
FIGURE 1.
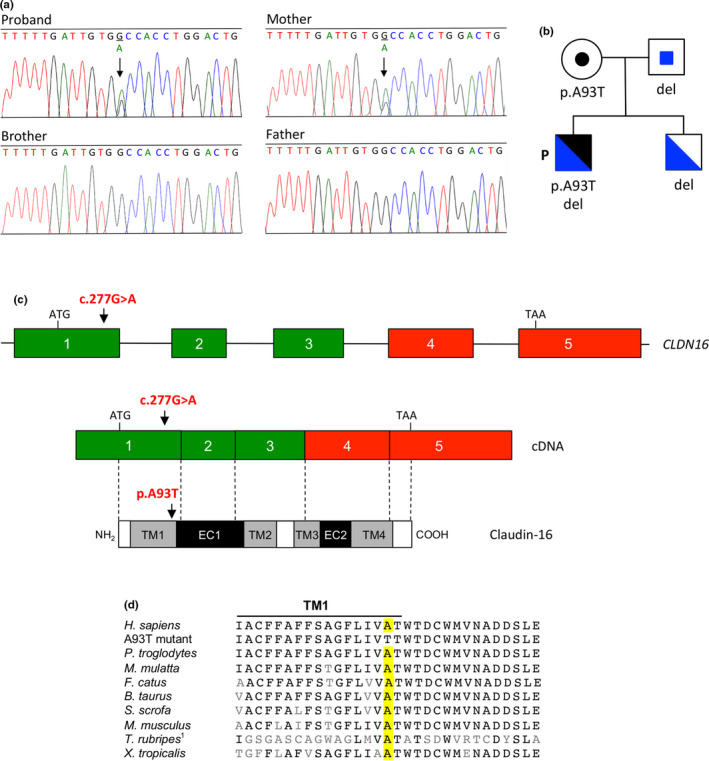
Identification of novel CLDN16 heterozygous missense mutation c.277G>A; p.(Ala93Thr) in patient with FHHNC. (a) Electropherograms showing the heterozygous substitution in exon 1 of the patient and his mother. Arrows indicate the position affected by the mutation in the patient and his mother. The brother and father revealed the normal sequence. (b) Pedigree of the family showing segregation of both CLDN16 variants (missense mutation and deletion in blue and black, respectively). Circle, female individual; squares, male individual; P, proband; del, c.(840+25_?)del detected in the other allele. The patient inherited the missense mutation and the deletion from his mother and father, respectively. (c) Schematic representation of the CLDN16 gene, cDNA, and claudin‐16 protein. Arrows indicate the position affected by missense mutation c.277G>A; p.(Ala93Thr). Colored boxes represent the five coding exons and black lines indicate intron sequences; exons missing in the deletion mutant are in red. Exons and introns sizes are not at scale. The positions of the ATG start codon and the TAA stop codon are also shown. Exons are associated by dotted lines to the schematic representation of the claudin‐16 protein where transmembrane domains (TM1–TM4), extracellular segments (EC1 and EC2) and cytoplasmic regions (white boxes) are indicated. (d) Multiple sequence alignment of claudin‐16 or related proteins from different species showing evolutionary conservation of alanine 93. 1 T. rubripes does not contain the ortholog for mammalian claudin‐16. The sequence shown is that of claudin‐11B