting the pathogen, as would occur for a biotroph, is of benefit to a necrotroph i.e. an inverse gene-for-gene interaction [64,65]. For example, the wheat Tsn1 gene encodes a serine/threonine protein kinase-nucleotide binding site-leucine rich repeat domain containing gene, domains characteristic of R genes, which recognises the effector ToxA from both Pyrenophora tritici-repentis and Parastagonospora nodorum to confer susceptibility [66,67]. Alternatively necrotrophic effectors can target a susceptibility protein involved in plant cell metabolism to enhance virulence. For example, the small secreted protein, SsSSVP1, from Sclerotinia sclerotiorum, manipulates plant energy metabolism to facilitate infection by targeting a protein component of the plant mitochondrial respiratory chain [68]. Moreover, necrotrophic effectors may suppress initial defence responses; the integrin‐like protein SsITL, from S. sclerotiorum, suppresses host immunity at the early stage of infection [69] by targeting a chloroplast‐localised calcium‐sensing receptor to inhibit salicylic acid accumulation [70]. Functional characterisation of the three candidate effector genes will determine their role in virulence and if they have susceptibility targets or proteins, which act in an inverse gene-for-gene manner that could be removed from the germplasm to enhance resistance to N. ditissima.
Conclusions
Reference genes for RT-qPCR have been identified for the first time in N. ditissima. Therefore, RT-qPCR using the validated reference genes will enable a finer dissection of the temporal expression patterns of candidate genes in future studies to enable prioritisation of targets for functional characterisation. Gene expression analysis of three N. ditissima candidate virulence genes (g4542, g5809 and g7123) provided evidence of significant up-regulation during apple infection, making them good candidates for further functional characterisation to elucidate their role in N. ditissima pathogenicity.
Supporting information
(PDF)
Single peaks were observed at the melting temperature (°C) of the respective amplicons. actin—81.6, Btub—80.9, mips—82.7, EfTu—83.8, S8–88.0, 18sAMT—88.5.
(TIF)
Data derived from three technical replicates from three biological replicates.
(TIF)
The relative expression of three Neonectria ditissima candidate virulence genes using (A) the least stable reference genes S8 and Btub versus (B) the most stably expressed reference genes actin and mips. Data derived from three technical replicates from three biological replicates.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
The authors would like to thank Shamini Pushparajah and Brogan McGreal for providing the gDNA of N. ditissima and the remaining apple pathogens used in the gene specificity analysis. In addition they would like to thank Kar Mun Chooi and Mark Andersen for critical reading of the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
LMF, RWAS, BMF, PWS, MDT and JKB all received funding from The New Zealand Institute for Plant and Food Research Limited, Strategic Science Investment Fund, Project number: 12070. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.McCracken AR, Berrie A, Barbara DJ, Locke T, Cooke LR, Phelps K, et al. Relative significance of nursery infections and orchard inoculum in the development and spread of apple canker (Nectria galligena) in young orchards. Plant Pathol. 2003;52(5):553–66. [Google Scholar]
- 2.Beresford RM, Kim KS. Identification of regional climatic conditions favorable for development of European canker of apple. Phytopathology. 2011;101:135–46. 10.1094/PHYTO-05-10-0137 [DOI] [PubMed] [Google Scholar]
- 3.da Silva Campos J, Bogo A, Sanhueza RMV, Casa RT, da Silva FN, da Cunha IC, et al. European apple canker: morphophysiological variability and pathogenicity in isolates of Neonectria ditissima in southern Brazil. Ciência Rural. 2017;47(5):e20160288. [Google Scholar]
- 4.Cooke LR. The influence of fungicide sprays on infection of apple cv. Bramley's seedling by Nectria galligena. Eur J Plant Pathol. 1999;105(8):783–90. [Google Scholar]
- 5.Latorre BA, Rioja ME, Lillo C, Munoz M. The effect of temperature and wetness duration on infection and a warning system for European canker (Nectria galligena) of apple in Chile. Crop Protect. 2002;21(4):285–91. [Google Scholar]
- 6.Plante F, Hamelin RC, Bernier L. A comparative study of genetic diversity of populations of Nectria galligena and N. coccinea var. faginata in North America. Mycological Research. 2002;106(2):183–93. [Google Scholar]
- 7.Saville RJ. A review of our current knowledge of Neonectria ditissima and identification of future areas of research. East Malling, Kent: East Malling Research—A study funded by The Horticultural Development Company; 2014. [Google Scholar]
- 8.Gómez-Cortecero A, Saville RJ, Scheper RWA, Bowen JK, Agripino De Medeiros H, Kingsnorth J, et al. Variation in host and pathogen in the Neonectria/Malus interaction; toward an understanding of the genetic basis of resistance to European canker. Frontiers in Plant Science. 2016;7:1365 10.3389/fpls.2016.01365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.H.M. H. Patterns of splash dispersed conidia of Fusarium poae and Fusarium culmorum. Eur J Plant Pathol. 2002;108(1):73–80. [Google Scholar]
- 10.Ghasemkhani M, Garkava-Gustavsson L, Liljeroth E, Nybom H. Assessment of diversity and genetic relationships of Neonectria ditissima: the causal agent of fruit tree canker. Hereditas. 2016;153(1):7 10.1186/s41065-016-0011-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasnauskas A, Gelvonauskienė D, Gelvonauskis B, Bendokas V, Baniulis D. Resistance to fungal diseases of apple cultivars and hybrids in Lithuania. Agron Res. 2006;4:349–52. [Google Scholar]
- 12.Bus V, Scheper RWA, Walter M, Campbell R, Kitson B, Turner L, et al. Genetic mapping of the European canker (Neonectria ditissima) resistance locus Rnd1 from Malus ‘Robusta 5’. Tree Genet Genom. 2019;15:25. [Google Scholar]
- 13.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–22. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 14.Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques. 2004;37(1):112–9. 10.2144/04371RR03 [DOI] [PubMed] [Google Scholar]
- 15.Llanos A, François JM, Parrou J-L. Tracking the best reference genes for RT-qPCR data normalization in filamentous fungi. BMC Genomics. 2015;16(1):71 10.1186/s12864-015-1224-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholtz JJ, Visser B. Reference gene selection for qPCR gene expression analysis of rust-infected wheat. Physiol Mol Plant Pathol. 2013;81:22–5. [Google Scholar]
- 17.Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46(1):69–81. 10.1016/s0165-022x(00)00129-9 [DOI] [PubMed] [Google Scholar]
- 18.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3(7):research0034 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50. 10.1158/0008-5472.CAN-04-0496 [DOI] [PubMed] [Google Scholar]
- 20.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–15. 10.1023/b:bile.0000019559.84305.47 [DOI] [PubMed] [Google Scholar]
- 21.Scheper RWA, Frijters L, Fisher BM, Hedderley DI. Effect of freezing of Neonectria ditissima inoculum on its pathogenicity. New Zealand Plant Protection. 2015;68:257–63. [Google Scholar]
- 22.Dubin HJ, English H. Factors affecting apple leaf scar infection by Nectria galligena conidia. Phytopathology. 1974;64(9):1201–3. [Google Scholar]
- 23.Broggini GAL, Bus VGM, Parravicini G, Kumar S, Groenwold R, Gessler C. Genetic mapping of 14 avirulence genes in an EU-B04 × 1639 progeny of Venturia inaequalis. Fungal Genet Biol. 2011;48(2):166–76. 10.1016/j.fgb.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 24.Fitzgerald A, van Kan JAL, Plummer KM. Simultaneous silencing of multiple genes in the apple scab fungus, Venturia inaequalis, by expression of RNA with chimeric inverted repeats. Fungal Genet Biol. 2004;41(10):963–71. 10.1016/j.fgb.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 25.Beresford RM, Royle DJ. Relationships between leaf emergence and latent period for leaf rust (Puccinia hordei) on spring barley, and their significance for disease monitoring. Journal of Plant Diseases and Protection. 1988;95(4):361–71. [Google Scholar]
- 26.Dubin HJ, English H. Epidemiology of European apple canker in California. Phytopathology. 1975;65:542–50. [Google Scholar]
- 27.Scheper RWA, Vorster L, Turner L, Campbell RE, Colhoun K, McArley D, et al. Lesion development and conidial production of Neonectria ditissima on apple trees in four New Zealand regions. New Zealand Plant Protection. 2019;72:123–34. [Google Scholar]
- 28.Gambino G, Perrone I, Gribaudo I. A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem Anal. 2008;19(6):520–5. 10.1002/pca.1078 [DOI] [PubMed] [Google Scholar]
- 29.Schaffer RJ, Friel EN, Souleyre EJ, Bolitho K, Thodey K, Ledger S, et al. A genomics approach reveals that aroma production in apple is controlled by ethylene predominantly at the final step in each biosynthetic pathway. Plant Physiol. 2007;144(4):1899–912. 10.1104/pp.106.093765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng CH, Scheper RWA, Thrimawithana AH, Bowen JK. Draft genome sequences of two isolates of the plant-pathogenic fungus Neonectria ditissima that differ in virulence. Genome Announcements. 2015;3(6):e01348–15. 10.1128/genomeA.01348-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gómez-Cortecero A, Harrison RJ, Armitage AD. Draft genome sequence of a European isolate of the apple canker pathogen Neonectria ditissima. Genome Announcements. 2015;3(6):e01243–15. 10.1128/genomeA.01243-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 34.Sperschneider J, Dodds PN, Gardiner DM, Singh KB, Taylor JM. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol Plant Pathol. 2018;19(9):2094–110. 10.1111/mpp.12682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack K. The Pathogen-Host Interactions database: additions and future developments. Nucleic Acids Res. 2015;43 Database Issue:D645–55. 10.1093/nar/gku1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang N, Ling H, Liu F, Su Y, Su W, Mao H, et al. Identification and evaluation of PCR reference genes for host and pathogen in sugarcane-Sporisorium scitamineum interaction system. BMC Genomics. 2018;19(1):479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perdomo H, García D, Gené J, Cano J, Sutton DA, Summerbell R, et al. Phialemoniopsis, a new genus of Sordariomycetes, and new species of Phialemonium and Lecythophora. Mycologia. 2013;105(2):398–421. 10.3852/12-137 [DOI] [PubMed] [Google Scholar]
- 41.Morales LT, González-García LN, Orozco MC, Restrepo S, Vives MJ. The genomic study of an environmental isolate of Scedosporium apiospermum shows its metabolic potential to degrade hydrocarbons. Stand Genomic Sci. 2017;12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim HK, Yun SH. Evaluation of potential reference genes for quantitative RT-PCR analysis in Fusarium graminearum under different culture conditions. The Plant Pathology Journal. 2011;27(4):301–9. [Google Scholar]
- 43.Che Omar S, Bentley MA, Morieri G, Preston GM, Gurr SJ. Validation of reference genes for robust qRT-PCR gene expression analysis in the Rice Blast fungus Magnaporthe oryzae. PLOS ONE. 2016;11(8):e0160637 10.1371/journal.pone.0160637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwan YM, Meon S, Ho CL, Wong MY. Selection of reference genes for quantitative real-time PCR normalization in Ganoderma-infected oil palm (Elaeis guineensis) seedlings. Australasian Plant Pathology. 2016;45(3):261–8. [Google Scholar]
- 45.Warzybok A, Migocka M. Reliable reference genes for normalization of gene expression in cucumber grown under different nitrogen nutrition. PLoS One. 2013;8(9):e72887 10.1371/journal.pone.0072887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen C, Xie T, Ye S, Jensen AB, Eilenberg J. Selection of reference genes for expression analysis in the entomophthoralean fungus Pandora neoaphidis. Brazilian Journal of Microbiology. 2016;47(1):259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majumder AL, Chatterjee A, Ghosh Dastidar K, Majee M. Diversification and evolution of L‐myo‐inositol 1‐phosphate synthase 1. FEBS Lett. 2003;553(1–2):3–10. 10.1016/s0014-5793(03)00974-8 [DOI] [PubMed] [Google Scholar]
- 48.Michell RH. Inositol derivatives: evolution and functions. Nature Reviews Molecular Cell biology. 2008;9(2):151–61. 10.1038/nrm2334 [DOI] [PubMed] [Google Scholar]
- 49.Donahue JL, Alford SR, Torabinejad J, Kerwin RE, Nourbakhsh A, Ray WK, et al. The Arabidopsis thaliana Myo-inositol 1-phosphate synthase1 gene is required for Myo-inositol synthesis and suppression of cell death. The Plant Cell. 2010;22(3):888–903. 10.1105/tpc.109.071779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hazra A, Nandy P. Myo-inositol 1-phosphate synthase–the chosen path of evolution. BioTechnologia. 2016;97(2):95–108. [Google Scholar]
- 51.Zhai H, Wang F, Si Z, Huo J, Xing L, An Y, et al. A myo-inositol-1-phosphate synthase gene, IbMIPS1, enhances salt and drought tolerance and stem nematode resistance in transgenic sweet potato. Plant Biotechnol J. 2016;14(2):592–602. 10.1111/pbi.12402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C, Chen K, Su T, Zhang B, Li G, Pan J, et al. Myo-inositol-1-phosphate synthase (Ino-1) functions as a protection mechanism in Corynebacterium glutamicum under oxidative stress. MicrobiologyOpen. 2019;8(5):e00721 10.1002/mbo3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye C, Bandara WMS, Greenberg ML. Regulation of inositol metabolism is fine-tuned by inositol pyrophosphates in Saccharomyces cerevisiae. J Biol Chem. 2013;288(34):24898–908. 10.1074/jbc.M113.493353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cryptococcus Xue C. and beyond—inositol utilization and its implications for the emergence of fungal virulence. PLoS Path. 2012;8(9):e1002869 10.1371/journal.ppat.1002869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wenneker M, de Jong PF, Joosten NN, Goedhart PW, Thomma BPHJ. Development of a method for detection of latent European fruit tree canker (Neonectria ditissima) infection in apple and pear nurseries. Eur J Plant Pathol. 2017;148:631–5. [Google Scholar]
- 56.Bradshaw RE, Guo Y, Sim AD, Kabir MS, Chettri P, Ozturk IK, et al. Genome‐wide gene expression dynamics of the fungal pathogen Dothistroma septosporum throughout its infection cycle of the gymnosperm host Pinus radiata. Mol Plant Pathol. 2016;17(2):210–24. 10.1111/mpp.12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toruño TY, Stergiopoulos I, Coaker G. Plant-pathogen effectors: Cellular probes interfering with plant defenses in spatial and temporal manners. Annual Reivew of Phytopathology. 2016;54:419–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian J, Gao Y, Wáng Y, Wu Y, Wāng Y, Zhao Y, et al. Selection and evaluation of appropriate reference genes for RT-qPCR normalization of Volvariella volvacea gene expression under different conditions. BioMed Research International. 2018;2018:6125706 10.1155/2018/6125706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Mitchell HD, Markillie LM, Gaffrey MJ, Orr G, Schilling J. Reference genes for accurate normalization of gene expression in wood-decomposing fungi. Fungal Genetics and Biology. 2019;123:33–40. 10.1016/j.fgb.2018.11.005 [DOI] [PubMed] [Google Scholar]
- 60.Pathan EK, Ghormade V, Deshpande MV. Selection of reference genes for quantitative real-time RT-PCR assays in different morphological forms of dimorphic zygomycetous fungus Benjaminiella poitrasii. PLOS ONE. 2017;12(6):e0179454 10.1371/journal.pone.0179454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oliver RP, Solomon PS. New developments in pathogenicity and virulence of necrotrophs. Curr Opin Plant Biol. 2010;13(4):415–9. [PubMed] [Google Scholar]
- 62.van Kan JAL. Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 2006;11(5):247–53. 10.1016/j.tplants.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 63.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444(7117):323–9. 10.1038/nature05286 [DOI] [PubMed] [Google Scholar]
- 64.Stergiopoulos I, Collemare J, Mehrabi R, De Wit PJGM. Phytotoxic secondary metabolites and peptides produced by plant pathogenic Dothideomycete fungi. FEMS Microbiology Reviews. 2013;37(1):67–93. 10.1111/j.1574-6976.2012.00349.x [DOI] [PubMed] [Google Scholar]
- 65.Oliver RP, Friesen TL, Faris JD, Solomon PS. Stagonospora nodorum: from pathology to genomics and host resistance. Annu Rev Phytopathol. 2012;50:23–43. [DOI] [PubMed] [Google Scholar]
- 66.Liu Z, Friesen TL, Ling H, Meinhardt SW, Oliver RP, Rasmussen JB, et al. The Tsn1–ToxA interaction in the wheat–Stagonospora nodorum pathosystem parallels that of the wheat–tan spot system. Genome. 2006;49(10):1265–73. 10.1139/g06-088 [DOI] [PubMed] [Google Scholar]
- 67.Faris JD, Zhang Z, Lu H, Lu S, Reddy L, Cloutier S, et al. A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proceedings of the National Academy of Sciences. 2010;107(30):13544–9. 10.1073/pnas.1004090107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lyu X, Shen C, Fu Y, Xie J, Jiang D, Li G, et al. A small secreted virulence-related protein Is essential for the necrotrophic interactions of Sclerotinia sclerotiorum with Its host plants. PLoS Path. 2016;12(2):e1005435 10.1371/journal.ppat.1005435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu W, Wei W, Fu Y, Cheng J, Xie J, Li G, et al. A secretory protein of necrotrophic fungus Sclerotinia sclerotiorum that suppresses host resistance. PLoS One. 2013;8(1):e53901 10.1371/journal.pone.0053901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang L, Yang G, Ma M, Liu X, Li B, Xie J, et al. An effector of a necrotrophic fungal pathogen targets the calcium-sensing receptor in chloroplasts to inhibit host resistance. Mol Plant Pathol. 2020;21(5):686–701. 10.1111/mpp.12922 [DOI] [PMC free article] [PubMed] [Google Scholar]