

Abstract
Background:
Key obstacles in human iNKT cell translational research and immunotherapy include the lack of robust protocols for dependable expansion of human iNKT cells and the paucity of data on phenotypes in post-expanded cells.
Methods:
We delineate expansion methods utilizing IL-2, IL-7, and allogeneic feeder cells and anti-CD2/CD3/CD28 stimulation by which to dependably augment Th2 polarization and direct cytotoxicity of human peripheral blood CD3+Vα24+Vβ11+ iNKT cells.
Results:
Gene and protein expression profiling demonstrated augmented Th2 cytokine secretion (IL-4, IL-5, IL-13) in expanded iNKT cells stimulated with anti-CD2/CD3/CD28 antibodies. Cytotoxic effector molecules including granzyme B were increased in expanded iNKT cells after CD2/CD3/CD28 stimulation. Direct cytotoxicity assays using unstimulated expanded iNKT cell effectors revealed α-galactosyl ceramide (α-GalCer)-dependent killing of the T-ALL cell line Jurkat. Moreover, CD2/CD3/CD28 stimulation of expanded iNKT cells augmented their (α-GalCer-independent) killing of Jurkat cells. Co-culture of expanded iNKT cells with stimulated responder cells confirmed contact-dependent inhibition of activated CD4+ and CD8+ responder T cells.
Discussion:
These data establish a robust protocol to expand and novel pathways to enhance Th2 cytokine secretion and direct cytotoxicity in human iNKT cells, findings with direct implications for auto-immunity, vaccine augmentation and anti-infective immunity, cancer immunotherapy, and transplantation.
Keywords: NKT cells, iNKT cells, regulatory T cells, transplantation, immunotherapy, leukemia, co-stimulation, cytotoxicity
Graphical Abstract
Introduction
Natural killer T (NKT) cells are innate regulatory lymphocytes that also participate in tumor immune editing [1–9]. Unlike human adaptive regulatory T cells, invariant NKT (iNKT) cells have a conserved Vα24-Jα18-Vβ11 TCR [10], are not restricted by classic major histocompatibility complex (MHC) antigens, and can be activated by glycolipids such as α-galactosyl ceramide (α-GalCer) presented by CD1d [11]. iNKT cells can regulate both self and allogeneic tolerance [10]; their lack of canonical MHC restriction allows allo-regulation across histocompatibility barriers, as we first reported in murine transplants [12–14].
Preventing graft-versus-host disease (GVHD) while maintaining graft-versus-tumor (GVT) activity remains a “holy grail” of allogeneic hematopoietic cell transplantation (HCT). iNKT cells regulate GVHD while maintaining GVT [12–16]. We defined that iNKT cell-derived T-helper type 2 (Th2) cytokines facilitate this process indirectly, by maintaining myeloid populations that expand naturally occurring Foxp3+ Treg [13, 14]. This finding has since been confirmed by others [17–20]. Large-scale clinical studies have also demonstrated strong associations of graft iNKT cell content [21, 22] and post-HCT donor iNKT cell reconstitution [23, 24] with reduced GVHD [21], relapse [24], and mortality [21, 23, 24], supporting potential roles for therapeutically expanded human iNKT cells in immunotherapy [4, 7, 9, 25–29]. Notably, early CD4+ iNKT cell expansion, CD161 expression, and IL-4 and IFN-γ secretion capacity were identified as positive predictive biomarkers [22–24].
Two significant challenges in iNKT cell immunotherapeutics include 1) the paucity of circulating iNKT cells; and 2) poor understanding of key functions in therapeutically expanded (as opposed to freshly isolated) human iNKT cells. One of the most salient translational aspects of our current approach is the lack of up-front sorting of iNKT cells (allowing PBMC-derived APCs to present α-GalCer to expand iNKT cells through day 7), allowing a robust log-fold expansion and low failure rate of expansions as compared to other existing protocols [30–36]. Murine iNKT cells expand using α-GalCer with IL-2 and IL-15 (two cytokines sharing a receptor β-chain, CD122) [37, 38]; IL-15 also expands NK cells [39]. Although CD122 is well-documented on both CD4neg and CD4+ iNKT cell subsets, IL-7 receptor-α (CD127) is mainly expressed on the CD4+ subset and drives human iNKT cell differentiation [40, 41]. We chose rhIL-7-driven over conventional rhIL-15-based expansions with specific intent to optimize a CD4+ iNKT cell expansion. The rationale include: 1) human CD4+ iNKT cells are thymically produced [40] and many therapeutic applications are envisioned in settings of thymic dysfunction (e.g. post-transplant), 2) human CD4+ iNKT cells display greater propensity for Th2 cytokine secretion [42–44] which we and subsequently others have shown regulates systemic inflammation in murine models [10, 13, 16] and supports therapeutic application [45], and 3) the CD4+ subset is dominant in most cell therapy sources [40, 46].
We report highly reproducible and robust expansion of human peripheral blood iNKT cells, which we have functionally characterized post-expansion. Further, we delineate a single mechanism to simultaneously enhance the expression of Th2 cytokines alongside other cytokines (a phenotype associated with iNKT cell alloregulatory potential) and to enhance killing capacity of expanded iNKT cells as compared to unstimulated cells following therapeutic expansion.
Materials and Methods
iNKT cell expansion
iNKT cell expansion media was RPMI 1640® (Cellgro, Manassas, VA, USA) with 10 mM HEPES (HyClone, Logan, UT, USA), 0.02 mg/mL gentamicin (Life Technologies, Grand Island, NY, USA), and 10% human AB serum (CellGro®, Manassas, VA, USA). Peripheral blood apheresis units were obtained from de-identified blood donors at St. Jude Blood Donor Center, under St. Jude IRB-exempted protocols. PBMCs were isolated by Ficoll-Paque Plus® density-gradient (GE Healthcare, Piscataway, NJ, USA). 2 × 108 PBMCs at 1 × 106 cells/mL were stimulated with 100 ng/mL α-GalCer (KRN7000; Funakoshi, Tokyo, Japan), 100 U/mL recombinant human (rh)IL-2 (Proleukin®, Prometheus, San Diego, CA, USA) and 0.4 ng/mL rhIL-7 (Sigma-Aldrich, St. Louis, MO, USA or R&D Systems, Minneapolis, MN, USA) (or in some expansions 0.4 ng/mL IL-15, R&D Systems, Minneapolis, MN, USA) for 7 days and CD3+Vα24+ iNKT cells sorted using a BD FACSAria-II® (BD Instruments, Santa Clara, CA, USA) to > 95% purity (cell yields > 105) or > 90% purity (cell yields between 104 - 105).
Sorted CD3+Vα24+ iNKT cells were cultured at 1 × 103 - 5 × 104 cells/mL in T25 or T75 flasks (MidSci, Valley Park, MO, USA) at a 1:50 ratio of iNKT cells to allogeneic irradiated PBMCs (5000 cGy using a 137Cs source) and stimulated with 50 ng/mL purified NA/LE anti-CD3 (BD Pharmingen, San Jose, CA, USA), adding 100 U/mL rhIL-2 and 0.4 ng/mL or 4 ng/mL rhIL-7 on day 8. From day 7 sort, all expansions were of maximum 5 × 106 iNKT cells; expansions with sort yields greater than 5 × 106 cells were extrapolated from results for 5 × 106 iNKT cells. On day 14, cells were replated in fresh media containing 100 U/mL rhIL-2 and 0.4 ng/mL or 4 ng/mL rhIL-7 at 1 × 103 - 5 × 104 cells/mL. The expansion from day 14 was limited to 20 × 106 cells. The expansion factor at day 7 or day 21 for CD4+, CD8+, and DN iNKT cell subsets was calculated by dividing the absolute number of cells on that day by the absolute number at day 0 [22].
Gene expression profiling
Day 0 CD3+Vα24+ iNKT cells were sorted to > 90% purity from St. Jude Blood Center apheresis products. Day 21 CD3+Vα24+ and CD3+Vα24neg cells were sorted to > 94% purity from expansions and cultured 24 hours with anti-CD2/CD3/CD28 loaded or unloaded beads (T cell Activation/Expansion Kit, Miltenyi Biotec, Auburn, CA, USA, according to manufacturer’s instructions) and RNA prepared using the Qiagen RNeasy Micro® kit (Qiagen Inc., Valencia CA, USA). RNA quality was measured with either the Bioanalyzer (Agilent) or the LabChip GX (Perkin Elmer). All samples had a RIN > 8.8.
Total RNA was converted into cDNA using the NuGEN WTA Pico v2® system (NuGEN Technologies Inc., San Carlos, CA, USA), fragmented and biotin-labeled using the Encore® Biotin module v2 (NuGEN), and hybridized overnight at 45°C to an Affymetrix GeneChip PrimeView® human gene expression array (Affymetrix Inc., Santa Clara, CA, USA). Microarrays were scanned using an Affymetrix GeneChip 3000 7G instrument (Affymetrix Inc.), and signals summarized using the RMA algorithm [47]. Differential expression between groups was identified using Gene Set Enrichment Analysis [48], with a false discovery threshold < 0.05. Canonical pathways were obtained from the Molecular Signature Data Base (Broad Institute), Ingenuity Pathways (Ingenuity Inc., now Qiagen) and Metacore (GeneGO, Inc., now Thompson Reuters). To estimate the cell type proportion of the day 21 CD3+Vα24neg samples from the bulk gene expression profiles, we used CIBERSORT [49] with the reference gene signature matrix LM22 (purified cell) containing of 547 genes that accurately distinguish 22 mature human hematopoietic populations. Since the gene signature matrix (LM22) requires HUGO gene symbols as gene identifiers, we aggregated the Microarray gene expression data by calculating the mean expression for each gene name after converting the probe ID to gene symbols. A Pearson’s correlation coefficient was generated comparing each Microarray expression matrix to the estimated cell mixture from CIBERSORT.
Flow cytometry
Staining reagents included PE-Cy7 anti-Vα24Jα18 TCR (clone 6B11), eFluor®450 anti-CD8a (SK1), AF700 anti-CD3 (OKT3), PerCp-Cy5.5 anti IFNγ (4S.B3), AF488 anti-TNFα (Mab11), PE-Cy7 anti-IL-13 (85BRD), eFluor660 anti-IL-10 (JES3–9D7), PE-Cy7 anti-CD127 (eBioRDR5), BB515 anti-CCR5 (4B4) (all from eBioscience, San Diego, CA, USA); BV421 anti-Va24 (6B11), BV711 anti-CD8a (SK1), APC anti-CD3 (HIT3a), APC-Cy7 anti-CD4 (RPA-T4), FITC anti-CD56 (MEM-188), PerCp-Cy5.5 anti-CD161 (HP-3G10), both APC-Fire750 and PE anti-CD45RO (UCHL1), both BV605 and PerCp-Cy5.5 anti-CD45RA (HI100), BV605 anti-CD127 (A019D5), AF488 anti-perforin (dG9), PerCp-Cy5.5 anti-CCR7 (G043H7), PE anti-FasL (NOK-1), APC-Cy7 anti-PD-1 (EH12.2H7), BV605 anti-CD25 (BC96), APC anti-CD215 (JM7A4), PE anti-TRAIL (RIK-2), APC-Cy7 anti-CXCR3 (G025H7), PE-Dazzle594 anti-CCR4 (L291H4), APC anti-CCR2 (K036C2), BV605 anti-CCR3 (5E8), PE-Cy7 anti-CXCR4 (12G5), BV605 anti-IL-2 (MQ1–17H12), PE-Dazzle594 anti-IL-4 (MP4–25D2) (all from Biolegend, San Diego, CA, USA); PE anti-Vβ11 TCR (C21) and FITC anti-CD62L (DREG56) (Beckman Coulter, Brea, CA, USA); BUV395 anti-CD4 (RPA-T4) and V450 anti-granzyme B (GB11, BD Pharmingen, San Diego, CA, USA); PE and APV-vio770 anti-Vβ11 (REA559; Miltenyi, Auburn, CA, USA). To exclude dead cells, Ghost 510® reagent (Tonbo, San Diego, CA, USA) was used in analyses and 4’, 6-diamidino-2-phenylindole (DAPI) (Santa Cruz Technologies, Santa Cruz, CA) in sorting. Vβ TcR spectratyping was by IOTest Beta Mark TcR Vβ Repertoire Kit (Beckman Coulter).
For intracellular staining, following 24-hour stimulations, cells were stained with Ghost 510® (30 minutes, 4°C), washed, fixed and permeabilized using FoxP3 Fixation/Permeabilization Kit (Biolegend), and stained with antibodies (30 minutes, 20°C). Data were acquired by 4-laser LSR® Fortessa (BD Instruments) and analyzed with FlowJo® v10 (TreeStar, Ashland, OR, USA). A threshold of 100 gated cells was required to report.
Proliferation and apoptosis
10uM BrdU was added to the expansions on day 7 and day 21 for 8 hrs. BrdU incorporation was quantified by flow cytometry. The following surface antibodies were stained in Annexin Binding Buffer (BD Biosciences) with BUV395 anti-CD4 (RPA-T4), BV711 anti-CD8α (SK1), BV421 anti-Vα24Jα18 TCR (6B11), and PE anti-Vβ11 TCR (REA559). Cells were washed, fixed, and permeabilized using BrdU staining buffer (Invitrogen), and DNA was degraded with 100U/mL/sample DNase (Qiagen) for 30 minutes at 37°C. For nuclear staining, AF647 anti-Ki67 (11F6) and AF488 anti-BrdU (3D4; both Biolegend) were incubated for 30 minutes at 4°C. Data were acquired by 4-laser LSR® Fortessa (BD Instruments) and analyzed with FlowJo® v10 (TreeStar). To study subset death versus expansion during iNKT cell expansion, BrdU was plotted against both Ki67 and Ghost 510® live-dead cell dye (Tonbo).
HLA analysis
Day 21 iNKT cells and Vα24-N cells were re-sorted and cryopreserved. Corresponding source PBMCs and day 7 PBMC feeders were cryopreserved. DNA was extracted using Illumina MiSeq® paired-end sequencing-by-synthesis [50]. Briefly, HLA class 1 exons were amplified (9 reactions), pooled, conjugated to adaptors, barcoded, and sequenced. Data were uploaded to BaseSpace® (Illumina, San Diego, CA) and FASTQ files were transferred to the GeMS-Analyzer® software (Scisco, Seattle, WA) to determine HLA type. The typing report was edited with GeMS-HLA® (Scisco).
Cytokine and chemokine profiling
Day 21 expanded iNKT cells and Vα24-N cells (4 × 105 /well in a 96-well plate) were sorted and stimulated 24 hours with anti-CD2/CD3/CD28-loaded or unloaded beads (Miltenyi Biotec). Supernatant cytokines were assayed using Milliplex MAP® 9-plex kit (Millipore, Billerica, MA, USA) and Multiplex-xMap® (Millipore). Samples were assayed in duplicate or triplicate.
Cytotoxicity
Sorted, cryopreserved day 21 CD3+Vα24+ iNKT cells were thawed, washed, and stimulated for 24 hours with anti-CD/CD3/CD28 antibody loaded beads versus control beads or media alone. K562 and Jurkat targets were labeled with 51Cr (0.1 mCi) (Perkin Elmer, Waltham, MA), washed, loaded with 100 ng/mL α-GalCer, and co-cultured with either α-GalCer (50 ng/mL) or media. Stimulated day 21 iNKT cell effectors (E) were incubated with targets (T) cells at E:T ratios between 8:1 and 0.0625:1 for 18 hours at 37°C. Culture supernatant (100 mL) was transferred to Luma® plates (ThermoFisher) and β-emission quantitated using a liquid scintillation counter (HIDEX Sense®, Turku, Finland). Assays were in duplicate or triplicate. Percent cytotoxicity was calculated from maximum and spontaneous release as (CPMexperimental –CPMspontaneous) / (CPMmaximum – CPMspontaneous) x 100.
Mixed Leukocyte Reaction (MLR)
Unsorted PBMC responders cryopreserved from the original iNKT cell expansion unit were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) and cocultured for 5 days with anti-CD2/CD3/CD28-loaded or unloaded beads at a bead : cell ratio of 1:2. Day 21 expanded iNKT cells at responder: iNKT cell ratio of 1:1 were added in cis- or trans-culture in a Transwell plate (1.0mm pore, Corning). Following staining and FACS analysis, data were analyzed using FCS Express (Ne Novo® v6) proliferation model. Average divisions (Avg Div) was defined as log2 (Division Index). Avg Div across assays was standardized as [Avg Div with iNKT cells (1:1) / Avg Div with anti-CD2/CD3/CD28 stimulation alone (1:0)]. Percent divided was calculated as [(percent divided with iNKT cells) (percent divided with anti-CD2CD3/CD28 stimulation alone)] x 100.
Statistical analyses
Statistical significance was analyzed between two groups by 2-tailed Mann-Whitney U, and 3 or more groups by Kruskal-Wallis with Dunn’s multiple comparisons using Prism® 6.0 (GraphPad, Inc., La Jolla, CA). Cell proliferation, cell death, cytokine concentrations (Luminex), cytotoxicity, and various receptor expressions (Figures 5A and 6B and Supplemental Figures 1C and 4C) comparisons were by 2-way ANOVA with Dunnett’s multiple comparisons (Prism® 7.0, GraphPad) with a Tukey correction for multiple comparisons. MLR comparisons were by unpaired t test with Welch’s correction. P < 0.05 was considered significant.
Figure 5: Expanded iNKT cells and Vα24-N cells share chemokine, chemokine receptor, and cytokine profiles after anti-CD2/CD3/CD28 stimulation, and the dominant cytokine-secreting iNKT cell subset is CD4+.

(A) Median and IQR ± range of the CCR4, CCR5, CCR7, CXCR3, and CXCR4 fluorescence intensity (FI) from day 0 iNKT cells (white), day 21 iNKT cells (red) and day 21 Vα24-N cells (blue) (N = 4) stimulated 24 hours with unloaded bead control (U), or anti-CD2/CD3/CD28-loaded beads (C).
(B) Mean cytokine and chemokine concentrations measured by Luminex® in culture supernatants from day 21 sorted iNKT cells (red circles) and day 21 sorted Vα24-N cells (blue triangles) (N = 6) stimulated 24 hours with media control (Stim M), unloaded bead control (Stim U), or anti-CD2/CD3/CD28-loaded beads (Stim C). Cytokine levels were compared to either media or unloaded bead controls.
(C) Representative dot plot overlays of day 21 iNKT cells stimulated with unloaded beads as a control (U; in blue) and anti-CD2/CD3/CD28-loaded beads (C; in dark red) showing intracellular cytokine expression for IL-2, IFN-γ, TNF-α, IL-4, IL-13, and IL-10 (left panel). Mean percentage of CD4/DN/CD8 subsets within the cytokine+ iNKT cells are shown in the right panel (N = 4). P < 0.05 is represented by *.
Figure 6: CD2/CD3/CD28 stimulation significantly enhances granzyme B expression and expanded iNKT cell direct cytotoxicity.

(A) mRNA expression of key cytotoxicity genes. Heat-map of log2 signal change relative to mean signal in day 0 iNKT cells, day 21 iNKT cells, and day 21 Vα24-N cells stimulated for 24 hours with media control (M), unloaded bead control (U), or anti-CD2/CD3/CD28 antibody-loaded beads (C). Red, increased relative expression; blue, decreased relative expression.
(B) Representative flow plots (top panel) showing perforin and granzyme B expression compared to isotype (black). Representative histograms and median and IQR ± range FI for granzyme B (middle panel) and perforin (lower panel) in day 21 expanded cells (N = 8) following 24-hour stimulation with media control (M, grey), unloaded bead control (U, blue), anti-CD2/CD3/CD28 antibody-loaded beads (C, red), or rhIL-2 control (IL-2, purple) compared to isotype control (pink dashed line).
(C) Representative histograms (left) and median and IQR ± range FI (right) for TRAIL (upper panels) and FasL/CD178 FI (lower panels) in day 21 expanded cells (N = 4) following a 24-hour stimulation with unloaded bead control (U, blue), anti-CD2/CD3/CD28 antibody-loaded beads (C, red) compared to isotype control (pink dashed line).
(D) Representative histograms of CD1d expression on K562 and Jurkat cells. Red, Isotype control; blue, anti-CD1d.
(E) Mean ± SEM percentage cytotoxicity in 51Cr release assay (N = 4) against K562 (upper panels) and Jurkat (lower panels) at specified effector : target (E:T) ratios for expanded and sorted day 21 iNKT cells stimulated 24 hours with media control (grey), rhIL-2 (purple) (left panels), unloaded bead control (blue), or anti-CD2/CD3/CD28 antibody-loaded beads (red) (right panels). Unloaded (open circles) or α-GalCer-loaded (triangles) targets were compared in each assay. Individual significant differences in treatment versus vehicle at the same E:T ratio are as follows: α) media + α-GalCer; β) IL-2 + α-GalCer; χ) anti-CD2/CD3/CD28; δ) unloaded beads + α-GalCer; ε) anti-CD2/CD3/CD28 + α-GalCer.
Results
iNKT cells from random healthy blood donors can be robustly and reproducibly expanded using IL-2 and IL-7 in tandem.
The expansion of iNKT cells was quantified at day 0, 7, 14 and 21 to determine if iNKT cells could be consistently expanded to clinically therapeutic levels. Based on a literature search (Table 1), initial experiments assessed whether greater yields of iNKT cells could be achieved using an IL-2/IL-7 or an IL-2/IL-15 based protocol (N = 4). Total iNKT cell yields at day 7 did not differ between IL-7 and IL-15 (Supplemental Figure 1A), and both methods appear to preferentially expand CD4+ iNKT cells within 7 days (Supplemental Figure 1B). However, we also investigated the expression of a number of surface markers (Supplemental Figure 1C). CD56, CD161, CD45RA, CD45RO, and chemokine receptors CD62L, CXCR3, CXCR4, CCR2, CCR3, CCR4, and CCR7 did not show a significant difference in expression with either strategy. Among cytokine receptors, both IL-7 and IL-15 significantly augmented iNKT cell CD25 (IL-2 receptor α) and CD215 (IL-15 receptor α) expression as compared to day 0 (non-expanded) iNKT cells. Considering that IL-2/IL-7-based expansion induced a significantly greater increase in CD215 as compared to IL-2/IL-15 and that CD215 is an important receptor in iNKT cell development and homeostasis, an IL-2/IL-7 expansion protocol was chosen for further study. A low dose IL-7 protocol (0.4ng/mL) was developed that expanded iNKT cells between day 0 and day 7 12-fold (median (IQR): day 0: 1.8×105 (1.7×104 - 6.0×105) vs day 7: 2.1×106 (1.5×105 - 1.2×107); N = 16, P < 0.5) (Supplemental Figure 2A, left panel). However, iNKT cells sort purified at day 7 failed to significantly expand through day 21 using the low dose IL-7 protocol [median (IQR): day 7 post sort: 1.0×106 (7.8×104 - 4.4×106); day 14: 7.0×105 (7.2×103 - 4.9×106); day 21: 8.3×105 (1.8×104 - 2.1×107); N = 16] (Supplemental Figure 2A, right panel). One obstacle in human iNKT cell immunotherapeutics is the wide variability in starting frequency of iNKT cells and frequent non-expanding units (expansion failures) [51, 52]. Stratifying expansion data by day 7 iNKT cell sort yield into 4 categories (104-105, N = 4; 105-106, N = 4; 106-107, N = 5; >107, N = 3) revealed that expansion failure after day 7 occurred irrespective of iNKT cell yield at day 7 sort (Supplemental Figure 2B). Between day 0 and day 21, the percentage of CD4+ iNKT cells increased at the expense of the DN subset, while CD8+ iNKT cells remained rare throughout expansion (Supplemental Figure 2C, left panel), and only the CD4+ iNKT cell subset was significantly expanded over day 0 at day 21 (Supplemental Figure 2C, right panel). The cumulative results suggested that, after iNKT cell isolation at day 7, additional cytokine support is required to support dependable iNKT cell expansion. The significant decrease in expression of IL-7Rα (CD127) following 7 days of IL-7 exposure supported a mechanistic explanation for the requirement for increased IL-7 concentrations after day 7 of expansions. Based upon this preliminary data, modifications were made to utilize a 10-fold higher concentration of IL-7 (4 ng/mL) post-sort.
Table 1.
INKT cell expansion protocols
| First Author | aGalCer/PHA | iNKT cell isolation | Cytokine | Feeders/APC |
|---|---|---|---|---|
| Watarai | aGalCer | No pre-enrichment | IL-2 | Auto APC (GM-CSF + IL-4) |
| Schmid | KRN7000,PBS44, PBS57 | No pre-enrichment | IL-2 | Auto PBMC |
| Moreira-Teixeira | aGalCer or OCH | No pre-enrichment | TGFb, +/− Rapamycin | Auto PBMC |
| Nishi | aGalCer | No pre-enrichment | IL-2, IL-15 and IL-7 | Auto PBMC |
| Van Der Vliet | aGalCer | No pre-enrichment | IL-12 or IL-7 | Auto PBMC |
| Van der Vliet | aGalCer | No pre-enrichment or Mag bead | IL-15 or IL-7 +dexamethasone | Auto PBMC or Auto mo-DC |
| van der Vliet | aGalCer | Monocyte depletion | IL-7 and/or IL-15 | B cells or EBV transformed B-LCLs or moDC (IL4+GM-CSF) |
| Li | aGalCer | Neg fraction CD14-Mag Beads | IL-2 | CD14+ fraction (GM-CSF + IL-4) |
| East | aGalCer | CD3+CD161+Mag Beads | IL-2 | Artificial APC |
| Exley | aGalCer | FACS sorting | IL-2 | Auto Irradiated PBMC |
| O’Reilly | PHA | Mag beads + sorting | IL-2 | Allo PBMC |
| Zeng | PHA | Mag beads + sorting | IL-2 | Plate bound anti-CD3 or Allo feeders |
| Takahashi | aGalCer | Mag beads | IL-2 | Auto Mo-DC (IL-4 GM-CSF) |
| Tian | aGalCer | Microbeads | IL-2 | Auto PBMC, transduced K562 line |
| Song | aGalCer | Mag beads | IL-2 | APC (GM-CSF +IL-4 + LPS) |
| Metelitsa | aGalCer | Mag beads | IL-2 | Auto Vα24-neg fraction |
| Huijts | aGalCer | Mag beads | IL-2 | Mature Mo-DC |
| Trujillo-Ocampo | aGalCer | Mag beads | IL-2 | Allo monocyte derived DCs |
| Rogers | aGalCer | Mag beads at Day 12 | IL-2 | Auto PBMC |
| Mavers | aGalCer | Mag beads | IL-2 or IL-2 + IL-15 | Auto PBMC |
| Moreno | aGalCer | Mag beads | IL-2, IL-7, IL-15 | Auto PBMC |
| Moreno | aGalCer | Mag beads | IL-2, rhIL-7, IL-15 | Mature M3CD1d-DC |
| Motohashi | aGalCer | No pre-enrichment | IL-2 | Auto PBMC |
| Kunii | aGalCer | No pre-enrichment | IL-2 | Auto PBMC |
| Yamasaki | aGalCer | No pre-enrichment | IL-2 | Auto PBMC |
| Exley | None | CliniMACS | IL-2 | anti-CD3 coated 24 well plates + auto PBMC |
Between day 0 and day 7, iNKT cell expansion was 23-fold (median using absolute number yields (IQR), day 0: 1.5 × 105 (7.1 × 104 – 3.1×105) vs day 7: 3.5 × 106 (1.5 × 106 – 7.9 × 106); N = 47, P < 0.05) (Figure 1A, upper left panel). The expansion was reflected in the increased percentage of CD3+Vα24+ iNKT cells on day 7 vs day 0 [median (IQR), day 0: 0.2% (0.1 – 0.3%) vs day 7: 5.1% (2.2 – 8.7%); N = 47] (Figure 1A, upper right panels). iNKT cells sorted at day 7 demonstrated significant subsequent expansion, with median 7.5-fold expansion between days 7 and 14 [median (IQR) absolute number, day 7 post-sort: 1.6 × 106 (4.1 × 105 – 4.0 × 106); day 14: 1.2 × 107 (1.5 × 106 – 3.8 × 107), N = 47, P < 0.05]. Median 4-fold expansion was achieved from days 14 to 21 (Figure 1A, lower left panel) [median (IQR), day 21: 4.8 × 107 (8.5 × 106 – 2.2 × 108); N = 47, P < 0.05]. Day 7 CD3+Vα24+ post-sort purity was > 95% when day 7 sort yield >105 (Figure 1A, lower right panels). Median fold-increase in iNKT cells between day 0 and day 21 was 320-fold. Absolute number CD3+Vα24+ iNKT cells at each time point is given in Figure 1B (left panel). To assess expansion failures, we stratified day 21 yield by day 7 iNKT cell sort yield in 4 categories (104-105, 105-106, 106-107, >107) (Figure 1B, right panel). Median fold-expansion between day 0 and day 21 correlated with the number of iNKT cells recovered at day 7 sort (fold expansion: 104-105: 0.30, N = 4; 105-106: 132, N = 16; 106-107: 457, N = 21; >107: 750, N = 6). Notably, fold-expansion between days 7 and 21 was not significantly different between categories (fold expansion: 105-106: 28; 106-107: 42; >107: 23) in 89% (N = 42) of expansions. iNKT cells failed to significantly expand between days 7 and 21 in 5 expansions (11%) (day 7 category 104-105: N = 4 and 105-106: N = 1). The data suggest that this approach robustly expands iNKT cells as long as they expand from 2 × 108 PBMC sufficient to allow sorting of at least 105 iNKT cells at day 7. These data provide a useful framework to predict how well iNKT cells will expand early in the procedure when aiming to generate large quantities for therapeutic applications.
Figure 1: Ex vivo expansion results in CD3+Vα24+Vβ11+, predominantly CD4+ iNKT cells.

(A) Median and IQR ± range absolute number CD3+Vα24+ iNKT cells at day 0 and day 7 (N = 47) (left upper panel) and representative flow plots of CD3 and Vα24 expression (right upper panel). Median and IQR ± range absolute number CD3+Vα24+ iNKT cells after day 7 sort, including days 14, and 21 (N = 47) (left lower panel) with representative flow plots showing CD3 and Vα24 expression pre- and post-sort on gated CD3+ cells on day 7 (right lower panel).
(B) Left panel, median absolute number CD3+Vα24+ iNKT cells at day 7, day 14, and day 21 stratified by day 7 sort yield categories 104-105 (orange, N = 4), 105-106 (yellow, N = 16), 106-107 (cyan, N =21), and >107 (magenta, N = 6). Right panel, sub analysis of expansion data by day 7 sort categories showing median and IQR ± range absolute number CD3+Vα24+ iNKT cells at specified time points in the expansion protocol (S, day 7 post-sort).
(C) Median and IQR ± range percentage (left panel) with representative flow plots of CD4 and CD8 expression in gated CD3+Vα24+ cells on days 0, 7, and 21 (N = 36). Median and IQR ± range absolute number of CD4+, DN, and CD8+ CD3+Vα24+ iNKT cells on days 0, 7, and 21 (left panel; N = 36). P < 0.05 is represented by *.
Expanded iNKT cells are predominantly CD4+ with memory phenotype.
Changes to the expanding iNKT cell phenotype were monitored at each timepoint. Day 0 iNKT cells were Vβ11+ [median (IQR): 99.6% (93.5% - 100%)] (data not shown) and either CD4+ [CD4+CD8neg, median (IQR): 26.2% (17.7% - 50.6%)] or double negative [DN, CD4negCD8neg, median (IQR): 64.1% (41.4% - 74.7%)], with a minimal CD8+ fraction [CD4negCD8+, median (IQR): 4.9% (2.5% - 11.5%] (Figure 1C, lower left panel). The CD4+ iNKT cell fraction significantly expanded by day 21 [median (IQR): 92.1% (67.1% - 95.8%), P < 0.05], with a progressive decrease in DN iNKT cells [median (IQR): 4.1% (1.7% - 18.9%), P < 0.05], and CD8+ iNKT cells [median (IQR): 2.8% (0.6% - 6.1%)]. Though all iNKT cell subsets expanded well between day 0 and day 21, the CD4+ subset showed the most significant expansion between day 7 and day 21, suggesting that this high dose IL-7 protocol supports proliferation of the CD4+ iNKT cell subset (Figure 1C, right panel).
Prior publications suggest that the ex vivo expansion factor of peripheral blood stem cell (PBSC)-derived CD4neg iNKT cells associates with increased co-expansion of CD4+ iNKT cells [22]. To explore this association, we stratified expansions by day 0 DN iNKT cell content (<50%, N = 10; >50%, N = 26) (Supplemental Figure 3A). The presence of >50% DN day 0 iNKT cell content associated with significant expansion of all subsets from days 7 to 21 (Supplemental Figure 3A, right panel).
The NK cell maturation marker CD161 (NKR-P1A) is expressed on iNKT cells early in thymic development and maintained in adult peripheral blood [40, 42, 51–53]. Day 21 expanded iNKT cells were CD161+CD56neg (Supplemental Figure 3B, left and middle panels). Unexpanded iNKT cells at day 0 express the IL-7Rα chain (CD127), which is downregulated in response to IL-7 based expansion, supporting the need for increased IL-7 levels in culture to maintain expansion (Supplemental Figure 3B, right panel). In agreement with previous reports [40, 46, 51–54], all subsets of unexpanded (day 0) and day 21 expanded iNKT cells exhibited a memory phenotype (CD45RO+CD45RAneg) (Supplemental Figure 3C).
CD3+Vα24neg cells are memory phenotype conventional T cells deriving from the primary expansion unit which co-expand with iNKT cells and share post-activation upregulated gene expression with expanded iNKT cells.
A CD3+Vα24neg (Vα24-N) cell population (Figure 2A, left panel) was consistently expanded from day 7 sorted CD3+Vα24+ iNKT cells [median (IQR), day 14: 3.8 × 106 (4.5 × 105 – 1.2 ×107) vs day 21: 3.8 × 107 (7.6 × 106 – 1.3×108); N = 47, P < 0.05] (Figure 2A, right panel). Vα24-N cells were Vβ11low [median (IQR): 3.4% (0.4% - 7.9%) (data not shown)], CD4+ [52% (35.7% – 74.1%)] or CD8+ [44% (20.1% - 57.5%)] (Figure 2B), and CD161negCD56neg (Figure 2C, upper panels). Like day 21 iNKT cells, day 21 Vα24-N cells were CD127low (Figure 2C, upper left panel) and CD45RO+CD45RAneg (Figure 2C, lower panels).
Figure 2: A CD45RO+ CD3+Vα24negVβ11neg cell fraction expands in parallel with iNKT cells.

(A) Representative flow plot of Vα24 and CD3 expression delineating the CD3+Vα24neg population at day 21 (left panel, red arrow). Median and IQR ± range absolute number CD3+Vα24neg cells at days 7, 14 and 21 (right panel, N = 47).
(B) Median and IQR ± range percentage (left panel) and representative flow plots of CD4 and CD8 expression (right panel) in gated CD3+Vα24neg cells at day 21 (N = 47).
(C) Median and IQR ± range and representative histogram of the CD161 fluorescence intensity (FI) (top left panel), CD56 FI (top middle panel), CD127 FI (top right panel), CD45RO FI (bottom left panel), and CD45RA FI (bottom right panel) in gated CD3+Vα24negCD4+CD8neg cells (CD4+ Vα24-N, yellow), CD3+Vα24negCD4negCD8neg cells (DN Vα24-N, blue), and CD3+Vα24negCD4negCD8+ cells (CD8+ Vα24-N, red) at day 21 (N = 47). Isotype control is shown in white or black line. P < 0.05 is represented by *.
The source and identity of Vα24-N cells were investigated. Class I HLA typing at 3 loci (HLA-A, B, and C) of sorted day 21 Vα24-N cells confirmed that these expanded Vα24-N cells derive from the iNKT cell source unit and not from allogeneic PBMC feeders (Supplemental Figure 4A). Vβ spectratyping revealed Vα24-N cells possess diverse T cell receptors (Supplemental Figure 4B) whereas expanded iNKT cells possess the canonical invariant Vβ11 chain (Supplemental Figure 4C). CIBERSORT analysis of overall gene expression profiles for sorted Vα24-N cells, both before and after activation with anti-CD2/CD3/CD28 loaded beads, support that these cells represent a mixed population of memory CD4 and CD8 T cells and a fraction of γδ−T cells (Supplemental Figure 4D). Pearson correlations of each sorted mixture with the Microarray mixture were all above 0.68 indicating the genetic signatures used from the reference gene signature matrix LM22 (547 genes distinguishing 22 mature human hematopoietic populations) matched up with the signatures found in each of the Vα24-N RNA mixtures/samples.
The expansion method preferentially expands CD4+ iNKT cells through enhanced proliferation.
We applied 8-hr BrdU incorporation assays at day 7 pre-sort and again at day 21 of the iNKT cell expansion procedure in combination with Ki-67 and Ghost 510® live-dead marker to examine subset proliferation versus cell death. On day 7 pre-sort, there was a significant proliferation of iNKT cells as compared to Vα24-N cells (Q2 and Q3 as compared to Q1 in Figure 3A, top right panel), further supporting that the Vα24-N cells expand post-sort under the influence of expansion support. On day 21, all subsets of iNKT cells and Vα24-N cells proliferate at a similar rate (Q2 and Q3 as compared to Q1 in Figure 3A, lower right panel). Notably, the Q2 but not the Q3 fraction was dominant, indicating prior but not ongoing proliferation at the time of assay. There was no significant difference in cell death between any subset of iNKT cells at either day 7 (Figure 3B, top panels) or day 21 (Figure 3B, bottom panels).
Figure 3: iNKT cell subset proliferation and minimized cell death during expansion.

(A) Representative flow plots of total iNKT cells (day 7 pre-sort and day 21) demonstrating the quadrant set for quantification of proliferation using both Ki67 and BrdU (left panel). Median and IQR ± range percentage (right panels) from all quadrants (Q1–4) are shown for day 7 pre-sort cells (upper right panel) and day 21 expanded cells (lower right panel) from gated CD3+Vα24+ total iNKT cells (grey), CD4+ iNKT cells (yellow), DN iNKT cells (blue), CD8+ iNKT cells (red), and CD3+ Vα24-N cells (white) (N = 4).
(B) Representative Ghost 510 histogram overlays of total iNKT cells (red) with total Vα24-N cells (blue) for both day 7 pre-sort and day 21 of expansion (left panel) with the gate used for quantifications shown in pink. Median and IQR ± range percentage for Ghost 510 positivity for total iNKT cells (grey), CD4+ iNKT cells (yellow), DN iNKT cells (blue), CD8+ iNKT cells (red), and Vα24-N cells (white) (right panel; N = 4).
Expanded iNKT cells and CD3+Vα24neg cells express similar chemokine receptor and developmental gene expression profiles.
To assess the changes induced in iNKT cells via expansion and the role of TCR-downstream activation in their phenotype, we began by comparing sorted day 21 iNKT cells and Vα24-N cells to freshly isolated iNKT cells (day 0 iNKT cells) (Figure 4A) by mRNA profiling, with and without CD2/CD3/CD28 stimulation. Principal component analysis demonstrated clustering in three distinct groups: 1) freshly isolated day 0 iNKT cells clustered together, distinct from expanded iNKT cells and Vα24-N cells; 2) CD2/CD3/CD28-stimulated day 21 iNKT cells and Vα24-N cells clustered closely together; but were distinct from 3) unstimulated day 21 iNKT cells and Vα24-N cells (Figure 4B). Profiling 19,892 genes, CD2/CD3/CD28 stimulation induced >2 log2 fold change in gene expression in both day 21 iNKT cells or Vα24-N cells (302 genes, orange) with a high correlation between iNKT cells and Vα24-N cells (r2 = 0.810) not seen with control stimuli (r2 = 0.005) (Figure 4C).
Figure 4: Expanded iNKT cells and Vα24-N cells share gene expression profiles.

(A) Representative gates used to sort day 0 CD3+Vα24+ iNKT cells (left panel) and day 21 (right panel) CD3+Vα24+ iNKT cell and CD3+Vα24neg Vα24-N cell populations.
(B) Principal component analysis (PCA) from gene expression of sorted day 0 iNKT cells (squares) (N = 3 per condition), day 21 iNKT cells (circles) (N = 4 per condition), and Vα24-N cells (triangles) (N = 4 per condition) stimulated 24 hours with media control (Stim M, yellow), unloaded bead control (Stim U, blue), or anti-CD2/CD3/CD28-loaded beads (Stim C, red).
(C) Mean fold gene upregulation (log2) over mean background in day 21 iNKT cells vs Vα24-N cells given Stim C vs Stim U (C/U, day 21) (right panel) and Stim U vs Stim M (U/M, day 21) (left panel). Stimulations are as defined in B. Correlation coefficient (r2) is indicated above each panel.
(D) Normalized enrichment score (NES) for pathway activity identified by gene set enrichment analysis (GSEA) in sorted day 0 iNKT cells given Stim C vs Stim U (iNKT C/U, day 0) vs day21 iNKT cells given Stim C vs Stim U (iNKT C/U, day 21). Stimulations are as defined in B. Blue, pathways significantly up- or down-regulated at day 21 but not at day 0; orange, pathways significantly up- or down-regulated at day 0 but not at day 21; magenta, pathways significantly up- or down-regulated at both day 0 and day 21; grey, pathways not significantly altered at day 0 or day 21.; and green, conventional T cell activation pathways,
(E) Biocarta NKT pathway gene expression profile. Heat map of log2 signal change in day 0 iNKT cells (N = 3), day 21 iNKT cells (N = 4), and Vα24-N cells (N = 4) stimulated under conditions identical to those in B. Red, increased relative expression; blue, decreased relative expression.
In unexpanded (day 0) iNKT cells, stimulated gene expression profiles varied significantly across donors indicating significant variability in day 0 Th1/Th2 polarization (Supplemental Figure 5A, left panel). Among the 302 genes upregulated at least 4-fold by stimulation in expanded iNKT cells or Vα24-N cells were those for canonical Th1 (IFNG, LTA) and Th2 pathways (IL5, IL13). Following CD2/CD3/CD28 stimulation, day 21 expanded iNKT cells and Vα24-N cells showed significantly increased Th2-type gene expression. Additionally, upregulated were other cytokines (IL1A, IL3, IL8, IL9, IL21, LIF), chemokines (CCL1, CCL3/CCL3L1, CCL4/CCL4L1, CXCL10), growth factors (CSF2, CSF1), receptors (IL2RA, IL17RB, CCR7) and activation-associated (TNFRSF8, TNFRSF9, TNFRSF4) or signaling genes (NAMPT, CD38); this profile was highly consistent across random donors following CD2/CD3/CD28 stimulation (Supplemental Figure 5A). Day 21 Vα24-N cells differed from iNKT cells by increased expression of Th17 cytokines (IL17F, IL22, IL26), and IL10 (Supplemental Figure 5A).
Expansion followed by CD2/CD3/CD28 stimulation enhances expression of key Th1 and Th2 cytokines and chemokine receptors on iNKT cells as compared to unexpanded iNKT cells.
Gene set enrichment analysis (GSEA) of CD2/CD3/CD28-stimulated unexpanded and day 21 expanded iNKT cells revealed 69 upregulated pathways (FDR < 0.05, normalized enrichment score/NES ≥ +1.8 at both days 0 and 21) (Figure 4D and Supplemental Table 1). Shared activated pathways included the Biocarta NKT and multiple cytokine, T cell activation, and inflammatory pathways (magenta) (Supplemental Table 1). Twenty-three pathways upregulated only at day 0 included those associated with T cell regulation, interferon signaling, and Th17 differentiation (orange) (Supplemental Table 2). Over 200 pathways were upregulated only at day 21 (blue) (Supplemental Table 3); the majority associated with carbohydrate metabolism and proliferation, likely attributable to the IL-7-based expansion procedure, a known inducer of glucose transporter 1 upregulation in T cells [55, 56]. Notably, activation pathways normally upregulated in conventional T cell CD2/CD3/CD28 stimulation were not upregulated (green) in CD2/CD3/CD28-stimulated iNKT cells, at either day 0 or 21.
Expanded iNKT cells and CD3+Vα24neg cells express similar chemokine receptor and developmental gene expression profiles.
Changes to chemokine receptor expression were investigated to determine if expansion could alter iNKT cell trafficking compared to unexpanded iNKT cells. Biocarta NKT pathway analysis revealed increased CCR7 and decrease in CCR3, CCR5, and CXCR3 expression in day 21 iNKT cells and Vα24-N cells after CD2/CD3/CD28 stimulation (Figure 4E). By flow cytometry (Figure 5A), increases in multiple chemokine receptors including CCR7 was seen. These data suggest that expansion alters the chemokine receptor program of human iNKT cells and that CD2/CD3/CD28 stimulation may be utilized to alter iNKT cell trafficking potential post-expansion. They also highlight the (well-documented) importance of protein assessment in iNKT cells, which are known to regulate gene expression by post-transcriptional regulation of translation; thus, mRNA profiling may not directly correlate with protein expression.
The Metelitsa group has published that CD62L expression may be critical to in vitro and in vivo anti-tumor activity of iNKT cells expanded using K562-derived APCs transduced to express co-stimulatory molecules [57]. In those studies, the CD62L+ subset demonstrated higher expression of ZBTB16/PLZF, LEF1, and GATA3 (a key LEF1–regulated protein), and low CD244, HAVCR2/TIM3, and LAG3 gene expression. Using our expansion procedure, we consistently observed high baseline gene expression of ZBTB16/PLZF, SELL/CD62L, LEF1, GATA3, CD244, HAVCR2/TIM3, and LAG3 in both unexpanded and expanded iNKT cells, with no significant change with stimulation (Supplemental Figure 5B). Within this gene set associated with T cell survival, terminal generation, and exhaustion, gene expression was comparable for day 0 and day 21 iNKT cells and Vα24-N cells, with the exception that Vα24-N cells had 4-fold lower expression of the iNKT cell development-associated gene ZBTB16/PLZF than day 21 iNKT cells. In addition, day 21 iNKT cells had 5-fold increased expression of HAVCR2/TIM3 than Day 0 iNKT cells (Supplemental Figure 5B). Despite high RNA expression of SELL/CD62L, we found no cell-surface expression of CD62L on unstimulated day 21 iNKT cells but did observe increased CD62L surface protein expression at day 21 with activation as compared to unactivated cells (Supplemental Figure 5C).
Ex vivo expansion enhances Th2 cytokine secretion in human iNKT cells following CD2/CD3/CD28 stimulation as compared to unstimulated expanded iNKT cells.
We confirmed at the protein level secretion of a diverse array of cytokines from sorted day 21 iNKT cells and Vα24-N cells with and without CD2/CD3/CD28 stimulation. CD2/CD3/CD28 stimulation robustly augmented IL-2, IFN-γ, TNF-α, IL-4, IL-13, IL-10, GM-CSF, CCL3, and CCL4 secretion from both day 21 iNKT cells and Vα24-N cells (Figure 5B) as compared to unstimulated expanded cells. Of note, anti-CD2/CD3/CD28 induced IL-10 secretion by expanded iNKT cells, and Vα24-N cells released even higher concentrations of IL-10 (Figure 5B). A similar pattern was confirmed and subset analysis performed using monensin-blocked CD2/CD3/CD28 stimulation prior to intracellular cytokine staining by flow cytometry (Figure 5C). Notably, the dominant cytokine-positive subset among day 21 expanded iNKT cells was the CD4+ subset, followed by the DN subset (Figure 5C, right panel). These data demonstrate that therapeutically expanded, CD2/CD3/CD28-stimulated iNKT cells simultaneously secrete Th1 and Th2-type cytokines and IL-10, a phenotype associated with immunoregulatory potential [10, 13, 16].
CD2/CD3/CD28 stimulation enhances cytotoxic pathways and tumor target killing.
To determine if expansion altered the cytolytic and anti-tumor capacity of iNKT cells, we looked at changes in RNA expression of select cytolytic effector molecules after stimulation at day 0 and day 21. GZMB (Granzyme B) expression was consistently increased after CD2/CD3/CD28 stimulation in both unexpanded day 0 and expanded day 21 iNKT cells (Figure 6A). FASLG (Fas-ligand) and costimulatory TNFSF9 (41BBL) increased only in day 21 expanded cells (Figure 6A, center and right panels), suggesting that expansion itself enhanced underlying capacity to express some cytotoxicity-associated genes. Flow cytometry confirmed that stimulation significantly increased granzyme B protein levels in day 21 expanded cells (Figure 6B). Notably, TRAIL and FasL/CD178 expression were not increased by anti-CD2/CD3/CD28 stimulation of day 21 iNKT cells (Figure 6C).
We then examined direct cytotoxicity of expanded day 21 sorted iNKT cells against key hematolymphoid tumors in vitro under various conditions. K562 (CML-derived AML cell line) does not express significant CD1d, the key Class I-like antigen-presenting molecule for iNKT cells (Figure 6D, left panel), whereas Jurkat (T-ALL) does express CD1d (Figure 6D, right panel). Using 51Cr-loaded K562 and Jurkat cells, unsorted day 21 expanded cells demonstrated dose-dependent killing of Jurkat (data not shown). iNKT cells were not able to kill K562. To exclude target cell killing by the Vα24-N cells in the expansion product, day 21 Vα24+ iNKT cells were re-sorted (> 97% purity) and their cytotoxicity assayed against 51Cr-loaded targets with or without α-GalCer target loading. α-GalCer loaded target cells augmented dose-dependent cytotoxicity of day 21 unstimulated iNKT cells against Jurkat (P < 0.05) (Figure 6E, bottom panels), but not against K562 (Figure 6E, top panels). However, CD2/CD3/CD28 stimulation induced a dose-dependent increase in iNKT cell cytotoxicity against Jurkat cells, achievable independent of α-GalCer loading (P < 0.05) (Figure 6D, bottom right panel).
Expanded iNKT cells regulate activated T cell proliferation in a contact-dependent manner.
Finally, the regulatory capacity of expanded iNKT cells was investigated in a bead-stimulated MLR model. The addition of unsorted day 21 expanded iNKT cells to 5-day transwell MLR assays in cis- (direct contact) with autologous pre-activated PBMC significantly reduced both the percentage (Figure 7A) and proliferation (Figures 7B and 7C) of activated CD4+ T cells. Transwell separation of iNKT cells from effectors with a membrane pore size sufficient to allow cytokine diffusion reversed this suppressor effect (Figures 7A, 7B, and 7C), supporting contact-and not purely cytokine-dependent suppressor activity. The percentage (Figure 7D) and proliferation (Figures 7E and 7F) of activated CD8+ T cells was similarly reduced by the addition of expanded iNKT cells co-cultured in cis to the responder PBMC but not in trans (Figures 7D, 7E, and 7F).
Figure 7: Expanded iNKT cells suppress activated CD4+ and CD8+ responder T cells in a contact-dependent manner.

(A) Left panel, representative CD25 and CD45RA expression on gated CD4+ CFSE+ responder cells stimulated for 5 days. Black, unloaded bead stim; red, anti-CD2/CD3/CD28-loaded bead stim. Right panel, mean ± SEM percentage gated CD25+ activated cells among CD4+ responder cells (N = 4). Responder : iNKT cell ratios: 1:0, responders alone; 1:1, responders with iNKT cells in direct contact at 1:1; 1:1 TW, responders and iNKT cells at 1:1 but separated by transwell.
(B) Average divisions (left panel) and % Divided (right panel) for gated CFSE+CD4+CD25+ responder cells (N = 4) cultured with expanded iNKT cells, normalized to CD2/CD3/CD28-stimulated responders alone. Dashed line represents responders alone.
(C) Representative proliferation of CFSE+CD4+CD25+ responder cells. Green cross-hatch, generation 0; blue, generation 1; orange, generation 2. TW, representative proliferation plot with iNKT cells separated from responders via transwell.
(D) Left panel, representative CD25 and CD45RA expression on control CD8+ gated CFSE+ responder cells stimulated for 5 days. Black, unloaded bead stim; red, anti-CD2/CD3/CD28-loaded bead stim. Right panel, mean ± SEM percentage gated CD25+ activated cells among CD8+ responder cells (N = 4). Responder : iNKT cell ratios: 1:0, responders alone; 1:1, responders with iNKT cells in direct contact at 1:1; 1:1 TW, responders and iNKT cells at 1:1 but separated by transwell.
(E) Average divisions (left panel) and % Divided (right panel) for gated CFSE+CD8+CD25+ responder cells (N = 4) cultured with expanded iNKT cells, normalized to CD2/CD3/CD28-stimulated responders alone. Dashed line represents responders alone.
(F) Representative proliferation of CFSE+CD8+CD25+ responder cells. TW, representative proliferation plot with iNKT cells separated from responders via transwell. P < 0.05 is represented by *.
Discussion
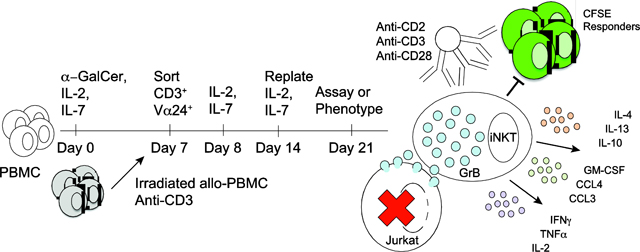
To address existing obstacles to iNKT cell immunotherapy, we have rigorously optimized expansion of human iNKT cells, phenotyped expanded cells, and delineated a specific post-expansion stimulation mechanism to simultaneously enhance multiple therapeutic iNKT cell functions. We have demonstrated through these approaches that the expanded iNKT cells have the capacity to robustly secrete cytokines upon TCR stimulation, as well as to kill hematolymphoid targets. Additionally, at least one subset of the expansion demonstrates the capacity to regulate a proliferative response in autologous responder effector T cells (Graphical abstract).
These studies represent N > 45 random donors over the course of 1.5 years. While other groups have reported human iNKT cell expansion (Table 1), the most common approaches isolate iNKT cells before expansion and stimulate iNKT cells dominantly with IL-2 (± IL-7 and/or IL-15) [29, 32, 35, 57–63]. Variations have included no or delayed iNKT cell enrichment [26–28, 30, 33, 36, 42, 64–66], artificial or transduced APCs [57, 58, 63, 67], complete lack of IL-2 [31, 33, 65], or phytohemagglutinin rather than α-GalCer stimulation [68, 69]. Of these, few studies have reported robust serial expansion capacity such as demonstrated here (prior studies generally report on N ≥ 10 sources and/or non-random selected expansions), poor reproducibility of expansions over multiple units [36, 64] or, where reproducible, expansion efficiencies up to 1 log-fold less robust than what we have reported [35]. Moreover, only one study has reported a means to improve anti-tumor killing by expanded iNKT cells [57]. The latter report, focused on iNKT cell targeting to specific tumors, was limited by very few expansions, did not specify the frequency of expansion failures, and did not assay iNKT cell regulatory capacity [57]. In summary, the approach in this report results in more robust and reproducible expansion of NKT cells [median 4.8 × 107 (IQR 8.5 × 106 – 2.2 × 108) from 2 × 108 starting PBMC] than prior reported techniques as well as improved uniformity in key functional phenotypes for immunotherapy. Given that the average cellular therapy product contains 2 ×1010 nucleated cells, the yield we report lends itself to translational and clinical application. The current protocol preferentially expands the CD4+ subset of iNKT cells, which are known to express significant resting levels of IL-7Rα and demonstrates that this subset is also dominant among cytokine-secreting subsets. Notably, the presence of a high fraction of CD4+ iNKT cells secreting both Th1 and Th2 cytokines upon activation does not inhibit the ability of these cells to directly kill tumor targets. The potential for harnessing these functions via transduction with activating chimeric antigen receptor (CAR) constructs is an ongoing area of investigation.
Expanded iNKT cells robustly and simultaneously secrete both Th2 and Th1 cytokines, as seen in freshly isolated human [10, 70, 71] and murine iNKT cells [13, 16, 17]. Through gene expression profiling and confirmatory functional assays, we delineated a straightforward post-expansion stimulation that enhances secretion of Th2 cytokines alongside other cytokines as well as regulatory IL-10 by expanded iNKT cells. Of note is the much more uniform ability to stimulate Th2-type (IL-4, IL-5, IL-13) cytokine secretion in expanded as compared to freshly isolated iNKT cells, underscoring that expansion not only quantitatively enhances iNKT cell numbers but also qualitatively modifies iNKT cell phenotypes.
CD2/CD3/CD28 stimulation of expanded iNKT cells enhanced cytotoxic molecules promoting DNA fragmentation and apoptosis, including granzyme B [72, 73]. Stimulation also augmented direct cytotoxicity, partially overcoming the previously reported requirement for α-GalCer target loading for efficient iNKT cell killing [33, 61, 68]. In keeping with knowledge that lymphoid malignancies are optimal targets for non-transduced iNKT cells [34, 60, 74], Jurkat but not the myeloid leukemia K562 was a target of expanded iNKT cell killing. Target killing also correlated with expression of CD1d by the cell lines.
To focus on rigorous characterization of iNKT cells pre- and post-expansion, we elected not to separately assay iNKT cell subset functions [68, 70]. However, robust expansions afforded by our current understanding will allow future CD4+, CD8+, and DN iNKT cell-specific studies to identify Th1- or Th2-polarizing, regulatory, and/or cytotoxic subsets [68, 70, 71, 75]. Our current data demonstrate, however, that CD4-dominant expansions of iNKT cells retain strong direct anti-tumor cytolytic effector function and suggest that CD4+ iNKT cells represent the dominant secretors of cytokines among expanded iNKT cells. Expanded iNKT cells regulate activation and proliferation of conventional CD4+ and CD8+ T cells. Previous reports of human expanded iNKT cell suppressor activity required rapamycin [76, 77]; however day 21 expanded iNKT cells suppressed conventional T cell proliferation without any further manipulation [35]. We concede that we have not separately sorted iNKT cells from Vα24-N cells to determine which subset is suppressive, but minimally the data show that the Vα24-N subset co-expanded in the product do not inhibit the regulatory capacity of iNKT cells against stimulated responders. To mimic the potential therapeutic usage planned for these cells, the responders in these assays were matched to the iNKT cell unit, excluding the possibility of an allo-response in abrogating responder proliferation. As has been reported [77], this suppressor activity was iNKT cell contact-dependent and not purely cytokine-dependent. Whether this contact-dependence indicates that expanded iNKT cells suppress conventional T cells directly or through a suppressive intermediary population, as we and subsequently others have shown [14, 45, 78], is an area of ongoing investigation.
The Vα24-N cells co-expanding after isolation of iNKT cells at day 7 is intriguing. The cumulative data suggests that the Vα24-N cells represent a mixed memory subset T cell population, based on the presence of both CD4+ and CD8+ single-positive but no substantial DN cells (Figure 2B), the expression of CD45RO (Figure 2C), the fact that this population demonstrates a stochastic TCR Vβ spectratype (Supplemental Figure 5B), and CIBERSORT analysis supporting all strong gene expression correlation with these subsets as well as a subset of γδ T cells (Supplemental Figure 5D), with TCRγδ expression further confirmed on a subset of Vα24-N cells by FACS analysis (data not shown). This population, however, demonstrates some important immunotherapeutic features including potent cytokine secretion upon activation and no significant evidence that they abrogate otherwise robust MLR suppression or cytotoxicity assays in vitro. Extensive characterization of these Vα24-N cells and their TCR-downstream signaling is underway as part of another report in preparation.
We report specific methods for robust ex vivo expansion of human Vα24+Vβ11+ human iNKT cells across serial donor sources and a straightforward and specific pathway to activate key effector functions in therapeutically expanded human iNKT cells. We envision application of these findings to therapeutically expand autologous or allogeneic human iNKT cells for such diverse research and therapeutic indications as vaccine development, chronic infectious diseases, auto-immunity, and cancer or peri-transplant immunotherapy.
Supplementary Material
Highlights:
Human iNKT cells robustly expand for cytotherapy via an IL-2/IL-7-based protocol.
iNKT cells expanded through this protocol retain regulatory capacity.
Post-expansion, CD2/CD3/CD28 stimulation enhances iNKT cell Th2 cytokine secretion.
CD2/CD3/CD28 stimulation also enhances expanded iNKT cell anti-tumor cytotoxicity.
Acknowledgments
We thank Jim Houston of the Department of BMTCT and the St. Jude Shared FACS Facility for FACS sorting and instrument support, Dr. Mark Exley for early discussions on reagents, and Drs. Helen Heslop, Nelson Chao, Randy Brutkiewicz, and John Koreth for their critiques. This work was funded by grants 5P30CA021765–36 (A.B.P.), R12/94–000 (Assisi Foundation) (A.B.P., K.A.), the V Scholar Award of the V Foundation for Cancer Research (A.B.P.), the Hyundai Scholar Award (A.B.P.), the American Lebanese Syrian Associated Charities (ALSAC) (A.B.P., K.A.), and the Batchelor Foundation for Pediatric Research (A.B.P., K.A., A.A.J.H.). This research was conducted in collaboration with and using the Biostatistics and Bioinformatics Shared Resource of the Sylvester Comprehensive Cancer Center, University of Miami.
Abbreviations:
- α-GalCer
α-galactosyl ceramide
- CFSE
carboxyfluorescein succinimidyl ester
- GVHD
graft-versus-host disease
- GVT
graft-versus-tumor
- HCT
hematopoietic cell transplantation
- iNKT
invariant NKT
- NKT
Natural killer T
- Th2
T-helper type 2
Footnotes
Conflict-of-interest Disclosures
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Metelitsa LS, Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans, Clinical immunology (Orlando, Fla.) 140(2) (2011) 119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brutkiewicz RR, Sriram V, Natural killer T (NKT) cells and their role in antitumor immunity, Critical reviews in oncology/hematology 41(3) (2002) 287–98. [DOI] [PubMed] [Google Scholar]
- [3].Crowe NY, Smyth MJ, Godfrey DI, A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas, J Exp Med 196(1) (2002) 119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Teng MW, Westwood JA, Darcy PK, Sharkey J, Tsuji M, Franck RW, Porcelli SA, Besra GS, Takeda K, Yagita H, Kershaw MH, Smyth MJ, Combined natural killer T-cell based immunotherapy eradicates established tumors in mice, Cancer research 67(15) (2007) 7495–504. [DOI] [PubMed] [Google Scholar]
- [5].Wilson SB, Delovitch TL, Janus-like role of regulatory iNKT cells in autoimmune disease and tumour immunity, Nat Rev Immunol 3(3) (2003) 211–22. [DOI] [PubMed] [Google Scholar]
- [6].Molling JW, Moreno M, van der Vliet HJ, van den Eertwegh AJ, Scheper RJ, von Blomberg BM, Bontkes HJ, Invariant natural killer T cells and immunotherapy of cancer, Clinical immunology (Orlando, Fla.) 129(2) (2008) 182–94. [DOI] [PubMed] [Google Scholar]
- [7].Smyth MJ, Wallace ME, Nutt SL, Yagita H, Godfrey DI, Hayakawa Y, Sequential activation of NKT cells and NK cells provides effective innate immunotherapy of cancer, J Exp Med 201(12) (2005) 1973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, Seeger RC, Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2, J Exp Med 199(9) (2004) 1213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, Ara T, Silverman AM, DeClerck YA, Seeger RC, Metelitsa LS, Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages, J Clin Invest 119(6) (2009) 1524–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bendelac A, Savage PB, Teyton L, The Biology of NKT Cells, Annual Review of Immunology 25(1) (2007) 297–336. [DOI] [PubMed] [Google Scholar]
- [11].Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI, Recognition of CD1d-restricted antigens by natural killer T cells, Nat Rev Immunol 12(12) (2012) 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pillai AB, George TI, Dutt S, Teo P, Strober S, Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation, J Immunol 178(10) (2007) 6242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pillai AB, George TI, Dutt S, Strober S, Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease, Blood 113(18) (2009) 4458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].van der Merwe M, Abdelsamed HA, Seth A, Ong T, Vogel P, Pillai AB, Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplantation, J Immunol 191(11) (2013) 5764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lowsky R, Takahashi T, Liu YP, Dejbakhsh-Jones S, Grumet FC, Shizuru JA, Laport GG, Stockerl-Goldstein KE, Johnston LJ, Hoppe RT, Bloch DA, Blume KG, Negrin RS, Strober S, Protective conditioning for acute graft-versus-host disease, The New England journal of medicine 353(13) (2005) 1321–31. [DOI] [PubMed] [Google Scholar]
- [16].Kohrt HE, Turnbull BB, Heydari K, Shizuru JA, Laport GG, Miklos DB, Johnston LJ, Arai S, Weng WK, Hoppe RT, Lavori PW, Blume KG, Negrin RS, Strober S, Lowsky R, TLI and ATG conditioning with low risk of graft-versus-host disease retains antitumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors, Blood 114(5) (2009) 1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schneidawind D, Pierini A, Alvarez M, Pan Y, Baker J, Buechele C, Luong RH, Meyer EH, Negrin RS, CD4+ invariant natural killer T cells protect from murine GVHD lethality through expansion of donor CD4+CD25+FoxP3+ regulatory T cells, Blood 124(22) (2014) 3320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang J, Gao L, Liu Y, Ren Y, Xie R, Fan H, Qian K, Adoptive therapy by transfusing expanded donor murine natural killer T cells can suppress acute graft-versus-host disease in allogeneic bone marrow transplantation, Transfusion 50(2) (2010) 407–17. [DOI] [PubMed] [Google Scholar]
- [19].Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, Negrin RS, Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism, Blood 117(11) (2011) 3220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kuwatani M, Ikarashi Y, Iizuka A, Kawakami C, Quinn G, Heike Y, Yoshida M, Asaka M, Takaue Y, Wakasugi H, Modulation of acute graft-versus-host disease and chimerism after adoptive transfer of in vitro-expanded invariant Valpha14 natural killer T cells, Immunology letters 106(1) (2006) 82–90. [DOI] [PubMed] [Google Scholar]
- [21].Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, McDonald D, Marin D, Milojkovic D, Pavlu J, Davis J, Rahemtulla A, Rezvani K, Goldman J, Roberts I, Apperley J, Karadimitris A, Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation, Blood 119(21) (2012) 5030–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rubio MT, Bouillie M, Bouazza N, Coman T, Trebeden-Negre H, Gomez A, Suarez F, Sibon D, Brignier A, Paubelle E, Nguyen-Khoc S, Cavazzana M, Lantz O, Mohty M, Urien S, Hermine O, Pre-transplant donor CD4-invariant NKT cell expansion capacity predicts the occurrence of acute graft-versus-host disease, Leukemia 31(4) (2017) 903–912. [DOI] [PubMed] [Google Scholar]
- [23].Rubio MT, Moreira-Teixeira L, Bachy E, Bouillie M, Milpied P, Coman T, Suarez F, Marcais A, Sibon D, Buzyn A, Caillat-Zucman S, Cavazzana-Calvo M, Varet B, Dy M, Hermine O, Leite-de-Moraes M, Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival, Blood 120(10) (2012) 2144–54. [DOI] [PubMed] [Google Scholar]
- [24].de Lalla C, Rinaldi A, Montagna D, Azzimonti L, Bernardo ME, Sangalli LM, Paganoni AM, Maccario R, Di Cesare-Merlone A, Zecca M, Locatelli F, Dellabona P, Casorati G, Invariant NKT cell reconstitution in pediatric leukemia patients given HLA-haploidentical stem cell transplantation defines distinct CD4+ and CD4-subset dynamics and correlates with remission state, J Immunol 186(7) (2011) 4490–9. [DOI] [PubMed] [Google Scholar]
- [25].Jenq RR, van den Brink MR, Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer, Nature reviews. Cancer 10(3) (2010) 213–21. [DOI] [PubMed] [Google Scholar]
- [26].Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, Shimizu N, Horiguchi S, Okamoto Y, Fujii S, Taniguchi M, Fujisawa T, Nakayama T, A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer, Clinical cancer research : an official journal of the American Association for Cancer Research 12(20 Pt 1) (2006) 6079–86. [DOI] [PubMed] [Google Scholar]
- [27].Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, Sakurai D, Taniguchi M, Nakayama T, Okamoto Y, Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma, Cancer science 100(6) (2009) 1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, Shimizu N, Ueno N, Yamamoto S, Taniguchi M, Motohashi S, Nakayama T, Okamoto Y, Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy, Clinical immunology (Orlando, Fla.) 138(3) (2011) 255–65. [DOI] [PubMed] [Google Scholar]
- [29].Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, Zeng W, Mizukami Y, Clark J, Nemer D, LeClair K, Canning C, Daley H, Dranoff G, Giobbie-Hurder A, Hodi FS, Ritz J, Balk SP, Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial, Clinical cancer research : an official journal of the American Association for Cancer Research 23(14) (2017) 3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nishi N, van der Vliet HJ, Koezuka Y, von Blomberg BM, Scheper RJ, Pinedo HM, Giaccone G, Synergistic effect of KRN7000 with interleukin-15, −7, and −2 on the expansion of human V alpha 24+V beta 11+ T cells in vitro, Hum Immunol 61(4) (2000) 357–65. [DOI] [PubMed] [Google Scholar]
- [31].van der Vliet HJ, Nishi N, Koezuka Y, von Blomberg BM, van den Eertwegh AJ, Porcelli SA, Pinedo HM, Scheper RJ, Giaccone G, Potent expansion of human natural killer T cells using alpha-galactosylceramide (KRN7000)-loaded monocyte-derived dendritic cells, cultured in the presence of IL-7 and IL-15, J Immunol Methods 247(1–2) (2001) 61–72. [DOI] [PubMed] [Google Scholar]
- [32].Moreno M, Molling JW, von Mensdorff-Pouilly S, Verheijen RH, von Blomberg BM, van den Eertwegh AJ, Scheper RJ, Bontkes HJ, In vitro expanded human invariant natural killer T-cells promote functional activity of natural killer cells, Clinical immunology (Orlando, Fla.) 129(1) (2008) 145–54. [DOI] [PubMed] [Google Scholar]
- [33].van der Vliet HJ, Molling JW, Nishi N, Masterson AJ, Kolgen W, Porcelli SA, van den Eertwegh AJ, von Blomberg BM, Pinedo HM, Giaccone G, Scheper RJ, Polarization of Valpha24+ Vbeta11+ natural killer T cells of healthy volunteers and cancer patients using alpha-galactosylceramide-loaded and environmentally instructed dendritic cells, Cancer research 63(14) (2003) 4101–6. [PubMed] [Google Scholar]
- [34].Das R, Bassiri H, Guan P, Wiener S, Banerjee PP, Zhong MC, Veillette A, Orange JS, Nichols KE, The adaptor molecule SAP plays essential roles during invariant NKT cell cytotoxicity and lytic synapse formation, Blood 121(17) (2013) 3386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Trujillo-Ocampo A, Cho HW, Herrmann AC, Ruiz-Vazquez W, Thornton AB, He H, Li D, Qazilbash MA, Ma Q, Porcelli SA, Shpall EJ, Molldrem J, Im JS, Rapid ex vivo expansion of highly enriched human invariant natural killer T cells via single antigenic stimulation for cell therapy to prevent graft-versus-host disease, Cytotherapy 20(8) (2018) 1089–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rogers PR, Matsumoto A, Naidenko O, Kronenberg M, Mikayama T, Kato S, Expansion of human Valpha24+ NKT cells by repeated stimulation with KRN7000, J Immunol Methods 285(2) (2004) 197–214. [DOI] [PubMed] [Google Scholar]
- [37].Matsuda JL, Gapin L, Sidobre S, Kieper WC, Tan JT, Ceredig R, Surh CD, Kronenberg M, Homeostasis of V alpha 14i NKT cells, Nat Immunol 3(10) (2002) 966–74. [DOI] [PubMed] [Google Scholar]
- [38].Chang CL, Lai YG, Hou MS, Huang PL, Liao NS, IL-15Ralpha of radiation-resistant cells is necessary and sufficient for thymic invariant NKT cell survival and functional maturation, J Immunol 187(3) (2011) 1235–42. [DOI] [PubMed] [Google Scholar]
- [39].Warren HS, Kinnear BF, Kastelein RL, Lanier LL, Analysis of the costimulatory role of IL-2 and IL-15 in initiating proliferation of resting (CD56dim) human NK cells, J Immunol 156(9) (1996) 3254–9. [PubMed] [Google Scholar]
- [40].Baev DV, Peng XH, Song L, Barnhart JR, Crooks GM, Weinberg KI, Metelitsa LS, Distinct homeostatic requirements of CD4+ and CD4-subsets of Valpha24-invariant natural killer T cells in humans, Blood 104(13) (2004) 4150–6. [DOI] [PubMed] [Google Scholar]
- [41].de Lalla C, Festuccia N, Albrecht I, Chang HD, Andolfi G, Benninghoff U, Bombelli F, Borsellino G, Aiuti A, Radbruch A, Dellabona P, Casorati G, Innate-like effector differentiation of human invariant NKT cells driven by IL-7, J Immunol 180(7) (2008) 4415–24. [DOI] [PubMed] [Google Scholar]
- [42].Takahashi T, Nieda M, Koezuka Y, Nicol A, Porcelli SA, Ishikawa Y, Tadokoro K, Hirai H, Juji T, Analysis of human V alpha 24+ CD4+ NKT cells activated by alpha-glycosylceramide-pulsed monocyte-derived dendritic cells, J Immunol 164(9) (2000) 4458–64. [DOI] [PubMed] [Google Scholar]
- [43].Lee PT, Benlagha K, Teyton L, Bendelac A, Distinct functional lineages of human V(alpha)24 natural killer T cells, J Exp Med 195(5) (2002) 637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim CH, Johnston B, Butcher EC, Trafficking machinery of NKT cells: shared and differential chemokine receptor expression among V alpha 24(+)V beta 11(+) NKT cell subsets with distinct cytokine-producing capacity, Blood 100(1) (2002) 11–6. [DOI] [PubMed] [Google Scholar]
- [45].Schneidawind D, Baker J, Pierini A, Buechele C, Luong RH, Meyer EH, Negrin RS, Third-party CD4+ invariant natural killer T cells protect from murine GVHD lethality, Blood 125(22) (2015) 3491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].D’Andrea A, Goux D, De Lalla C, Koezuka Y, Montagna D, Moretta A, Dellabona P, Casorati G, Abrignani S, Neonatal invariant Valpha24+ NKT lymphocytes are activated memory cells, Eur J Immunol 30(6) (2000) 1544–50. [DOI] [PubMed] [Google Scholar]
- [47].Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP, Exploration, normalization, and summaries of high density oligonucleotide array probe level data, Biostatistics (Oxford, England) 4(2) (2003) 249–64. [DOI] [PubMed] [Google Scholar]
- [48].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc Natl Acad Sci U S A 102(43) (2005) 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA, Robust enumeration of cell subsets from tissue expression profiles, Nature Methods 12(5) (2015) 453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nelson WC, Pyo CW, Vogan D, Wang R, Pyon YS, Hennessey C, Smith A, Pereira S, Ishitani A, Geraghty DE, An integrated genotyping approach for HLA and other complex genetic systems, Hum Immunol 76(12) (2015) 928–38. [DOI] [PubMed] [Google Scholar]
- [51].Montoya CJ, Pollard D, Martinson J, Kumari K, Wasserfall C, Mulder CB, Rugeles MT, Atkinson MA, Landay AL, Wilson SB, Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11, Immunology 122(1) (2007) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sandberg JK, Bhardwaj N, Nixon DF, Dominant effector memory characteristics, capacity for dynamic adaptive expansion, and sex bias in the innate Valpha24 NKT cell compartment, Eur J Immunol 33(3) (2003) 588–96. [DOI] [PubMed] [Google Scholar]
- [53].Prussin C, Foster B, TCR V alpha 24 and V beta 11 coexpression defines a human NK1 T cell analog containing a unique Th0 subpopulation, J Immunol 159(12) (1997) 5862–70. [PubMed] [Google Scholar]
- [54].van Der Vliet HJ, Nishi N, de Gruijl TD, von Blomberg BM, van den Eertwegh AJ, Pinedo HM, Giaccone G, Scheper RJ, Human natural killer T cells acquire a memory-activated phenotype before birth, Blood 95(7) (2000) 2440–2. [PubMed] [Google Scholar]
- [55].Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC, IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival, Blood 111(4) (2008) 2101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jacobs SR, Michalek RD, Rathmell JC, IL-7 is essential for homeostatic control of T cell metabolism in vivo, J Immunol 184(7) (2010) 3461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, Guo L, Xu X, Torikai H, Mo Q, Dotti G, Cooper LJ, Metelitsa LS, CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo, J Clin Invest (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mavers M, Simonetta F, Lee AW, Hirai T, Maas-Bauer K, Baker J, Alvarez M, Turkoz M, Meyer EH, Negrin RS, IL-2 Plus IL-15 Leads to Enhanced Ex Vivo Expansion of Human Invariant Natural Killer T Cells, Blood 130(Suppl 1) (2017) 3199–3199. [Google Scholar]
- [59].Exley MA, Hou R, Shaulov A, Tonti E, Dellabona P, Casorati G, Akbari O, Akman HO, Greenfield EA, Gumperz JE, Boyson JE, Balk SP, Wilson SB, Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR alpha-chain CDR3 loop, Eur J Immunol 38(6) (2008) 1756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Song W, van der Vliet HJ, Tai YT, Prabhala R, Wang R, Podar K, Catley L, Shammas MA, Anderson KC, Balk SP, Exley MA, Munshi NC, Generation of antitumor invariant natural killer T cell lines in multiple myeloma and promotion of their functions via lenalidomide: a strategy for immunotherapy, Clinical cancer research : an official journal of the American Association for Cancer Research 14(21) (2008) 6955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, Kronenberg M, Seeger RC, Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells, J Immunol 167(6) (2001) 3114–22. [DOI] [PubMed] [Google Scholar]
- [62].Li X, Tsuji M, Schneck J, Webb TJ, Generation of Human iNKT Cell Lines, Bio-protocol 3(6) (2013) e418. [PMC free article] [PubMed] [Google Scholar]
- [63].Moreno M, Molling JW, von Mensdorff-Pouilly S, Verheijen RH, Hooijberg E, Kramer D, Reurs AW, van den Eertwegh AJ, von Blomberg BM, Scheper RJ, Bontkes HJ, IFN-gamma-producing human invariant NKT cells promote tumor-associated antigen-specific cytotoxic T cell responses, J Immunol 181(4) (2008) 2446–54. [DOI] [PubMed] [Google Scholar]
- [64].Watarai H, Nakagawa R, Omori-Miyake M, Dashtsoodol N, Taniguchi M, Methods for detection, isolation and culture of mouse and human invariant NKT cells, Nat. Protocols 3(1) (2008) 70–78. [DOI] [PubMed] [Google Scholar]
- [65].Van Der Vliet HJ, Nishi N, Koezuka Y, Peyrat MA, Von Blomberg BM, Van Den Eertwegh AJ, Pinedo HM, Giaccone G, Scheper RJ, Effects of alpha-galactosylceramide (KRN7000), interleukin-12 and interleukin-7 on phenotype and cytokine profile of human Valpha24+ Vbeta11+ T cells, Immunology 98(4) (1999) 557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Schmid H, Schneidawind C, Jahnke S, Kettemann F, Secker KA, Duerr-Stoerzer S, Keppeler H, Kanz L, Savage PB, Schneidawind D, Culture-Expanded Human Invariant Natural Killer T Cells Suppress T-Cell Alloreactivity and Eradicate Leukemia, Frontiers in immunology 9 (2018) 1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].East JE, Sun W, Webb TJ, Artificial antigen presenting cell (aAPC) mediated activation and expansion of natural killer T cells, Journal of visualized experiments : JoVE (70) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].O’Reilly V, Zeng SG, Bricard G, Atzberger A, Hogan AE, Jackson J, Feighery C, Porcelli SA, Doherty DG, Distinct and overlapping effector functions of expanded human CD4+, CD8alpha+ and CD4-CD8alpha-invariant natural killer T cells, PLoS One 6(12) (2011) e28648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zeng SG, Ghnewa YG, O’Reilly VP, Lyons VG, Atzberger A, Hogan AE, Exley MA, Doherty DG, Human invariant NKT cell subsets differentially promote differentiation, antibody production, and T cell stimulation by B cells in vitro, J Immunol 191(4) (2013) 1666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kim CH, Butcher EC, Johnston B, Distinct subsets of human Valpha24-invariant NKT cells: cytokine responses and chemokine receptor expression, Trends in immunology 23(11) (2002) 516–9. [DOI] [PubMed] [Google Scholar]
- [71].Berzins SP, Smyth MJ, Baxter AG, Presumed guilty: natural killer T cell defects and human disease, Nat Rev Immunol 11(2) (2011) 131–42. [DOI] [PubMed] [Google Scholar]
- [72].Trapani JA, Sutton VR, Granzyme B: pro-apoptotic, antiviral and antitumor functions, Curr Opin Immunol 15(5) (2003) 533–43. [DOI] [PubMed] [Google Scholar]
- [73].Chowdhury D, Lieberman J, Death by a thousand cuts: granzyme pathways of programmed cell death, Annu Rev Immunol 26 (2008) 389–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bassiri H, Das R, Guan P, Barrett DM, Brennan PJ, Banerjee PP, Wiener SJ, Orange JS, Brenner MB, Grupp SA, Nichols KE, iNKT cell cytotoxic responses control T-lymphoma growth in vitro and in vivo, Cancer immunology research 2(1) (2014) 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cerundolo V, Silk JD, Masri SH, Salio M, Harnessing invariant NKT cells in vaccination strategies, Nat Rev Immunol 9(1) (2009) 28–38. [DOI] [PubMed] [Google Scholar]
- [76].Moreira-Teixeira L, Resende M, Devergne O, Herbeuval JP, Hermine O, Schneider E, Dy M, Cordeiro-da-Silva A, Leite-de-Moraes MC, Rapamycin combined with TGF-beta converts human invariant NKT cells into suppressive Foxp3+ regulatory cells, J Immunol 188(2) (2012) 624–31. [DOI] [PubMed] [Google Scholar]
- [77].Huijts CM, Schneiders FL, Garcia-Vallejo JJ, Verheul HM, de Gruijl TD, van der Vliet HJ, mTOR Inhibition Per Se Induces Nuclear Localization of FOXP3 and Conversion of Invariant NKT (iNKT) Cells into Immunosuppressive Regulatory iNKT Cells, J Immunol 195(5) (2015) 2038–45. [DOI] [PubMed] [Google Scholar]
- [78].Miyairi S, Hirai T, Ishii R, Okumi M, Nunoda S, Yamazaki K, Ishii Y, Tanabe K, Donor bone marrow cells are essential for iNKT cell-mediated Foxp3+ Treg cell expansion in a murine model of transplantation tolerance, Eur J Immunol 47(4) (2017) 734–742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.