

Abstract
Lung squamous carcinoma (LUSC) is a highly metastatic disease with a poor prognosis. Using an integrated screening approach, we found that miR-671–5p reduces LUSC metastasis by inhibiting a circular RNA (circRNA), CDR1as. Although the putative function of circRNA is through miRNA sponging, we found that miR-671–5p more potently silenced an axis of CDR1as and its antisense transcript, cerebellar degeneration related protein 1 (CDR1). Silencing of CDR1as or CDR1 significantly inhibited LUSC metastases and CDR1 was sufficient to promote migration and metastases. CDR1, which directly interacted with adaptor protein 1 (AP1) complex subunits and COPI proteins, no longer promoted migration upon blockade of Golgi trafficking. Therapeutic inhibition of the CDR1as/CDR1 axis with miR-671–5p mimics reduced metastasis in vivo. This report demonstrates a novel role for CDR1 in promoting metastasis and Golgi trafficking. These findings reveal a miRNA/circRNA axis that regulates LUSC metastases through a previously unstudied protein, CDR1.
Keywords: Non-coding RNA, metastasis, cancer, circular RNA, Golgi
INTRODUCTION:
Lung cancer is the leading cause of cancer-related deaths worldwide and is an intrinsically highly metastatic disease. Even patients who undergo surgery at early stages have a high risk of recurrence (1). Despite LUSC being amongst the most lethal cancers, the mechanistic underpinnings of LUSC metastasis are poorly understood.
Although discovery of molecular aberrations in lung adenocarcinomas has led to development of effective targeted therapies (2), corresponding “drivers” in lung squamous carcinomas (LUSC) have not materialized. Extensive molecular profiling through The Cancer Genome Atlas (TCGA) has revealed LUSC tumors have non-recurrent somatic mutations and are largely driven by copy number alterations and distinct transcriptional programs (3,4). Transcriptional profiling can stratify LUSC tumors into biologically distinct subtypes with different survival outcomes, indicating that regulation of gene expression in LUSC has clinically relevant implications (5,6).
Metastasis accounts for approximately 90% of cancer-related deaths (7), yet there is a surprising paucity of scientific evidence addressing the mechanisms that drive LUSC metastasis. The ability of cancer to spread is reliant on both cancer cell intrinsic and extrinsic factors. Gene expression programs within cancer cells facilitate egress from the tumor through blood or lymphatic vessels, extravasation, and colonization of distant organs (8). MicroRNAs (miRs) regulate an increasing number of metastasis-relevant pathways through inhibition of target genes (9). However, the function of each miR is highly dependent on cancer type and even subtype (10,11).
Recently it was discovered that circular RNAs (circRNAs) can regulate miR function. CircRNAs are a covalently closed RNA which form through an alternative splicing mechanism that links the end of a 3’ exon with an upstream 5’ exon (termed a backsplice junction) and represent a new class of long-non-coding RNAs (12). Some circRNAs act as miR sponges and contain multiple miR seed sequences (13,14), but circRNAs may also act as protein scaffolds (15) or sequester RNA binding proteins (RBPs) (16). Though expression of circRNAs is dysregulated in cancer, the repertoire of circRNA functions beyond miR-sponging is still unclear. CircRNAs are structurally stable and released into the circulation within extracellular vesicles making them appealing biomarkers (17). Thus, characterizing the mechanism of action for circRNAs will facilitate biomarker development and identify possible therapeutic targets. Currently, therapeutic targeting of circRNAs is unexplored.
In the current study, we evaluated whether miRs can regulate LUSC progression and discovered that miR-671–5p suppresses metastasis through the circRNA, CDR1as. By integrating miR and mRNA sequencing data from TCGA and profiling of metastatic LUSC models generated from in vivo passaging, we identified miR-671–5p as a key regulator of LUSC metastasis. Screening for miR targets revealed that miR-671–5p functions through CDR1as. While the proposed function of CDR1as is through sponging of miR-7 (13,14), we instead describe a mechanism based on its antisense transcript, cerebellar degeneration related protein 1 (CDR1). CDR1 was highly linked to metastatic gene expression signatures and was necessary and sufficient to promote LUSC metastasis. CDR1 is a cryptic protein with an unusual sequence (composed predominantly of hexapeptide repeats) and no known function (18,19). We found that CDR1 directly interacts with vesicular coat protein complexes and that inhibiting vesicular trafficking blocked CDR1-dependent migration. Using a targeted lipid nanoparticle to deliver miR-671–5p to target the CDR1as/CDR1 axis we inhibited metastatic spread in vivo. These findings identify a previously unknown regulatory axis in LUSC progression and expand the functional repertoire of circRNAs in metastasis.
MATERIALS AND METHODS:
Cell lines and maintenance.
SK-MES-1 and H520 cells were obtained from the ATCC. HEK293T cells were kindly provided by Antonio Amelio (University of North Carolina at Chapel Hill) and HCC2814 were obtained from University of Texas Southwestern Medical Center. SK-MES-1 cells were grown in MEM supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin Streptomycin, 1% Non-Essential Amino Acids, and 1 mM Sodium Pyruvate. H520 and HCC2814 cells were grown in RPMI with 10% Fetal Bovine Serum (FBS) and 1% Penicillin Streptomycin. HEK293T cells were grown in DMEM with 10% FBS and 1% Penicillin Streptomycin. UNCN3T cells and culture methods for them have been described in detail (20). All cell lines were tested to confirm the absence of mycoplasma and grown at 37°C in 5% CO2/95% air. Experiments were performed with cells at 60–80% confluence. Cells were used fewer than 10 passages from thaw for in vivo experiments and fewer than 20 passages from thaw for in vitro experiments.
Generation of metastatic sub-clones.
Cells were harvested from metastatic lesions by mechanical dissection in serum free media containing 0.125% collagenase III and 0.1% hyaluronidase under sterile conditions. Minced tissues were filtered through a sterile 40 μm filter. After centrifugation, cells were incubated with 0.25% trypsin for 20 min at 37°C, agitating every 5–7 min. Cells were then grown in complete medium. Cells were expanded and FACS was performed using FITC-conjugated anti-human HLA-A,B,C antibody (32294x, Pharmingen) to remove any contaminating mouse cells.
Transfection of miR-671–5p mimics, siRs and pDNA.
For in vitro experiments, miR-671–5p or miR control mimics were purchased from Life Technologies and siRNAs were purchased from Sigma. Transfections of siRs and miR-671 mimics were performed using RNAiMAX (Invitrogen) at a final concentration of 20 nM. For transfection of DNA, lipofectamine 2000 (Invitrogen) was used.
Animals, in vivo models and tissue processing.
Female athymic nude mice were purchased from the UNC Animal Studies Core. All animals used were between 6–10 weeks of age at the time of injection. For all animal experiments, cells were trypsinized, washed and resuspended in Hanks balanced salt solution (HBSS; Gibco) prior to injection. H520 or SK-MES-1 cells or related sub-clones were injected by an intra-pulmonary technique [1 × 106 cells in 50 μL 1:1 mixture of HBSS and BD Matrigel (BD Biosciences)]. For the intra-pulmonary injections, mice were anesthetized with ketamine (80 mg/kg) + xylazine (8 mg/kg) + acepromazine (1 mg/kg) and placed in the right lateral recumbency. Following sterile skin preparation, an incision parallel to the rib cage between ribs 10 and 11 was made to visualize the lung through the intact thoracic pleura. A 1 mL tuberculin syringe with a 30-g needle was used to inject the cell suspension directly into the lung parenchyma at the left lateral dorsal axillary line. After injection, the skin incision was closed using surgery clips and the mice were turned on the left lateral recumbency and observed until fully recovered. In all experiments 6–15 mice per group were used and mice were randomized before injection of cancer cells. For survival analyses, mice were monitored until they became moribund, then sacrificed. For cross-sectional analysis, once mice in any group became moribund, they were all sacrificed, necropsied, and tumors were harvested. At the time of sacrifice, tumor weights, number and location of lymphatic and distant metastases were recorded. Tissues used for immunohistochemistry analysis were fixed in 10% neutral buffered formalin, and embedded in paraffin. Tissues were stained with either Hematoxylin and Eosin, or Masson’s trichrome stain for histological evaluation. Tissues for RNA and protein extraction were snap frozen and stored at −80°C. RNA from tissues was isolated with TRIzol (Invitrogen) according to manufacturer’s instructions. Before RT-qPCR analysis, TRIzol isolated RNA was treated with DNase using DNA-free DNA removal kit (Invitrogen) according to manufacturer’s instructions. Luciferase-labeled tumor progression was monitored weekly using an IVIS Lumina optical imaging system and Nano-Glo Luciferase Assay substrate (Promega). Nano-Glo substrate was diluted in PBS and 250 μg/kg was administered 10 min before imaging.
Study Approval.
Animals were cared for according to the guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the U.S. Public Health Service policy on Human Care and Use of Laboratory Animals. All mouse studies were approved and supervised by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee. Tissue microarray (TMA) samples for lung squamous cell carcinoma and lung adenocarcinoma cancers were obtained and prepared following Institutional Review Board approval for UNC Chapel Hill.
Data and materials availability:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE19 partner repository with the dataset identifier PXD012286.
Supplementary Methods.
More detailed methods are available in the Supplementary Data and Methods. Sequences of shRs and RT-qPCR primers are listed in the Supplementary Data (Supplementary Table 1).
RESULTS:
Generation of highly metastatic LUSC models.
To develop metastatic models of LUSC, we chose SK-MES-1 and H520 human cell lines because they were recently found to be from aggressive mRNA subtypes associated with poor LUSC survival (5,6,21) and used an in vivo selection approach. We considered the highly prognostic value of lymph node (LN) metastases in lung cancer (1), including occult LN micro-metastases (22,23) and obtained sub-clones derived from LN metastases. While orthotopically injected parental SK-MES-1 cells metastasized to LNs in only 24% of mice, sub-clone SK-MES-LN1 (LN1) cells produced 100% LN metastases and very high rates of metastases to clinically relevant sites (e.g. adrenal glands and chest wall) (Supplementary Fig. 1A and B). While mice injected with SK-MES-1 lived >120 days, LN1-injected mice had substantially reduced survival (median 41 days, p<0.0001) (Supplementary Fig. 1C). Similarly, H520 and H520-LN3 (LN3), which was generated from 3 in vivo passages, had LN metastasis rates of 20% and 100%, respectively (Supplementary Fig. 1D and E). In vivo passaging of H520 significantly reduced survival of orthotopically injected mice from >60 days to 39.5 days mean survival (p<0.0001) (Supplementary Fig. 1F). Consistent with the human disease and the importance of LN metastasis (22,23), in both models, spontaneous development of LN metastasis significantly correlated with distant metastases (Spearman’s 2-sided t-test p<0.0001) (Supplementary Fig. 1G). Indeed, recent evidence supports that metastatic cells within LNs can efficiently seed distant sites (24–26). There were no observed differences in proliferation between parental cell lines and respective sub-clones (Supplementary Fig. 1H). While distant metastases were most commonly found on the chest wall and in the adrenal glands, we also observed distant metastases in the optic nerve, base of tongue, and pancreas (Supplementary Fig. 2A–E). Histological evaluation of LN1 orthotopic tumors revealed undifferentiated features, while LN3 showed central necrosis that is classically associated with LUSC. In both models orthotopic tumors had considerable intra-tumoral collagen, and LN metastases showed extra-capsular extension (Supplementary Fig. 3). Metastasis is associated with epithelial to mesenchymal transition (EMT) (27), to assess the role of EMT in the behavior of metastatic sub-clones, we profiled markers of EMT by qPCR. Relative to parental cell lines, both LN1 and LN3 had increased expression of SNAI2 and ZEB2, key transcriptional regulators of EMT (Supplementary Fig. 4). Taken together, the LN1 and LN3 models are highly metastatic and display typical features of human LUSC.
miR-671–5p inhibits LUSC metastasis.
miRs can have remarkable roles in regulating a repertoire of metastatic programs in many cancers (28). To screen miRs with potential roles in LUSC metastases, we analyzed miR expression data from 348 LUSC patients in TCGA. Using median cut-offs for each miR, 30 miRs were significantly associated with overall survival when individually considered (p<0.05) (Supplementary Table 2); therefore, this analysis was exclusively used as a screening tool to prioritize miR genes for experimental validation. Among these 30 genes, high expression was associated with poor survival (hazard ratio (HR)>1) in only 5 miRs, while for the remaining 25 miRs low expression was associated with poor survival (HR<1) (Fig. 1A and B). Altogether, these data suggest that suppression of miRs may generally lead to poorer outcomes in LUSC.
Figure 1. miR-671–5p inhibits LUSC metastasis.
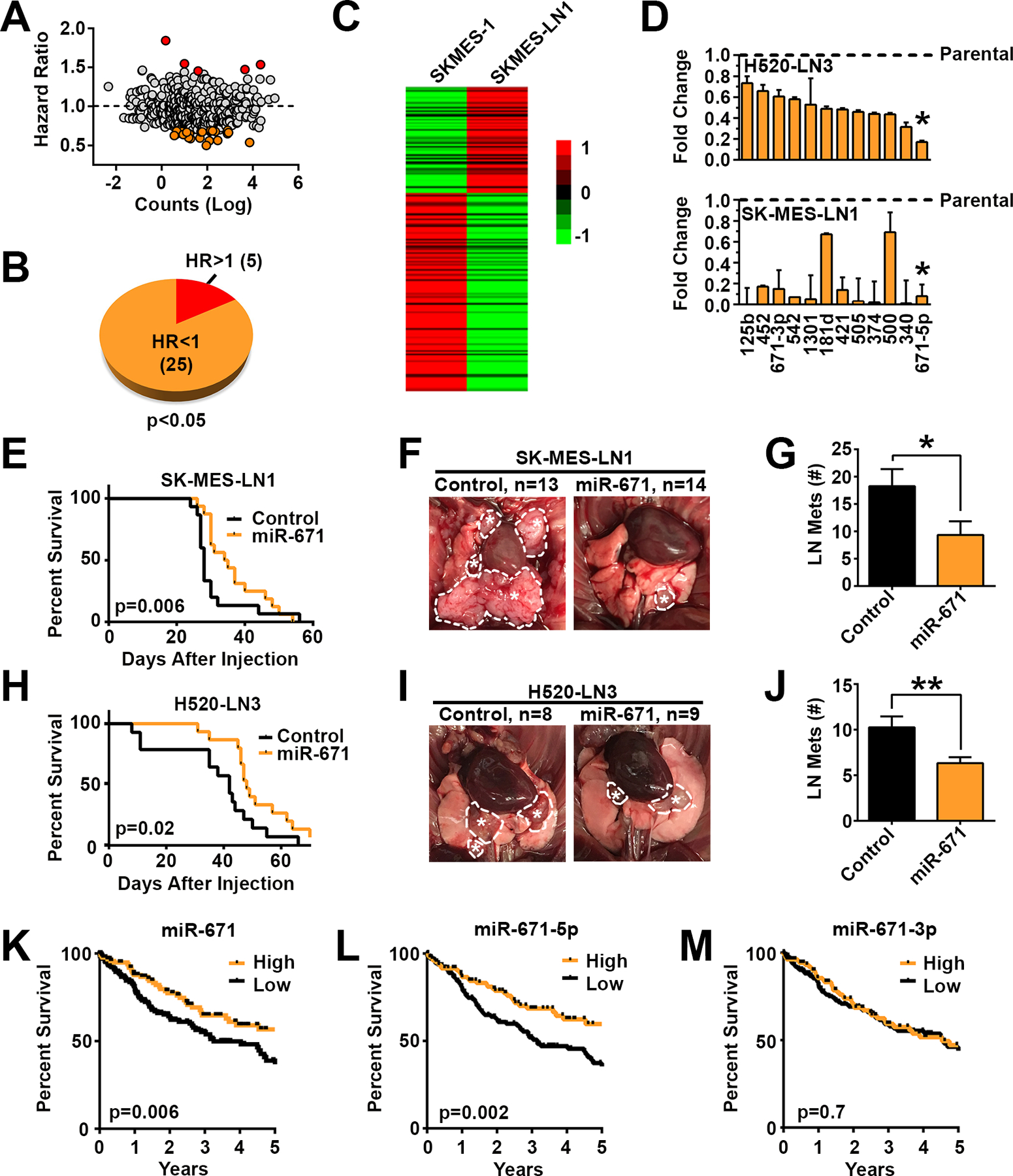
A, Hazard ratios for survival based on mean miR expression by abundance in TCGA. B, Screening of miRs significantly (p<0.05) associated, when individually considered, with increased survival (HR<1) or decreased survival (HR>1). C, Heatmap of Nanostring miR profiling of parental SK-MES-1 and LN1 sub-clone. D, Validation of miR expression in metastatic sub-clones compared with parental lines by qPCR. Asterisk indicates lead candidate. E, Survival plots for LN1 model when control miR or miR-671 is stably over-expressed. LN1 control n=15, miR-671 n=16. F, Representative LN metastasis (dotted circles with asterisk), and G, enumeration of LN metastases 23d after orthotopic injection with control miR or miR-671 overexpressing LN1. H, Survival of mice after injection of LN3 cells overexpressing control miR or miR-671, control n=14, miR-671 n=15. I, Representative images and J, enumeration of LN disease 31 d after injection with control or miR-671 LN3. K thru M, Overall survival in LUSC TCGA patients based on above/below mean expression of miR-671 gene and the two miR-671 isoforms, 5p and 3p. Survival based on the 5p and 3p isoforms. * p<0.05, ** p<0.01 by one-tailed Student’s t-test.
Next, we compared miR expression profiles between parental SK-MES-1 and the LN1 sub-clone using Nanostring data. Similar to our TCGA findings (Fig. 1A and B), a majority of miRs were down-regulated in the metastatic clones (Fig. 1C and Supplementary Table 3). Integrating miRs associated with survival and decreased in metastatic sub-clones, we selected 12 candidate miRs for validation (more details in Supplementary Methods). All miR candidates were decreased based on qPCR in both metastatic LN1 and LN3 sub-clones relative to their respective parental lines (Fig. 1D). Of these candidates, miR-671–5p was the most consistently reduced (<20% of parental expression) in both LN1 and LN3. In each model, stable over-expression of miR-671 at physiologic levels (4- to 7-fold increases over control miR) significantly increased survival and decreased metastatic burden (Fig. 1E–J), implicating miR-671 as a novel metastasis suppressor in LUSC. As seen when comparing metastatic and parental cell lines, no appreciable effect on proliferation was observed in miR-671 over-expressing cells (Supplementary Fig. 5). Survival analysis based on the expression of the miR-671 gene and miR-671–5p and −3p isoforms in TCGA revealed that miR-671–5p (HR=0.56, 95% CI 0.39 to 0.80, p=0.002), but not −3p (HR=0.92, 95% CI 0.64 to 1.33, p=0.7), was strongly associated with LUSC survival (Fig. 1K–M). The expression of miR-671–5p and −3p was significantly correlated (Spearman’s 2-sided t-test p<0.0001, R=0.19), but the expression of miR-671–5p was far more dynamic and significantly higher than −3p (Supplementary Figure 6A and B).
miR-671–5p silences the CDR1as/CDR1 axis.
To systematically identify biologically relevant miR-671–5p target genes in LUSC, we leveraged a previously described approach to integrate a linear regressions model of TCGA data and in silico predictions (11,29). The miR target prediction algorithm TargetScan (30) was used to predict miR-671–5p regulated genes (Supplementary Fig. 7A). Then, to enrich for genes relevant for LUSC biology, candidate genes had to meet 4 conditions based on TCGA analysis (n = 348): 1) inverse correlation with miR-671–5p (R < −0.25, FDR <0.00001), 2) survival relevance (p < 0.05 and HR > 1.5), 3) influence of miR-671–5p on target mRNA expression (> 25%), and 4) joint survival of target mRNA and miR-671 with more stringent criteria than for genes alone (p < 0.01 and HR > 2). Based on these 4 criteria, 13 mRNAs were considered clinically relevant candidate miR-671–5p targets in LUSC (Supplementary Table 4). To validate these putative target genes, qPCR was performed on LN1 and LN3 cells stably expressing miR-671 or miR-671–5p inhibitors. Two previously validated targets (FOXM1 (31) and CDR1as (32)) were included as positive controls. Of the genes tested, two genes (MSR1 and CD93) were lowly expressed in LN1 and LN3 cell lines and therefore excluded. Surprisingly, CDR1as and IL-16 were the only target genes to show the predicted directionality in all 4 validation experiments (Supplementary Fig. 7B–E). Effects of miR-671 manipulation on CDR1as were notably stronger than on IL-16 in most experiments. Ranking miR-671–5p target genes based on miR binding-site strength revealed that CDR1as, a circular antisense transcript of the CDR1 gene (32), had a Context++ score of −3.7, several fold lower than the next lowest score of −0.53 for ANTXR2 (Supplementary Fig. 7F). Moreover, the predicted miR-671–5p binding site in CDR1as was complementary at 21/23 bases, resembling an siRNA (Fig. 2A and B). We confirmed that miR-671–5p potently decreased CDR1as expression in both of our metastatic sub-clones (Fig. 2C). These data show that miR-671–5p strongly targets the non-coding RNA CDR1as. CDR1as was not detected in our initial screen because our target prediction was limited to linear, protein-coding mRNAs.
Figure 2. miR-671–5p silences the CDR1as/CDR1 axis.
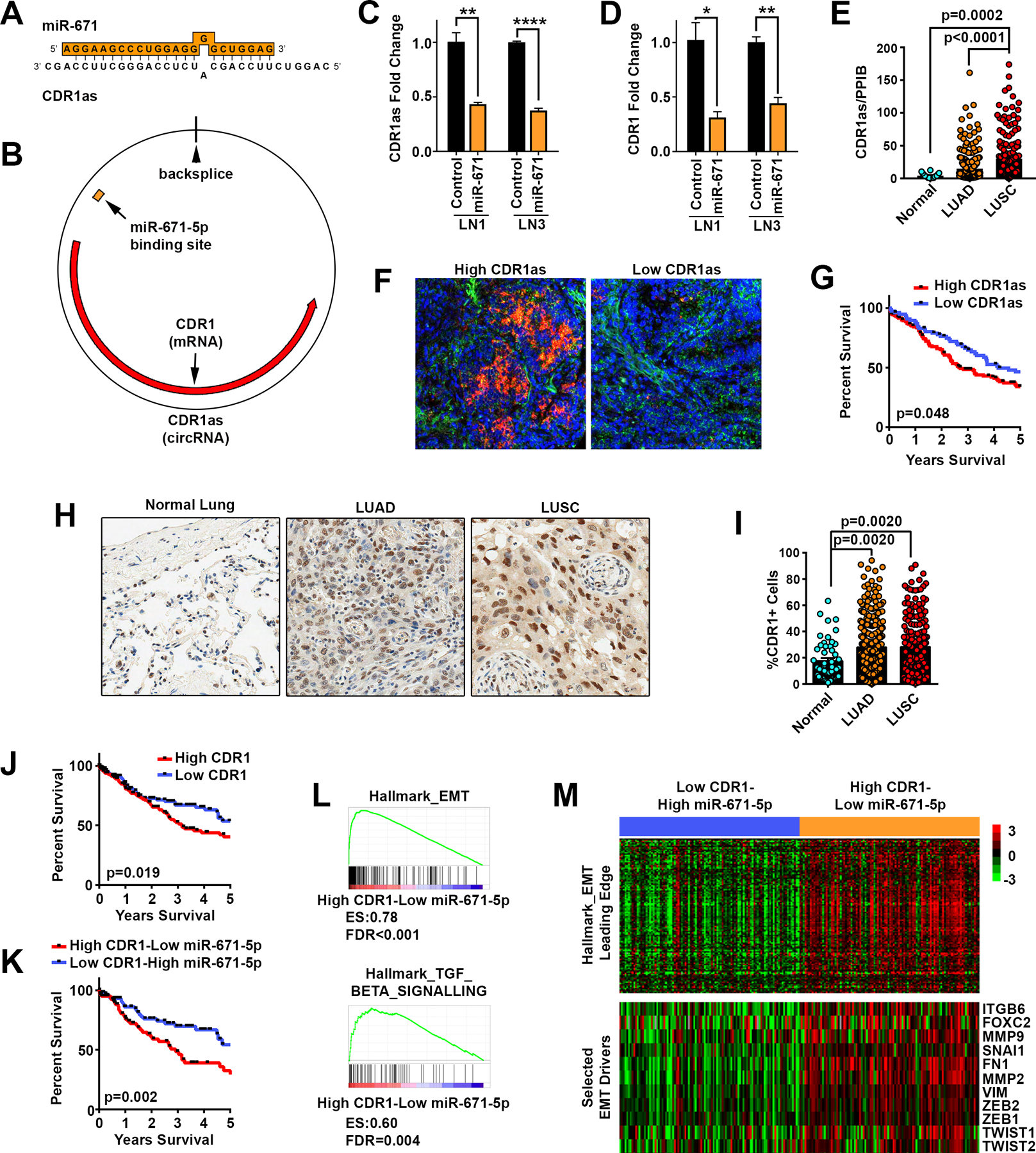
A, miR-671–5p binding site on CDR1as target gene. B, Orientation of miR-671–5p binding on the circular CDR1as and regions of complementarity between CDR1as and CDR1 mRNA. C and D, Transfection with 20 nM miR-671 mimics inhibited CDR1as and CDR1 RNA levels measured by qPCR at 48h after transfection, using divergent and stranded primers respectively. Expression was normalized to 18S, GAPDH, and TBP housekeeping genes (n=3). E, Quantification of CDR1as normalized to positive control PPIB in normal lung tissue (n=18), LUAD (n=266), and LUSC (n=191) tumors. Significance determined by ANOVA corrected for multiple testing using the FDR. F, Representative ISH for CDR1as (red) and positive control PPIB (green) in LUSC tissue microarrays. G, Survival of NSCLC patients based on High (n=144) or Low (n=77) CDR1as by ISH. H, Representative IHC for CDR1 in NSCLC tissue microarrays. I, Quantification of %CDR1+ cells per TMA core, normal lung tissue (n=50), LUAD (n=281), and LUSC (n=202) tumors. Significance determined by ANOVA corrected for multiple testing using the FDR. J, Survival of LUSC patients based on High or Low CDR1 mRNA expression in TCGA based on the median expression of CDR1. K, Joint (median-based) survival analysis of CDR1 and miR-671–5p. L and M, Gene set enrichment plots and heatmaps of 122 genes belonging to the hallmark EMT leading edge (top) and of 11 EMT selected markers (bottom), showing their upregulation in High CDR1/Low miR-671–5p LUSC tumors. * p<0.05, ** p<0.01, **** p<0.0001 by two-tailed Student’s t-test.
CDR1as (also known as ciRS-7), is a direct target of miR-671–5p as confirmed by luciferase assay (32) and Argonaut cross-linking and immunoprecipitation (AGO-CLIP) in mouse and human (33). CDR1as contains >70 repeats of the miR-7 seed sequence and is known to act as a miR-7 sponge (13,14,32). Unlike miR-671–5p, which acts through the endonuclease Ago2 (32), miR-7 binds more weakly through a canonical 7–8 nucleotide seed sequence (Supplementary Fig. 8A). Therefore, CDR1as can sequester miR-7 and associated RNA-induced silencing complexes (RISCs) leading to disinhibition of miR-7 targets (13,14). To determine whether miR-671–5p silencing of CDR1as can increase free miR-7 and lead to decreased miR-7 targets, we measured the levels of 9 validated miR-7 targets with a focus on those relevant to cancer biology (Supplementary Fig. 8B and C). Interestingly, we found no significant changes in miR-7 targets in LN1 or LN3 cells overexpressing miR-671. Of the 9 genes tested, none showed more than a 30% decrease in cells overexpressing miR-671. These findings suggest miR-671–5p’s role in LUSC metastasis may occur through CDR1as, although in a miR-7-independent manner. CDR1as has also been shown to increase the expression of the CDR1 mRNA, with which it shares a gene locus (32). CDR1 was discovered as a target of autoantibodies in paraneoplastic cerebellar degeneration (18,19). Although CDR1 is highly expressed in the brain and up-regulated in some cancer types, its biologic function is unknown. To confirm that miR-671–5p also reduced CDR1 expression in LUSC cells, we measured CDR1 by strand-specific reverse transcription followed by qPCR in cells transfected with miR-671–5p mimics. miR-671–5p strongly decreased CDR1 expression in both LN1 and LN3 sub-clones (Fig. 2D).
We next assessed the biological relevance of the CDR1as/CDR1 axis in lung cancer and metastases. Expression of CDR1as was measured using in situ hybridization (ISH) on a clinically-annotated tissue microarray containing duplicate biopsies of LUSC, LUAD and patient matched-normal lung samples (Fig. 2E–G). Notably, we found that CDR1as was 10-fold higher in LUSC tumors than matched-normal lungs (p=0.0002) and 2-fold higher compared to LUAD tumors (p<0.0001) (Fig. 2E). High CDR1as expression was associated with worse survival in non-small cell lung cancer (NSCLC) (HR=1.39, 95% CI 1.01 to 1.94, p=0.048) (Fig. 2G), suggesting a role in driving metastases. Immunohistochemistry (IHC) for CDR1 also showed a significant increase in both LUAD and LUSC tumors compared to normal lung (Fig. 2H and I). CDR1 staining was present in the nucleus, cytoplasm, and perinuclear space and its localization varied between patients and cells within an individual tumor (Supplementary Fig 9). High CDR1 expression was associated with worse survival in NSCLC (HR=1.43, 95% CI 1.02 to 2.01, p=0.0429). Similarly, in TCGA, high expression of CDR1 in LUSC patients was associated with significantly worse survival (HR=1.57, 95% CI 1.10 to 2.24, p=0.019) (Fig. 2J). To address the importance of the miR-671–5p/CDR1as/CDR1 axis in LUSC, we performed a dual survival analysis of miR-671–5p and CDR1 and found that Low miR-671–5p/High CDR1 expression in tumors was associated with worse survival than High miR-671–5p/Low CDR1 expression (HR=2.094, 95% CI 1.311 to 3.344, p=0.002) (Fig. 2K). Using TCGA to cluster LUSC tumors based on High CDR1/Low miR-671–5p and Low CDR1/High miR-671–5p expression, we identified 2,123 differentially expressed genes (Supplementary Fig. 10A, Supplementary Table 5). Gene set enrichment analysis (GSEA) in the High CDR1/Low miR-671–5p cohort revealed dramatic enrichment for signatures of EMT and signaling of TGFβ (a potent inducer of EMT) (Fig. 2L and M). These GSEA findings were powerfully matched by a gene ontology (GO) analysis of the genes upregulated in this cohort (Supplementary Fig. 10A, top portion), which showed that the most statistically significant GO term (p=1.10E-49) was ‘cell adhesion’ (Supplementary Fig. 10B and Supplementary Table 6); indeed EMT and cell-cell adhesion are intimately related (27). GSEA also identified a strong enrichment for genes upregulated when there is loss of the cell adhesion molecule E-Cadherin (CDH1) (Supplementary Fig. 10C). Consistent with these findings, CDR1 expression across >1,000 cancer cell lines strongly correlated (Spearman’s 2-sided t-test p<0.0001) with several EMT genes (Supplementary Fig. 10D, Supplementary Table 7). Taken together, these findings imply that CDR1 expression is closely tied to poor survival and the expression of metastatic gene signatures in LUSC, including EMT.
The CDR1as/CDR1 axis is necessary and sufficient for LUSC metastasis.
To determine whether inhibiting the CDR1as/CDR1 axis is necessary for the suppressive effect of miR-671–5p on LUSC metastasis, LN1 cells were stably transduced with shRNAs (shRs) against CDR1 or CDR1as. Inhibition of CDR1/CDR1as with several independent shRs (Supplementary Fig. 11A–C) significantly increased survival of orthotopically injected mice from 53 days (control shR) to between 78 and 136 days (CDR1 and CDR1as shRs, respectively) (Fig. 3A). In addition, inhibition of CDR1 or CDR1as markedly reduced LN metastases (Fig. 3B) in all 4 groups, and tumor burden as measured by luciferase expression (Fig. 3C and D). CDR1as shR2 showed the strongest inhibition of LUSC progression and metastasis, reducing LN mets by >75% (p=0.004) and tumor burden by 30-fold (p=0.0002) relative to control (Fig. 3B and C). To test whether CDR1 is sufficient to promote LUSC progression and metastasis, SK-MES-1 and LN1 cells were stably transduced with the CDR1 ORF. CDR1 overexpression in SK-MES-1 cells was sufficient to increase distant metastases to the level of the aggressive LN1 model (p<0.05), but had no significant effect on primary tumor size (Fig. 3E–G). CDH1 was decreased in tumors over-expressing CDR1, supporting a role for CDR1 in EMT (Supplementary Fig. 12). Taken together, the CDR1as/CDR1 axis was necessary and CDR1 alone was sufficient to drive LUSC metastasis. Importantly, overexpression of CDR1 was able to partially rescue the metastatic phenotype of CDR1as knockdown, showing that the biological function of CDR1as is at least in part dependent on its regulation of CDR1 (Fig. 3H and I).
Figure 3. The CDR1as/CDR1 axis is necessary and CDR1 is sufficient for LUSC metastasis.
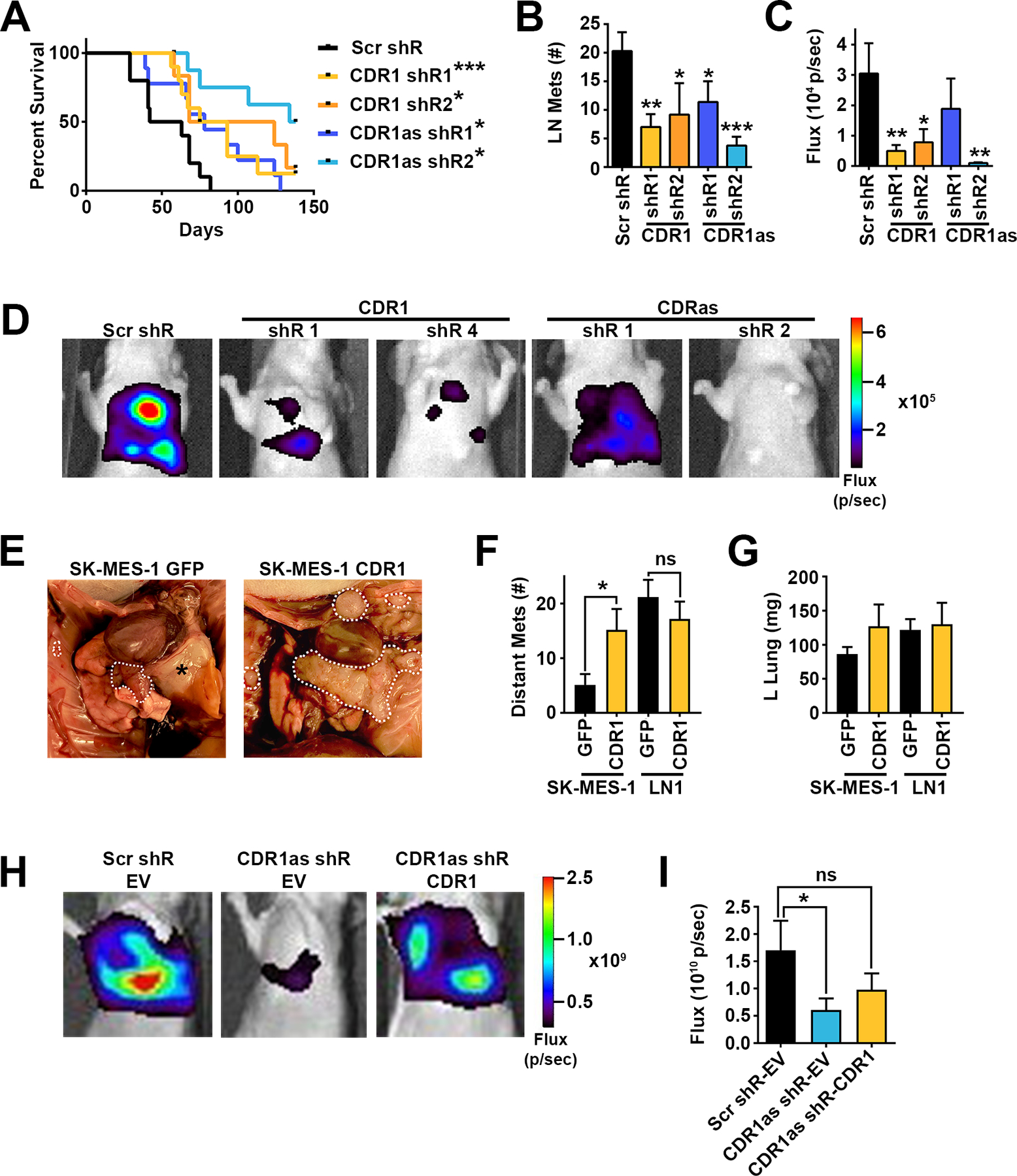
A, Kaplan Meyer survival curves of mice orthotopically injected with shR transduced SK-MES-LN1 cells. Scr shR n=10, CDR1 shR1 n=10, CDR1 shR2 n=6, CDR1as shR1 n=9, CDR1as shR2 n=9. * p<0.05, *** p<0.001 by log-rank test. B, LN metastases at time of death in mice orthotopically injected with shR transduced SK-MES-LN1 cells. C, Quantification of thoracic luciferase signal and D, representative luciferase images 3 weeks after orthotopic injection of shR transduced cells. * p<0.05, ** p<0.01, *** p<0.001 by ANOVA corrected for multiple testing using the FDR. E, Representative images of thoracic disease, F, quantification of distant metastases, and G, weight of tumor bearing left lung in mice orthotopically injected with SK-MES-1-GFP, SK-MES-1 CDR1, LN1-GFP, or LN1-CDR1 (n=15 per group). * p<0.05 by Student’s t-test. H, Representative luciferase images and I, quantification of thoracic luciferase signal 3 weeks after orthotopic injection of shR and EV or CDR1 transduced SK-MES-LN1 cells. Scr-shR-EV n=9, CDR1as shR-EV n=10, CDR1as shR-CDR1 n=10. * p<0.05 by one-tailed Student’s t-test.
Therapeutic delivery of miR-671–5p inhibits metastasis.
In order to target the CDR1as/CDR1 axis therapeutically, we formulated lipid protamine hyaluronic acid nanoparticles (LPH-NPs) containing miR-671–5p mimics (Fig. 4A). A characteristic core structure was seen by transmission electron microscopy (TEM) (Fig. 4B) and LPH-NPs had a hydrodynamic diameter ~100 nm in size, as measured by dynamic light scattering (DLS), with a charge of +40 mV. LPH-NPs were decorated with the Sigma1R ligand amino-ethyl-anisamide (AE-AA) to facilitate uptake into tumor cells (34). Both LN3 and LN1 expressed Sigma1R on the cell surface as determined by biotinylation and isolation of cell surface proteins (Supplementary Fig. 13A). Also, SIGMAR1 expression in LUSC tumors was just over half that of EGFR (1820±1200 and 3630±7070, respectively), which is often amplified in LUSC (4), and more than 10 times higher than hormone receptors, ESR1 (90±140), PGR (30±40), and AR (20±40) (Supplementary Fig. 13B). Incorporation of AE-AA increased uptake of LPH-NPs carrying fluorescently tagged dsRNA (Fig. 4C). Given the higher expression of Sigma1R on the surface of LN3 cells (Supplementary Fig. 13), this model was chosen for further therapeutic experiments. LPH-NPs carrying fluorescently labeled dsRNA were used to measure the biodistribution of dsRNA in tumor bearing mice. Forty-eight hours after a single IV injection, dsRNA accumulated in H520-LN3 lung primary and metastatic tumors (Fig. 4D and E). Time-kinetic experiments revealed that miR-671–5p LPH-NPs decreased CDR1as expression by 60% relative to control LPH-NPs at 24h, and CDR1 positive cells by 70% at 48h (Supplementary Fig. 14A–C). Treatment with miR-671–5p NPs 3x per week for three weeks significantly decreased the number of LN metastases in H520-LN3 mice (Fig. 4F and G) without evident toxicity or weight loss (Supplementary Fig. 15). In tumors collected 24h after the final NP injection, the number of CDR1 positive cells was significantly decreased in LN metastases of mice treated with miR-671–5p NPs compared to controls (Fig. 4H and I). Interestingly, LN metastases had significantly higher numbers of CDR1 positive cells than primary lung tumors (Fig. 4I), consistent with CDR1 having a role in LUSC metastases.
Figure 4. Therapeutic delivery of miR-671–5p inhibits metastasis.
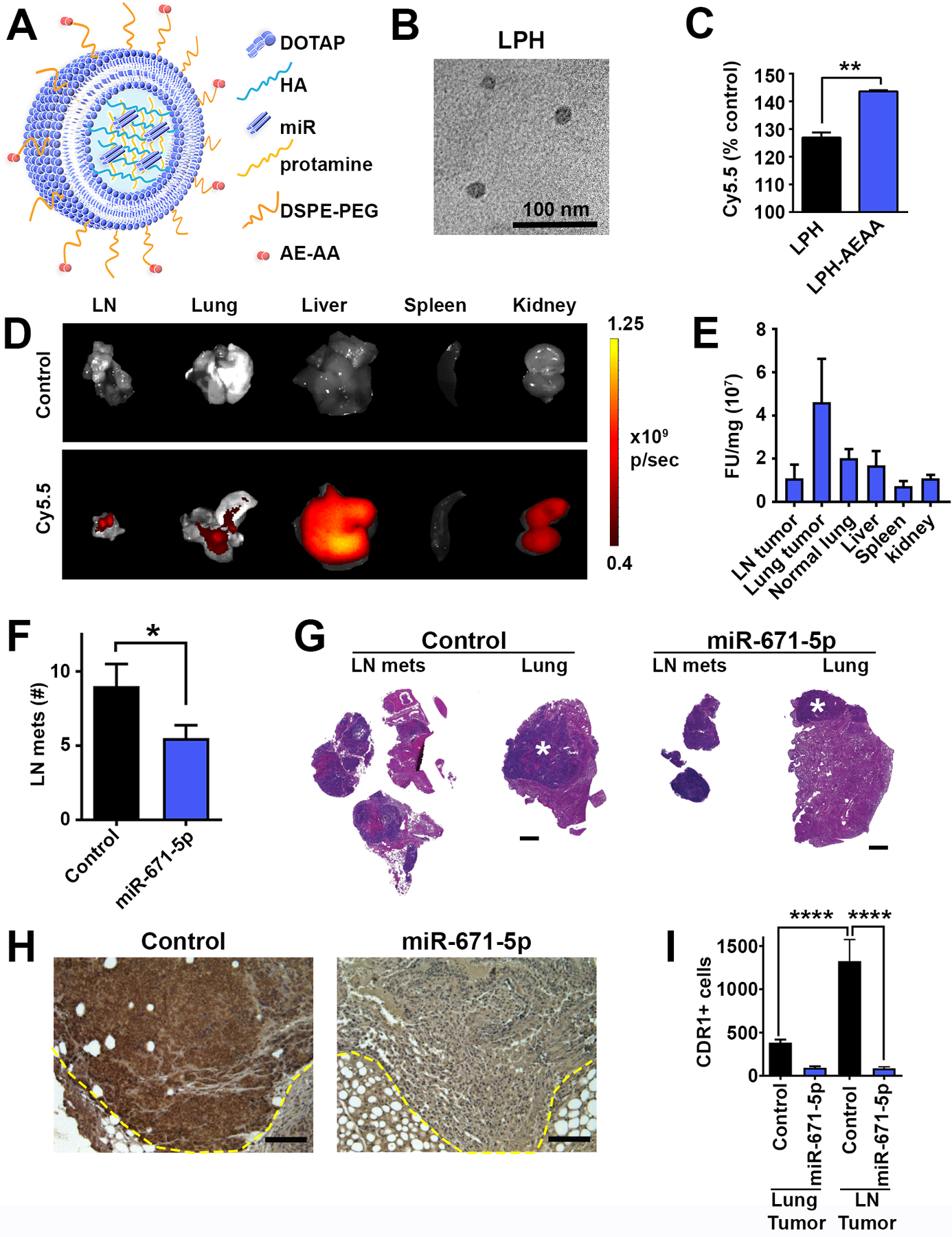
A, Graphic representation of lipid protamine hyaluronic acid (LPH) NPs, hyaluronic acid (HA), Aminoethyl anisamide (AE-AA). B, Transmission electron micrograph of LPH NPs. C, In vitro uptake of Cy5.5 labeled RNAs encapsulated in LPH NPs with or without SIG-1R ligand AE-AA in LN3 measured by flow cytometry, shown are the mean and SEM for three independent samples. ** p<0.01 by Student’s t-test. D and E, Mice orthotopically injected with H520-LN3 cells were administered Cy5.5 labeled RNA formulated in LPH NPs IV, 24h later organs were harvested for ex vivo quantification of fluorescence. D, Representative images and E, quantification of fluorescence normalized to tissue weight (n=2–3). Non-injected tumor bearing mice were used as control. F, Number of LN metastases after 8 IV injections of control miR or miR-671–5p NPs in H520-LN3 model. Control (n=11), miR-671–5p (n=10). * p<0.05 by one-tailed Student’s t-test. G, representative LN metastases and tumor bearing left lungs treated with control or miR-671–5p NPs. Asterisk indicates primary lung tumor. H, Representative IHC staining for CDR1 24h after final IV administration of control miR or miR-671–5p NPs; scale bar is 100 μm. I, quantification of CDR1 positive cells by IHC per high powered field 24h after final injection of miR control or miR-671–5p NPs. 25–30 fields per group, **** p<0.0001 by ANOVA corrected for multiple testing using the FDR.
CDR1 promotes migration and interacts with ER-Golgi network.
To characterize the cellular effects of CDR1 overexpression, we assessed proliferation and migration in our LUSC cell lines. While modest effects on proliferation and colony formation were observed (Supplementary Fig. 16A–C), overexpression of CDR1 in parental SK-MES-1 cells was sufficient to significantly drive chemotactic migration to the level of the LN1 sub-clone (Fig. 5A). Similarly, SK-MES-1-CDR1 cells moved with greater velocity (p<0.01) and traveled a greater distance (p<0.05) compared to SK-MES-1-GFP in a random migration assay on type 1 collagen (Fig. 5B). These experimental outcomes are aligned with the GO analysis of the genes upregulated in High CDR1/Low miR-671–5p, which shows that one of the most significant biological process of these patients is ‘positive regulation of cell migration’ (Supplementary Fig. 10B), intended as an increased frequency, rate or extent of migratory capabilities. To understand the mechanism by which CDR1 promotes metastasis, we identified protein interacting partners of CDR1 using epitope-mediated immunoprecipitation followed by mass spectrometry (IP-MS). A total of 34 proteins were associated with CDR1 in both 293T and LN1 cells stably expressing CDR1-Flag (Fig. 5C and D), and 19/34 proteins were localized to the Golgi apparatus and/or endoplasmic reticulum (Fig. 5C). It is noteworthy that EMT-driven Golgi compaction has recently been linked to promotion of lung adenocarcinoma metastasis (35). Several CDR1-interacting partners were validated by IP-WB (Fig. 5E). CDR1 interacted with coatomer protein I (COPI) complex subunits, COPA and COPE, which mediate transport between the ER and Golgi (36), as well as several members of the AP-1 complex (AP1G1, AP1G2, and AP1S1). The AP-1 complex serves as an adaptor for clathrin, traffics between the trans-Golgi network and endosomes, and is important for basolateral polarized sorting (36).
Figure 5. CDR1 promotes migration and interacts with ER-Golgi network.
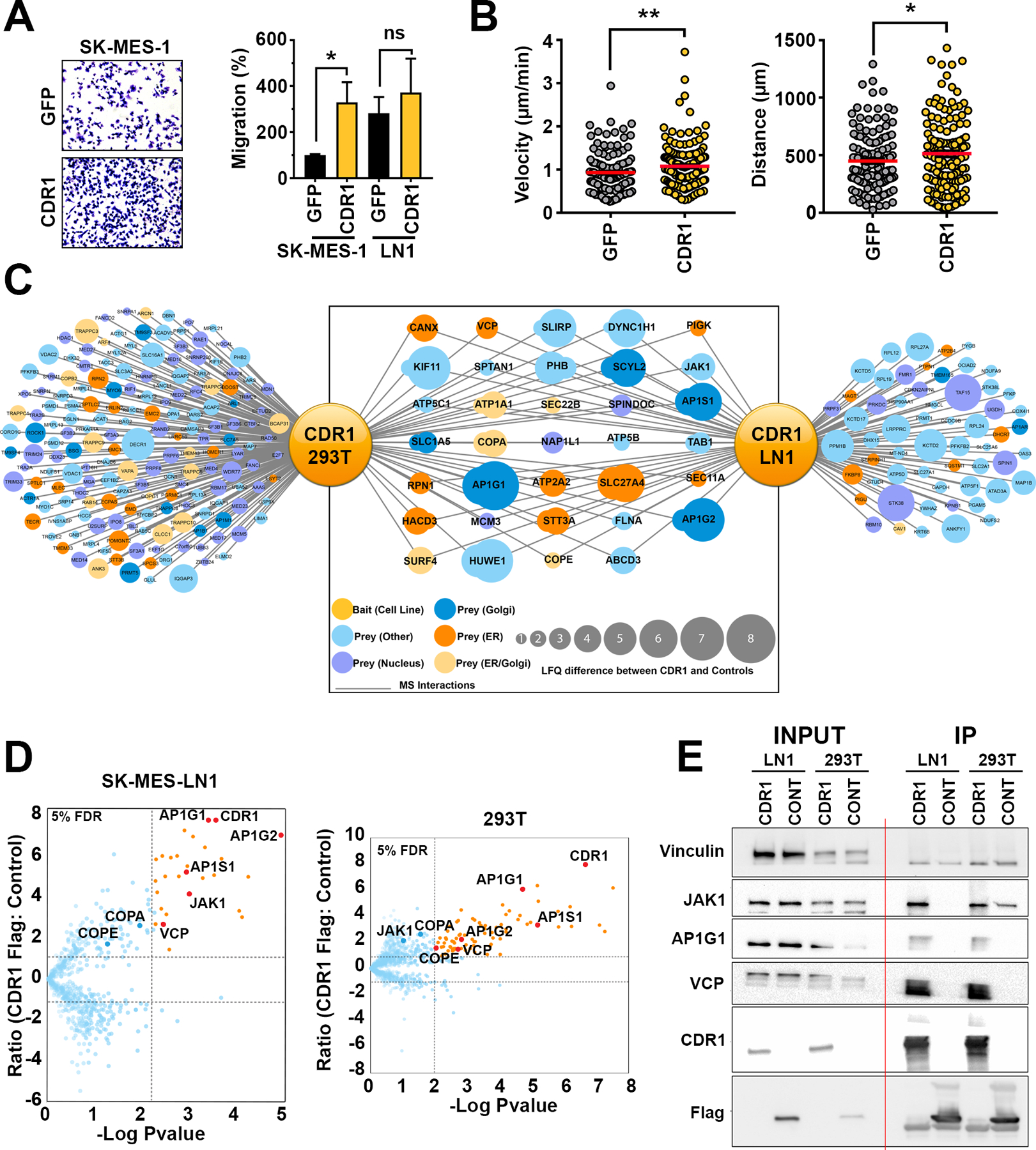
A, Representative images and quantification of trans-well migration of parental SK-MES-1 and LN1 cells stably overexpressing CDR1 or GFP control. Cell counts per high powered field were normalized to SK-MES-1 GFP control, shown are the mean and SEM of three independent experiments performed in duplicate. * p<0.05 by Student’s t-test. B, Velocity and distance of SK-MES-1 GFP (n=135) or CDR1 (n=145) cells analyzed by live cell tracking, pooled data from three independent experiments. * p<0.05 by one-tailed Student’s t-test. C, Network of CDR1 interacting partners determined by IP-MS. D, Volcano plots showing proteins enriched in CDR1-Flag immunoprecipitation (IP) compared to HcRED-Flag control in LN1 and 293T. E, Western blot validation of IP. Vinculin (VINC) was used as a loading control.
CDR1 promotes migration through Golgi trafficking.
To explore the interaction between CDR1 and vesicular proteins, we performed immunocytochemistry for CDR1. CDR1 staining was observed in the nucleoplasm, ER and Golgi structures (Fig. 6A) consistent with IP-MS results (Fig. 5C). Co-immunostaining for COPA and AP1G1 showed that CDR1 partially co-localized with COPA and AP1G1 in perinuclear structures consistent with Golgi (Fig. 6A, Supplementary Fig. 17A and Supplementary Table 8). CDR1 also partially colocalized with COPA and AP1G1 in HCC-2814 cells, which express intermediate endogenous levels of CDR1 (Supplementary Fig. 17B and Supplementary Table 8). Proximity ligation assays confirmed that CDR1 interacts with COPA and AP1G1 in LN1 and LN3 cells at endogenous levels (Fig. 6B). Consistent with CDR1 regulation of Golgi-mediated processes, air-liquid interface (ALI) cultures of the human bronchial epithelial cell line (HBEC) UNCN3T overexpressing CDR1 displayed a striking increase in mucin production (Supplementary Fig. 18). Furthermore, use of brefeldin A (BFA) to inhibit Arf1-GTPase, which is required for AP-1 and COPI membrane anchorage (37), abolished CDR1-dependent migration and haptotaxis (Fig. 6C and D). Similarly, in epistasis experiments, silencing of COPA but not AP1G1 blocked CDR1-dependent migration (Fig. 6E), suggesting that COPA is functionally downstream of CDR1-mediated migration.
Figure 6. CDR1 driven migration is dependent on vesicular trafficking.
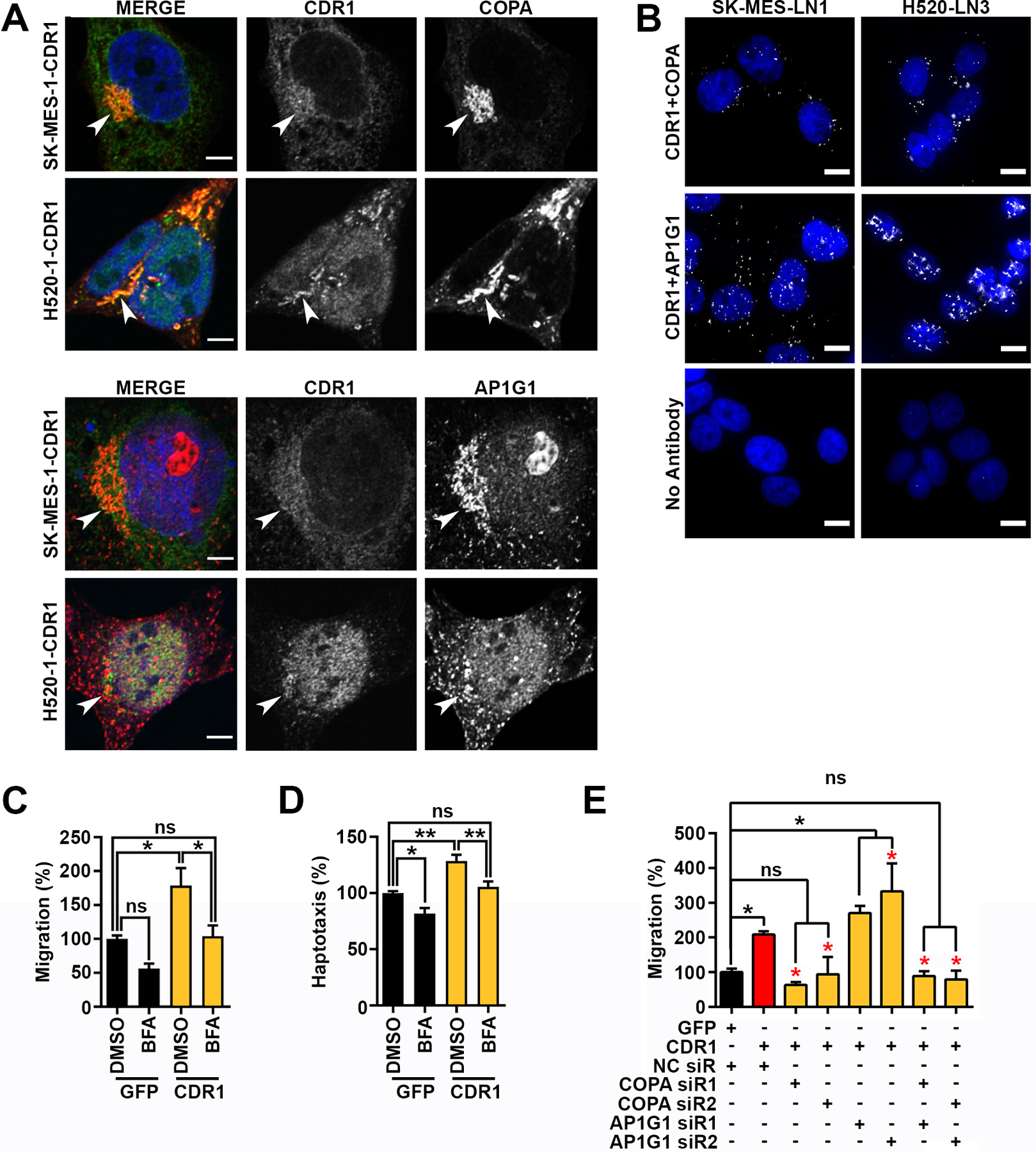
A, Localization of COPA or AP1G1 (red) and CDR1 (Green) in SK-MES-1 CDR1 or H520-CDR1. Shown is a single slice of a z-stack, arrows indicate perinuclear Golgi structures, scale bar is 5 μm. B, Proximity ligation assays showing close proximity of CDR1 with COPA or AP1G1 at endogenous levels in SK-MES-LN1 and H520-LN3 cells, scale bar is 10 μm. C and D, quantification of C, trans-well migration or D, haptotaxis of SK-MES-1 GFP or CDR1 with vehicle DMSO or 1 μg/mL Brefeldin A (BFA), shown are the mean and SEM of three experiments performed in duplicate. E, quantification of trans-well migration of SK-MES-1 CDR1 48 hours post transfection with negative control (NC), COPA, or AP1G1 siRs, shown are the mean and SEM of three experiments performed in duplicate. Black asterisks indicate significance from GFP NC siR, red asterisks indicate significance from CDR1 NC siR. * p<0.05, ** p<0.01 by ANOVA corrected for multiple testing using the FDR.
Considering that Golgi-complex reorientation is essential during cell migration (38), we tested whether CDR1 expression altered Golgi orientation during a scratch assay. Consistent with a pro-migratory phenotype, SK-MES-1-CDR1 cells invaded the scratch in significantly greater numbers than SK-MES-1-EV cells (Fig 7A and B). The orientation of the Golgi in cells at the scratch edge was scored as either on the side of the nucleus facing toward the scratch (toward) or on the side of the nucleus facing away from the scratch (away). Based on this analysis, a significantly higher proportion of CDR1 cells had Golgi oriented toward the scratch than EV cells (Fig. 7C), indicating that CDR1 increases Golgi orientation toward the direction of migration.
Figure 7. CDR1 increases Golgi reorientation and trafficking kinetics.
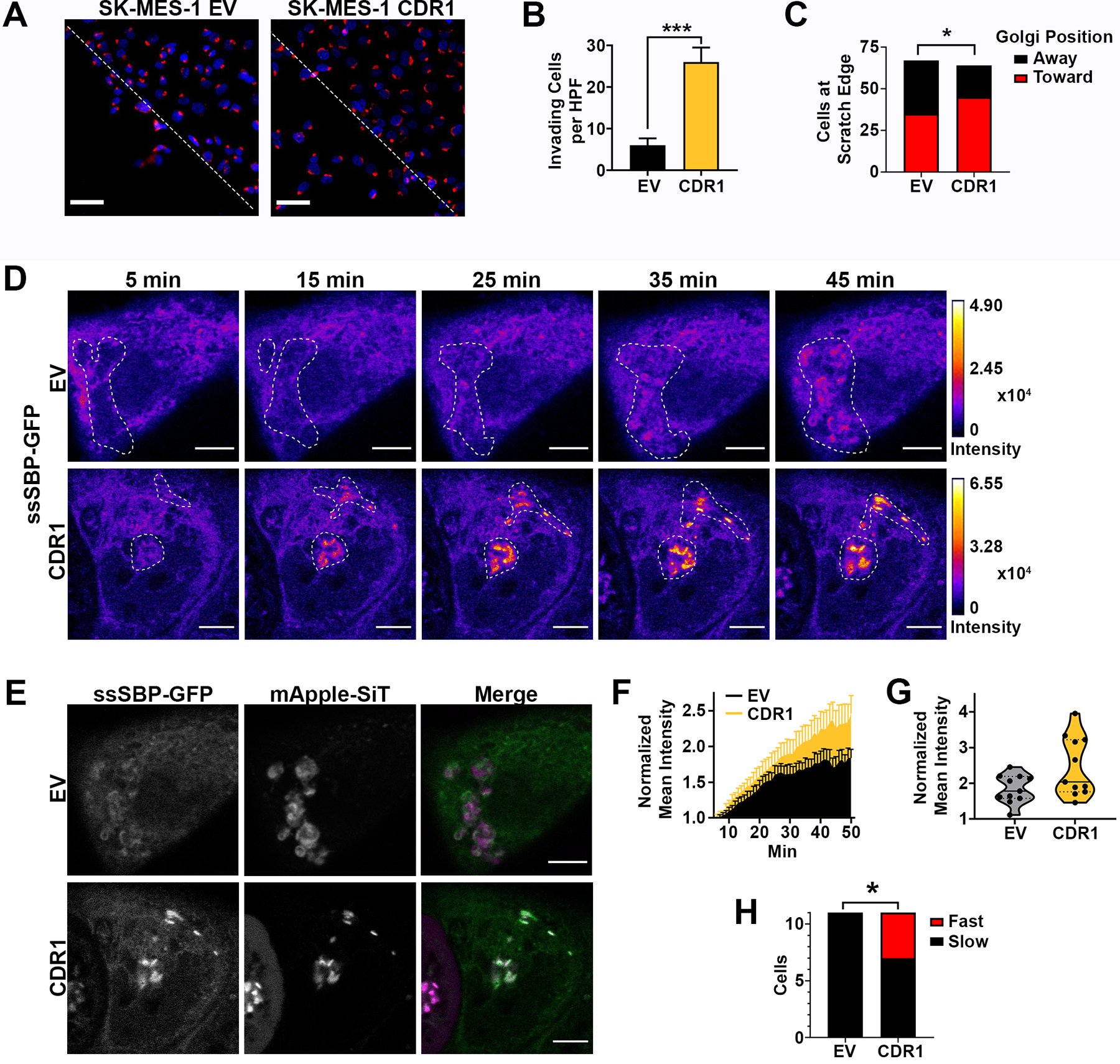
A, Representative images of SK-MES-1 EV and CDR1 cells 12 hours after scratch was formed, red is Golgi marker GM130. Dotted line indicates scratch border, scale bar is 50 μm. B, Quantification of cells invading the scratch at 12h (n=6 fields per group), *** p<0.001 by two-tailed Student’s t-test. C, Quantification of Golgi orientation towards or away from the scratch in cells at the scratch edge, 64–67 cells per group, significance determined by two-tailed Chi-square test, * p<0.05. D, Representative time course of ssSBP-GFP trafficking from the ER to the Golgi beginning 5 min after biotin addition in SK-MES-1 EV and CDR1 cells. Images are colored by GFP fluorescence intensity, Golgi mask (dotted line) determined by mApple-SiT labeling. Scale bar is 5 μm. E, Representative images of ssSBP-GFP (Green) and Golgi marker mApple-siT (Fuchsia) 45 min after biotin addition in SK-MES-1 EV and CDR1 cells. Scale bar is 5 μm. F, Mean GFP intensity in the Golgi normalized to baseline value from 5–50 min after biotin addition in SK-MES-1 EV and CDR1 cells. Golgi position was determined by mApple-SiT marker. Shown are the mean and SEM of two independent experiments, 11 cells per group. G, Mean GFP intensity in the Golgi normalized to baseline value at 50 min post biotin addition in SK-MES-1 EV and CDR1 cells. Solid line is the mean, dotted lines represent quartiles, 11 cells per group. H, Proportion of cells with Fast (>3 Fold Baseline) or Slow (<3 Fold Baseline) trafficking at 50 min after biotin addition. Significance determined by two-tailed Chi-square test * p<0.05, 11 cells per group.
ER to Golgi trafficking is required for the secretion of pro-metastatic factors. To determine whether CDR1 overexpression has a functional effect on ER to Golgi trafficking, we utilized the retention with selective hooks (RUSH) assay (39) to quantitatively measure the rate of ER to Golgi trafficking. SK-MES-1-empty vector (EV) or -CDR1 cells were transfected with streptavidin binding protein (SBP) fused to a secreted soluble GFP (ssSBP-GFP) and streptavidin fused to the invariant chain of the MHC (Ii) (Str-Ii), which acts as an ER hook. ssSBP-GFP was retained in the ER until the addition of biotin, which released ssSBP-GFP to traffic into the Golgi. Fluorescent images were captured every min starting 5 min after biotin addition for 45 min. GFP accumulation in the Golgi, labeled with mApple-Sialyltransferase (mApple-SiT), was quantified (Fig. 7D and E, Supplementary Video 1 and 2). Both SK-MES-1-EV and -CDR1 showed an accumulation of GFP in the Golgi over the imaging period (Fig. 7F). We observed a small population of fast trafficking cells (>3 fold from baseline at 50 min after biotin addition) (Fig. 7G). There was a significantly higher proportion of “fast” trafficking cells in CDR1 (4/11 cells) compared to EV cells (0/11 cells) (p<0.05) (Fig. 7H). These data demonstrate that CDR1 can promote ER to Golgi trafficking. Together, these findings represent the first functional characterization of CDR1 and identify it as a key driver of LUSC metastatic biology, which is dependent on Golgi trafficking.
DISCUSSION:
Therapeutic strategies to inhibit metastatic spread in aggressive cancers, such as LUSC, are urgently needed. To identify key regulators of LUSC metastasis, we screened miRs associated with patient survival and metastatic phenotypes and identified miR-671–5p as an anti-metastatic miR. Expression of miR-671–5p was strongly associated with patient survival and genetic overexpression or therapeutic delivery of miR-671–5p was capable of inhibiting metastasis. The circRNA, CDR1as, was strongly inhibited by miR-671–5p as was its antisense transcript CDR1. This CDR1as/CDR1 axis was associated with worse survival in LUSC patients and inhibition of CDR1as or CDR1 decreased tumor burden and metastases. Overexpression of CDR1 was sufficient to promote metastatic spread and rescue loss of CDR1as.
These findings are consistent with miR profiling studies that found miR-671–5p among miRs associated with increased survival in stage I NSCLC (40). In agreement with previous studies, we found that miR-671–5p targeted the circular RNA CDR1as (32,33). However, CDR1as was not detected by a bioinformatic screen for miR-671–5p targets. This highlights a weakness of standard miR target prediction approaches, which can overlook miR-noncoding RNA interactions.
We found that CDR1 promotes metastasis through Golgi orientation and trafficking. Intrinsic functions of Golgi trafficking are critical for metastasis (41) and cell migration (38). We report that CDR1 overexpression can increase cancer cell motility, chemotaxis, and haptotaxis and epistasis experiments revealed that this gain-of-function is dependent on Golgi trafficking. Golgi trafficking can also promote metastasis through extrinsic mechanisms by secretion of pro-invasive and pro-angiogenic factors (42). These secreted factors require processing and trafficking in the ER-Golgi. Using non-transformed human bronchial cells, we found that CDR1 expression increased mucin secretion. CDR1 overexpression in LUSC cells facilitated Golgi orientation towards the direction of migration and increased the rate of ER to Golgi trafficking, suggesting CDR1 may also have effects on the secretome and extracellular state. In addition to the Golgi, CDR1 also localized to the nucleus by immunostaining and interacted with several nuclear proteins in our IP-MS analysis. Further investigation of the role of CDR1 in the nucleus is warranted.
Understanding of the cross-talk between circRNA and miRs is rapidly expanding. Though circRNAs were discovered decades ago, they were initially dismissed as splicing errors (43). The renaissance of circRNAs began with the discovery that circRNAs are abundant and functional in mammals (13,14). These early reports indicated that circRNAs could function as miR-sponges, and suggested that this mechanism could influence cancer biology. In particular, CDR1as acts as a sponge for the tumor suppressor miR-7 (13,14). CDR1as is highly expressed in the brain and fully-body knock-out models of CDR1as resulted in a brain specific decrease in miR-7, increase in miR-7 targets, and mild neuropsychological deficits, suggesting that sponging of miR-7 by CDR1as stabilizes rather than inhibits miR-7 (33). In neurons, excess miR-7 can also enhance the silencing of CDR1as by miR-671–5p (44). Importantly, no adverse effects of CDR1as deletion were found outside the CNS, making CDR1as a highly appealing therapeutic target for cancer.
There are >70 miR-7 seed matches decorating CDR1as, which is an unusually high amount. For most circRNAs there are no more miR binding sites than expected by chance (45). Other known functions for circRNAs include acting as a decoy for RBPs and serving as protein scaffolds. However, little is known about mechanisms for circRNAs in cancer beyond miR-sponging (17). CDR1as is also an unusual circRNA in that it is a natural antisense transcript (NAT) (32). NATs can increase or decrease expression of their complimentary mRNA. In some cases, this is through complimentary base-pairing (46). Given that the entire CDR1 mRNA is complementary to CDR1as, CDR1as likely stabilizes CDR1 directly through base pairing. Consistent with our findings in LUSC, the function of CDR1as in melanoma is also miR-7 independent (47). Interestingly, in melanoma, CDR1 was not found to be expressed and CDR1as instead functions through an RBP-mediated mechanism to inhibit metastasis (47). Thus, CDR1as functions in a context-dependent manner to enact different, even divergent, cancer phenotypes.
The repeated miR-7 seed sequence on CDR1as is highly conserved. Interestingly, this seed sequence corresponds to the hexapeptide repeat that is present in CDR1. Therefore, it is unclear whether protein sequence, miR seed, or both drive evolutionary conservation. We found miR-671–5p’s role in suppressing metastasis occurred through a CDR1as/CDR1 axis and that CDR1 alone was sufficient to promote metastasis. CDR1 is highly expressed in the brain and acts as an onconeural antigen in paraneoplastic disorders (18,19). The predicted structure of CDR1 is an α-helix with a 30 aa segment of β-sheet conformation at its C-terminus. Repeated sequences and α-helices are characteristic of a wide range of proteins, including structural proteins, RNA polα, and fungal surface proteins (18,19). Therefore, it is difficult to discern the function of CDR1 by sequence alone. Based on direct protein-protein interaction and immunostaining, we determined that CDR1 localized to multiple cellular compartments and was associated with COPI and AP1 vesicles. Furthermore, epistasis experiments revealed that inhibition of the COPI subunit COPA blocked CDR1-dependent migration. Given the importance of secretion and retrograde transport in neurotransmission and the high expression of CDR1 in the brain, these findings have important implications in neurobiology.
In conclusion, we have found that miR-671–5p inhibits LUSC metastases by targeting the poorly understood CDR1as/CDR1 axis. Although previously only known to be associated with paraneoplastic cerebellar degeneration (18,19), we found that CDR1 promotes metastases via increased cell migration through Golgi trafficking. To our knowledge, this is one of few reports to show a circRNA driving metastasis in vivo and the first for CDR1as. In addition, we describe the first therapeutic targeting of a circRNA. These findings reveal a complex regulatory network used to promote LUSC metastases and may have important implications in the understanding of the progression of other cancer types. Further, these findings reveal a broader functional repertoire for circRNAs which can be targeted through RNAi-based strategies.
Supplementary Material
Significance:
This study shows that circular RNA CDR1as promotes lung squamous migration, metastasis, and Golgi trafficking through its complimentary transcript, cerebellar-degeneration related protein 1 (CDR1).
Acknowledgments:
The authors acknowledge members of the Pecot lab for helpful discussions and feedback. The authors would like to especially thank Drs. Yongjuan Xia and Nana Feinberg from the UNC Translational Pathology Lab for their help with in situ hybridization, immunohistochemistry, and processing of the tissue microarrays and the UNC Animal Histopathology Core. Confocal microscopy was performed with equipment and assistance from the UNC Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine. We also thank Dr. Antonio Amelio for providing the Fluorescent-Nanoluciferase plasmids and Drs. Gianpietro Dotti and Hongwei Du for GFP-luciferase labeling of cell lines. The authors acknowledge the following financial support: The UNC Translational Pathology Laboratory is supported in part by grants from the NCI (5P30CA016080-42), NIH (U54-CA156733), NIEHS (3P30 EOS010126-17), UCRF, and NCBT (2015-IDG-1007). C. V. Pecot was supported in part by the National Institutes of Health (NIH) R01CA215075, a Mentored Research Scholar Grants in Applied and Clinical Research (MRSG-14-222-01-RMC) from the American Cancer Society, the Jimmy V Foundation Scholar award, the UCRF Innovator Award, the Stuart Scott V Foundation/Lung Cancer Initiative Award for Clinical Research, the University Cancer Research Fund, the Lung Cancer Research Foundation, the Free to Breathe Metastasis Research Award and the Susan G. Komen Career Catalyst Award. A. Belanger was supported by a grant from the National Heart, Lung and Blood Institute of the NIH under award number HL007106-38, an ASCO Young Investigator Award and by the Lung Cancer Initiative of North Carolina. S. H. Azam and I. A. Windham were supported in part by a grant from the National Institute of General Medical Sciences under award 5T32 GM007092 and GM119999, respectively. E. B. Harrison was supported by a grant from the National Cancer Institute of the NIH under award number T32CA196589 and by the Lung Cancer Initiative of North Carolina. S. Cohen and I. A. Windham were supported in part by an NIH R35 grant award number GM133460. S. H. Randell was supported in part by NIH 5P30DK065988. The UNC Microscopy Services Laboratory, Lineberger Comprehensive Cancer Center Animal Histopathology and Animal Studies Cores are all supported in part by an NCI Center Core Support Grant (CA016086) to the UNC Lineberger Comprehensive Cancer Center. The UNC Flow Cytometry Core Facility is also supported in part by the North Carolina Biotech Center Institutional Support Grant 2012-IDG-1006.
Footnotes
Conflict of interest: The authors declare no competing interests.
References:
- 1.Rami-Porta R, Ball D, Crowley J, Giroux DJ, Jett J, Travis WD, et al. The IASLC Lung Cancer Staging Project: proposals for the revision of the T descriptors in the forthcoming (seventh) edition of the TNM classification for lung cancer. J Thorac Oncol 2007;2:593–602 [DOI] [PubMed] [Google Scholar]
- 2.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama 2014;311:1998–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res 2010;16:4864–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porrello A, Leslie PL, Harrison EB, Gorentla BK, Kattula S, Ghosh SK, et al. Factor XIIIA-expressing inflammatory monocytes promote lung squamous cancer through fibrin cross-linking. Nat Commun 2018;9:1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 8.Chiang AC, Massagué J. Molecular basis of metastasis. New Engl J Med 2008;359:2814–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 2013;15:546–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 2011;17:1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun 2013;4:2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014;32:453–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384. [DOI] [PubMed] [Google Scholar]
- 14.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333. [DOI] [PubMed] [Google Scholar]
- 15.Du WW, Fang L, Yang W, Wu N, Awan FM, Yang Z, et al. Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ 2017;24:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014;56:55–66 [DOI] [PubMed] [Google Scholar]
- 17.Kristensen L, Hansen T, Venø M, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 2018;37:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dropcho EJ, Chen Y-T, Posner JB, Old LJ. Cloning of a brain protein identified by autoantibodies from a patient with paraneoplastic cerebellar degeneration. P Natl Acad Sci USA 1987;84:4552–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y-T, Rettig WJ, Yenamandra AK, Kozak CA, Chaganti R, Posner JB, et al. Cerebellar degeneration-related antigen: a highly conserved neuroectodermal marker mapped to chromosomes X in human and mouse. P Natl Acad Sci USA 1990;87:3077–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, et al. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol-Lung C 2009;296:L82–L91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu D, Pang Y, Wilkerson MD, Wang D, Hammerman PS, Liu JS. Gene-expression data integration to squamous cell lung cancer subtypes reveals drug sensitivity. Brit J Cancer 2013;109:1599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusch VW, Hawes D, Decker PA, Martin SE, Abati A, Landreneau RJ, et al. Occult metastases in lymph nodes predict survival in resectable non-small-cell lung cancer: report of the ACOSOG Z0040 trial. J Clin Oncol 2011;29:4313–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubuschok B, Passlick B, Izbicki JR, Thetter O, Pantel K. Disseminated tumor cells in lymph nodes as a determinant for survival in surgically resected non-small-cell lung cancer. J Clin Oncol 1999;17:19–24 [DOI] [PubMed] [Google Scholar]
- 24.Brown M, Assen F, Leithner A, Abe J, Schachner H, Asfour G, et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018;359:1408–11 [DOI] [PubMed] [Google Scholar]
- 25.Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EF, Jones D, et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018;359:1403–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leslie PL, Chao YL, Tsai YH, Ghosh SK, Porrello A, Van Swearingen AED, et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat Commun 2019;10:4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs--the micro steering wheel of tumour metastases. Nat Rev Cancer 2009;9:293–302 [DOI] [PubMed] [Google Scholar]
- 29.Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot CV, et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013;23:186–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan X, Fu Y, Chen L, An S, Lee W, Lai Y, et al. miR-671–5p promotes epithelial-to-mesenchymal transition by downregulating FOXM1 expression in breast cancer. Cancer Res 2015;75:3062- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. miRNA‐dependent gene silencing involving Ago2‐mediated cleavage of a circular antisense RNA. EMBO J 2011;30:4414–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piwecka M, Glazar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017;357 [DOI] [PubMed] [Google Scholar]
- 34.Banerjee R, Tyagi P, Li S, Huang L. Anisamide‐targeted stealth liposomes: A potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer 2004;112:693–700 [DOI] [PubMed] [Google Scholar]
- 35.Tan X, Banerjee P, Guo HF, Ireland S, Pankova D, Ahn YH, et al. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J Clin Invest 2017;127:117–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonifacino JS, Lippincott-Schwartz J. Coat proteins: shaping membrane transport. Nat Rev Mol Cell Bio 2003;4:409. [DOI] [PubMed] [Google Scholar]
- 37.Guo X, Mattera R, Ren X, Chen Y, Retamal C, González A, et al. The adaptor protein-1 μ1B subunit expands the repertoire of basolateral sorting signal recognition in epithelial cells. Dev Cell 2013;27:353–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing M, Peterman MC, Davis RL, Oegema K, Shiau AK, Field SJ. GOLPH3 drives cell migration by promoting Golgi reorientation and directional trafficking to the leading edge. Mol Biol Cell 2016;27:3828–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, et al. Synchronization of secretory protein traffic in populations of cells. Nat Methods 2012;9:493–8 [DOI] [PubMed] [Google Scholar]
- 40.Lu Y, Govindan R, Wang L, Liu PY, Goodgame B, Wen W, et al. MicroRNA profiling and prediction of recurrence/relapse-free survival in stage I lung cancer. Carcinogenesis 2012;33:1046–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan X, Banerjee P, Guo H-F, Ireland S, Pankova D, Ahn Y-h, et al. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J Clin Invest 2017;127:117–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halberg N, Sengelaub CA, Navrazhina K, Molina H, Uryu K, Tavazoie SF. PITPNC1 recruits RAB1B to the Golgi network to drive malignant secretion. Cancer Cell 2016;29:339–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cocquerelle C, Mascrez B, Hetuin D, Bailleul BJTFJ. Mis-splicing yields circular RNA molecules. FASEB J 1993;7:155–60 [DOI] [PubMed] [Google Scholar]
- 44.Kleaveland B, Shi CY, Stefano J, Bartel DP. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 2018;174:350–62.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014;15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su W-Y, Li J-T, Cui Y, Hong J, Du W, Wang Y-C, et al. Bidirectional regulation between WDR83 and its natural antisense transcript DHPS in gastric cancer. Cell Res 2012;22:1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanniford D, Ulloa-Morales A, Karz A, Berzoti-Coelho MG, Moubarak RS, Sanchez-Sendra B, et al. Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 2020;37:55–70.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.