
FIGURE 2.
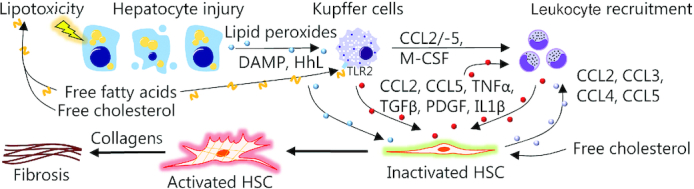
Initiation of NASH-related fibrosis. Nutrient overload results in lipotoxicity and hepatocyte injury leading to the release of a plethora of danger signals (e.g., DAMPs, HhL, and reactive oxygen species) that interact with HSCs and Kupffer cells (resident liver macrophages) in conjunction with free fatty acids and free cholesterol to promote inflammation (24–26). Monocytes are recruited from the circulation, in part, via CCL2, CCL5, and M-CSF released by Kupffer cells. Following differentiation into monocyte-derived macrophages, these infiltrating cells promote hepatic inflammation and recruitment of additional circulating leucocytes by contributing to the release of CCL2, CCL3, and CCL5 (27). Hepatocyte injury, Kupffer cells, and infiltrating macrophages facilitate activation of HSCs, which normally reside in a quiescent state in the healthy liver, but differentiate into ECM-producing myofibroblasts upon activation. HSC collagen production is promoted by TGFβ while PDGF promotes HSC proliferation and migration (27, 28). Macrophages also enhance HSC survival via TNFα and IL1β. In addition to this, HSCs themselves exert immunoregulatory effects by releasing CCL2, CCL3, CCL4, and CCL5, which recruit monocytes from the circulation, thereby sustaining their own activation (27). Thus, chronic inflammation and continual activation of HSCs ultimately result in liver fibrosis or even cirrhosis. CCL, chemokine (c-c motif) ligand; DAMP, damage-associated molecular pattern; ECM, extracellular matrix; HhL, hedgehog ligand; HSC, hepatic stellate cells; M-CSF, macrophage colony-stimulating factor; NASH, nonalcoholic steatohepatitis; PDGF, platelet-derived growth factor; TGFβ, transforming growth factor β; TLR2, Toll-like receptor 2.