
FIGURE 3.
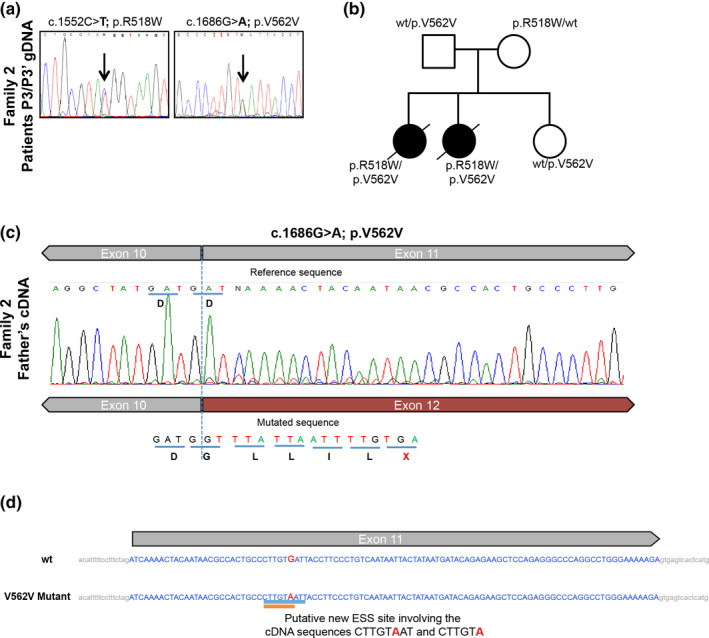
The silent variant p.V562 V was found in a second unrelated family (a) gDNA analysis of exons 9 and 11 in siblings P3 and P3′ (Family 2—F2), showing the variants in heterozygosity. (b) Subsequent/additional analysis on the parents and a non‐affected sister shows the segregation of the variants. (c) Sequencing chromatogram of cDNA‐derived PCR product comprising exons 9–13; at the boundaries of exon 10 and 11 it is possible to observe the second transcript, lacking exon 11 and with the premature stop codon. (d) Schematic representation of p.V562V localization on exon 11 and the effect on splicing based on in silico predictions (Human Splicing Finder—HSF and EX‐SKIP tools). EX‐SKIP compares the Exonic Splicing Enhancer (ESE)/Exonic Splicing Silencer (ESS) profile of a wild type (wt) and a mutated allele to determine if a specific exonic variant increases the chance of exon skipping. It calculates the total number of ESSs, ESEs, and their ratio. The V562V mutant is associated with a change in the ESS/ESE ratio which is compatible with a higher chance of exon skipping than in the wt allele. In addition, HSF (a tool to predict the effects of mutations on splicing signals or to identify splicing motifs in any human sequence) predicts that the V562V mutant leads to the creation of an ESS site. It involves the cDNA sequences CTTGTAAT (Zhang & Chasin, 2004) or CTTGTA (Goren et al., 2006), which might be associated with a potential alteration of splicing