

Abstract
The Arg/N-degron pathway targets proteins for degradation by recognizing their specific N-terminal residues or, alternatively, their non-N-terminal degrons. In mammals, this pathway is mediated by the UBR1, UBR2, UBR4, and UBR5 E3 ubiquitin ligases, and by the p62 regulator of autophagy. UBR1 and UBR2 are sequelogous, functionally overlapping, and dominate the targeting of Arg/N-degron substrates in examined cell lines. We constructed, here, mouse strains in which the double mutant [UBR1−/− UBR2−/−] genotype can be induced conditionally, in adult mice. We also constructed human [UBR1−/− UBR2−/−] HEK293T cell lines that unconditionally lack UBR1/UBR2. ATF3 is a basic leucine zipper transcription factor that regulates hundreds of genes and can act as either a repressor or an activator of transcription. Using the above double-mutant mice and human cells, we found that the levels of endogenous, untagged ATF3 were significantly higher in both of these [UBR1−/− UBR2−/−] settings than in wild-type cells. We also show, through chase-degradation assays with [UBR1−/− UBR2−/−] and wild-type human cells, that the Arg/N-degron pathway mediates a large fraction of ATF3 degradation. Furthermore, we used split-ubiquitin and another protein interaction assay to detect the binding of ATF3 to both UBR1 and UBR2, in agreement with the UBR1/UBR2-mediated degradation of endogenous ATF3. Full-length 24 kDa ATF3 binds to ∼100 kDa fragments of 200 kDa UBR1 and UBR2 but does not bind (in the setting of interaction assays) to full-length UBR1/UBR2. These and other binding patterns, whose mechanics remain to be understood, may signify a conditional (regulated) degradation of ATF3 by the Arg/N-degron pathway.
Graphical Abstract
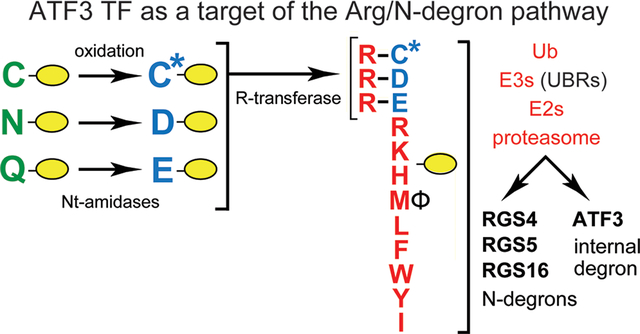
Regulated protein degradation protects cells from mis-folded, aggregated, or otherwise abnormal proteins and also controls the levels of proteins that evolved to be short-lived in vivo. The intracellular protein degradation is mediated largely by the ubiquitin (Ub)–proteasome system (UPS) and by autophagosome–endosome–lysosome pathways, with molecular chaperones being a part of both systems.1–8 The UPS is a set of pathways that have in common two classes of enzymes, E3–E2 Ub ligases and deubiquitylases. A Ub ligase recognizes a substrate protein through its degradation signal (degron) and conjugates Ub, a 9 kDa protein (usually in the form of a poly-Ub chain), to an amino acid residue of a targeted substrate (usually its internal lysine). The functions of deubiquitylases include deubiquitylation of Ub-conjugated proteins.1–11 The 26S proteasome, an ATP-dependent protease, binds to a poly-Ub of a ubiquitylated protein, unfolds the protein, and processively cleaves it to peptides that range in size from ∼3 to ∼25 residues.12–17
N-degron pathways (previously called “N-end rule pathways”) are proteolytic systems that have in common their ability to recognize proteins containing N-terminal (Nt) degrons called N-degrons, thereby causing degradation of these proteins by the 26S proteasome and/or autophagy in eukaryotes, and by ClpS-ClpAP in bacteria (Figure 1 and Figure S1).2,18–53 Determinants of an N-degron include a destabilizing Nt-residue of a protein, an internal Lys residue(s) that functions as a polyubiquitylation site, and a region used by the proteasome to initiate degradation.2,19,54,55
Figure 1.
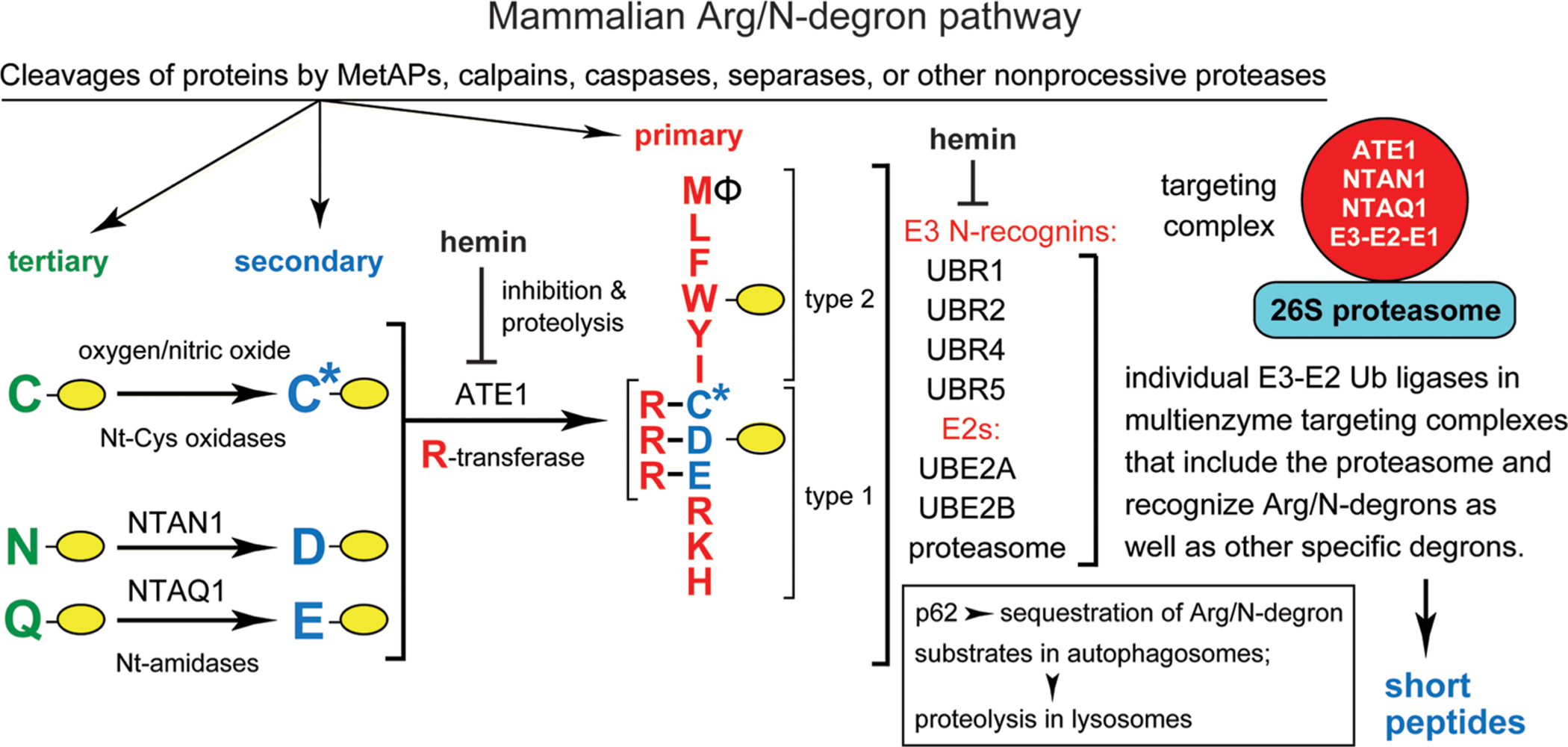
Mammalian Arg/N-degron pathway.2,24,26 Yellow ovals denote the rest of a protein substrate. Nt-residues are denoted by single-letter abbreviations. This pathway targets proteins for either proteasome-mediated degradation (via UBR1, UBR2, UBR4, and UBR5 E3s) or lysosome-mediated degradation (via p62). The cited E3 Ub ligases (N-recognins) of the pathway can recognize not only the indicated (destabilizing) Nt-residues but also specific non-N-terminal degrons. “Primary”, “secondary”, and “tertiary” refer to mechanistically distinct classes of destabilizing Nt-residues. NTAN1 and NTAQ1 are Nt-amidases that convert the tertiary destabilizing Nt-Asn and Nt-Gln to Nt-Asp and Nt-Glu, respectively. C* denotes oxidized N-terminal Cys, either Cys-sulfinate or Cys-sulfonate, produced in vivo through reactions that involve oxygen and nitric oxide. The ATE1 Arg-tRNA-protein transferase (arginyltransferase or R-transferase) conjugates Arg, a primary destabilizing residue, to Nt-Asp, Nt-Glu, and (oxidized) Nt-Cys. Hemin (Fe3+-heme) inhibits the enzymatic activity of R-transferase and accelerates its degradation in vivo. Hemin also binds to UBR1/UBR2 E3s and inhibits specific aspects of their activity. Type 1 and type 2 refer to two sets of primary destabilizing Nt-residues, basic (Arg, Lys, and His) and bulky hydrophobic [Leu, Phe, Trp, Tyr, Ile, and also Met, if the latter is followed by a bulky hydrophobic residue (Φ)], respectively. These Nt-residues are recognized by substrate-binding sites of the pathway’s E3 Ub ligases UBR1, UBR2, UBR4, and UBR5. Another N-recognin is p62, an autophagy-regulating protein distinct from Ub ligases. p62 can bind, in particular, to Nt-arginylated proteins and mediate their targeting for degradation through the autophagosome–lysosome pathway. UBR1 and UBR2 E3s are sequelogous to each other and to S. cerevisiae UBR1. In contrast, sequelogies between UBR1/UBR2 and UBR4 or UBR5 are confined largely to their ∼80-residue UBR domains, which recognize the N-terminal Arg, Lys, or His residues. Enzymes of the mammalian Arg/N-degron pathway (UBR1 or UBR2, UBE2A or UBE2B, the R-transferase ATE1, the Asn/Nt-amidase NTAN1, and the Gln/Nt-amidase NTAQ1) form a targeting complex.56 An analogous targeting complex mediates the S. cerevisiae Arg/N-degron pathway. A “generic” targeting complex (red circle on the right) is envisioned as a set of analogous complexes, only one of which (containing either UBR1 E3 or UBR2 E3) has already been discovered.56 The other (likely to exist but remaining to be identified) complexes of this pathway would contain either UBR4 or UBR5 E3s, together with pathway’s “upstream” enzymes such as R-transferase and Nt-amidases. A targeting complex apparently includes the 26S proteasome, as well,56 as shown in the diagram. Also indicated, in a targeting complex, is the E1 (Ub-activating) enzyme, a transient component of the complex that binds to E2. See the introduction for other details and additional references.
Eukaryotic N-degron pathways comprise the Arg/N-degron pathway (it recognizes, in particular, specific unacetylated Nt-residues), the Ac/N-degron pathway [it recognizes, in particular, the Nα-terminally acetylated (Nt-acetylated) Nt-residues], the Pro/N-degron pathway (it recognizes, in particular, the Nt-Pro residue), the Gly/N-degron pathway (it recognizes the Nt-Gly residue), and the fMet/N-degron pathway (it recognizes Nt-formylated proteins) (Figure 1 and Figure S1).2,18–20,24–28,30–44,46–48,50–52,56
Initially, most N-degrons are cryptic (pro-N-degrons). They are converted to active N-degrons either constitutively (for example, cotranslationally) or via regulated steps. Many nonprocessive proteases, including Met-aminopeptidases, caspases, calpains, separases, and cathepsins, act as initial targeting components of N-degron pathways, because cleavage of a protein can generate a C-terminal (Ct) fragment bearing an N-degron.2,21,40,50,57,58 Active N-degrons can also be formed through enzymatic Nt-acetylation, Nt-deamidation, Nt-oxidation, Nt-arginylation, Nt-leucylation, and Nt-formylation of specific proteins or their Ct fragments (Figure 1 and Figure S1).2,20,26,27,31,39,47,48 Recognition components of N-degron pathways are called N-recognins. They are E3 Ub ligases or other proteins, for example, mammalian p62 and bacterial ClpS, that can recognize N-degrons.2,6,26,43,46,49,59,60 At least some N-recognin E3s can recognize not only N-degrons but also specific non-N-terminal degrons in other proteins.2,44,61,62 In cognate sequence contexts, all 20 amino acids of the genetic code can function as destabilizing Nt-residues (Figure S1). Thus, many proteins in a cell are conditionally short-lived N-degron substrates, either as full-length proteins or as Ct fragments.
Regulated degradation of proteins and their natural fragments by N-degron pathways has been shown to mediate a multitude of biological processes, including the sensing of oxygen, nitric oxide (NO), heme, and short peptides; the control of subunit stoichiometries in protein complexes; the elimination of misfolded proteins and of proteins retrotrans-located to the cytosol from other compartments; a suppression of neurodegeneration and regulation of apoptosis; the control of DNA repair, transcription, replication, and chromosome cohesion/segregation; the regulation of chaperones, G proteins, cytoskeletal proteins, autophagy, gluconeogenesis, peptide transport, meiosis, circadian rhythms, fat metabolism, cell migration, adaptive and innate immunity (including inflammation), the cardiovascular system, neurogenesis, and spermatogenesis; and plant defenses against pathogens, plant cell differentiation, the sensing of oxygen and NO, and many other processes in plants (refs 2, 18, 21–28, 30–37, 39, 42–51, 53, 56, and 63–67 and references therein).
To keep notations uniform, human (Homo sapiens, hs) genetic terms (all-uppercase letters) are used throughout the paper to denote genes and proteins, including those of mouse (Mus musculus, mm) and yeast (Saccharomyces cerevisiae, sc). scUBR1 encodes the 225 kDa RING-type E3, the sole N-recognin of the S. cerevisiae Arg/N-degron pathway. Unmodified N-terminal Arg, Lys, His, Leu, Phe, Tyr, Trp, Ile, and Met (if Nt-Met is followed by a bulky hydrophobic residue) are “primary” destabilizing Nt-residues in that they can be bound by the type 1 and type 2 sites of scUBR1.2,24,33,68,69 In contrast, Nt-Asp and Nt-Glu are destabilizing because of their Nt-arginylation by scATE1 arginyltransferase (R-transferase).70 The resulting Nt-conjugated Arg can be bound by scUBR1. Nt-Asn and Nt-Gln are destabilizing because scNTA1 Nt-amidase converts them to Nt-arginylatable Nt-Asp and Nt-Glu (Figure S1G).52,71
In contrast to S. cerevisiae, the Arg/N-degron pathway of mammals and other animals is mediated by at least four E3 N-recognins: the 200 kDa UBR1 and UBR2, the 570 kDa UBR4 (p600, BIG), and the 300 kDa UBR5 (EDD1, HYD) (Figure 1).43,59,60,72–74 Another N-recognin of the mammalian Arg/N-degron pathway is p62, an autophagy-regulating protein distinct from E3 Ub ligases.6,46 hsUBR1 and hsUBR2 E3s are sequelogous (similar in sequence75)a to each other and to S. cerevisiae scUBR1. In contrast, sequelogies (sequence similarities75)a between, e.g., hsUBR1/hsUBR2 and hsUBR4 or hsUBR5 are confined largely to their ∼80-residue UBR domains, which recognize the N-terminal Arg, Lys, or His residues.2,68,69 In contrast to the dual specificity of the S. cerevisiae Asn/Gln/Nt-amidase scNTA1, animals and plants contain two Nt-amidases, the Nt-Asn-specific NTAN1 and the Nt-Gln-specific NTAQ1.51,66,76,77
At least in multicellular eukaryotes, Nt-arginylation encompasses not only Nt-Asp and Nt-Glu but also Nt-Cys, after its oxygen/NO-dependent oxidation to Nt-arginylatable Nt-Cys-sulfinate or Nt-Cys-sulfonate (Figure 1). Consequently, the Arg/N-degron pathway functions as a sensor of oxygen/NO in animals and plants, through the conditional, arginylation-dependent degradation of transcription factors and other regulators that bear the Nt-Cys residue.22,29,47,78,79 Five enzymes of the mammalian Arg/N-degron pathway (UBR1 or UBR2 E3, UBE2A or UBE2B E2, the R-transferase ATE1, the Asn/Nt-amidase NTAN1, and the Gln/Nt-amidase NTAQ1) form a targeting complex56 (Figure 1). An analogous targeting complex mediates the S. cerevisiae Arg/N-degron pathway.56
Homozygous inactivation of the human hsUBR1 gene [with retention of other Arg/N-recognins (Figure 1)] causes Johanson-Blizzard syndrome (JBS). Its symptoms include exocrine pancreatic insufficiency and inflammation, anatomical malformations, mental retardation, and deafness.2,80–82 mmUBR1−/− mice have a milder version of JBS.80 Phenotypes of mmUBR2−/− mice include infertility of males, owing to apoptosis of mmUBR2−/− spermatocytes.2,74 Mouse (or human) UBR1 and UBR2 E3s are 47% identical; their functions partly overlap.2,24,26 In contrast to the viability of mmUBR1−/− and mmUBR2−/− mouse strains, mice that lack both mmUBR1 and mmUBR2 die as midgestation embryos, with neural and cardiovascular defects.83
In the study presented here, we used the cre-lox technique84 to ask whether it is possible to generate viable adult [mmUBR1−/− mmUBR2−/−] mice, despite the 100% embryonic lethality of the [mmUBR1−/− mmUBR2−/−] genotype.83 As described below, the answer is yes, with telling qualifications, including the nearly 50% lethality of the adult-onset double-mutant genotype. We also used, here, the CRISPR-Cas9 method85–87 to construct [hsUBR1−/− hsUBR2−/−] human HEK293T cell lines that lack both hsUBR1 and hsUBR2.
ATF3 is a basic leucine zipper (bZIP) transcription factor (TF) that regulates hundreds of genes (including its own gene) and can act as either a repressor or an activator of transcription.88–97 Using the double-mutant mice and human cells described above, we found, here, that the levels of endogenous, untagged ATF3 were significantly higher in both of these [UBR1−/− UBR2−/−] settings than in wild-type cells. In some double-mutant mice, the upregulation of ATF3 was strikingly high. Furthermore, chase-degradation assays with [UBR1−/− UBR2−/−] and wild-type human cell lines showed that the Arg/N-degron pathway mediates a large fraction of ATF3 degradation. To the best of our knowledge, ATF3 is the first physiological substrate of the mammalian Arg/N-degron pathway that is targeted for degradation through an internal (remaining to be mapped) degron, as distinguished from an Arg/N-degron.
Another line of inquiry in this study involved the S. cerevisiae-based split-ubiquitin98,99 and E. coli-based BACTH (bacterial two-hybrid)100 protein interaction assays. We found that human (and mouse) ATF3 physically interacts with UBR1 and UBR2, in agreement with the UBR1/UBR2-mediated degradation of endogenous ATF3. The full-length 24 kDa ATF3 binds to ∼100 kDa fragments of 200 kDa UBR1 and UBR2 but does not bind (in the setting of interaction assays) to full-length UBR1 and UBR2. These and other ATF3 binding patterns, whose mechanics remain to be understood, may signify a conditional (regulated) degradation of ATF3 by the Arg/N-degron pathway.
MATERIALS AND METHODS
Antibodies and Other Reagents.
The following primary antibodies were used for immunoblotting (IB): anti-flag M2 mouse monoclonal antibody (Sigma, F1804), anti-ha rabbit polyclonal antibody (Sigma, H6908), anti-ATF3 rabbit polyclonal antibody (Santa Cruz, SC-188), anti-GAPDH 6C5 mouse monoclonal antibody to human/mouse glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz, SC-32233), anti-hsUBR1 A-5 mouse monoclonal antibody (Santa Cruz, SC-515753), and anti-hsUBR2 rabbit polyclonal antibody (Bethyl, A305-416-A). Anti-hsUBR1 and anti-hsUBR2 also recognized mmUBR1 and mmUBR2, respectively. Secondary antibodies for IB were the goat anti-mouse IRDye 800CW conjugate (Li-Cor, C60405-05), the goat anti-mouse HRP conjugate (Bio-Rad, 1706516), and the goat anti-rabbit HRP conjugate (Bio-Rad, 1706515). Protein bands on IB membranes were detected and quantified using an Odyssey-9120 instrument (Li-Cor, Lincoln, NE) for near-infrared signals, or by chemilumenescence, using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, RPN2232).
Human cells lines were grown in DMEM containing also 4.5 g/L glucose, l-glutamine, and sodium pyruvate (Corning, 10-013-CV). Growth media were supplemented with GlutaMAX (Gibco, 35050061) and penicillin-streptomycin (Genesee Scientific, 25-512). For propagation of cell cultures, cell monolayers were treated with trypsin-EDTA (VWR, 0154-0100). Transfections were carried out using GeneJuice Transfection Reagent (Sigma, 70967) and the manufacturer’s protocol. “Complete protease inhibitor cocktail” (Roche, 11697498001) was added to lysis buffers for the preparation of cell extracts. SDS–PAGE was carried out using 8%, 10%, 12%, or 4–12% NuPAGE Bis-Tris Gels (ThermoFisher). Other reagents were phenylmethanesulfonyl fluoride (PMSF) (Sigma, P7626), DNase I (Roche, 11284932001), glutathione-Sepharose-4B (GE Healthcare, 17075601), zirconia/silica beads (0.5 mm) (Biospec, 11079105Z), and restriction endonucleases, T4 DNA ligase, and Q5 DNA polymerase (New England Biolabs).
Animal Care and Treatments.
All animal care and procedures in this study were conducted according to relevant National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee (Protocol 1328), the Office of Laboratory Animal Research (OLAR) at the California Institute of Technology, where the study was carried out. Mice were housed at 22–24 °C and 30–70% humidity in a pathogen-free (barrier) facility at 22 °C, using a 13 h light/11 h dark cycle and PicoLab Rodent Diet 20 (LabDiet, 5053), ad libitum. Mice aged between 3 and 8 weeks were treated with tamoxifen (TM) (Sigma, T5648) by daily intraperitoneal injections (2 mg in 0.2 mL of sesame oil) over 5 days. Both injections with TM and post-TM work with these mice were carried out in conventional (nonbarrier) facilities. Mice were weighed weekly, starting 3 days before the first TM treatment.
Construction of [CaggCreERtm; mmUBR1−/− mmUBR2flox/flox] and [mmUBR1−/− mmUBR2flox/flox] Mouse Strains.
Gt(ROSA)26Sortm1(FLP1)Dym mice (expressing Flp recombinase) were obtained from Jackson Laboratory (Bar Harbor, ME). Another mouse strain, mmUBR2tm1a(KOMP)Mbp/+, was produced by us, using standard techniques,101,102 from a clone of embryonic stem (ES) cells that was generated by the NIH-supported Knockout Mouse Project (KOMP) and was obtained from the KOMP Repository (www.komp.org). In mmUBR2tm1a(KOMP)Mbp/+ mice, one copy of mmUBR2 was disrupted, downstream from exon 3, by insertion of the frt-En2SA-PIRES-LacZ-pA-loxP-PhBact-Neo-pA-frt-loxP-exon 4-exon 5-loxP segment that contained, among other genetic elements, two frt sites (recognized by Flp recombinase) and three loxP sites (recognized by Cre recombinase) (Figure S2).
Heterozygous matings of mmUBR2tm1a(KOMP)Mbp/+ and Gt-(ROSA)26Sortm1(FLP1)Dym mice led to the Flp-mediated excision of LacZ-Neo DNA between exons 3 and 4 of the mmUBR2 gene (Figure S2). The excision was verified using genomic DNA, oligonucleotide primers TV228 and TV229 (Table S2), and PCR, which yielded a 412 bp amplified DNA fragment that signified excision versus a predicted 7316 bp fragment before excision (Figure S3). Heterozygous matings of the resulting mmUBR2+/flox mouse strains produced mmUBR2flox/flox mice, in which both copies of mmUBR2 were “floxed” (containing loxP sites flanking exons 4 and 5) (Figure 2A,B and Figure S2).
Figure 2.
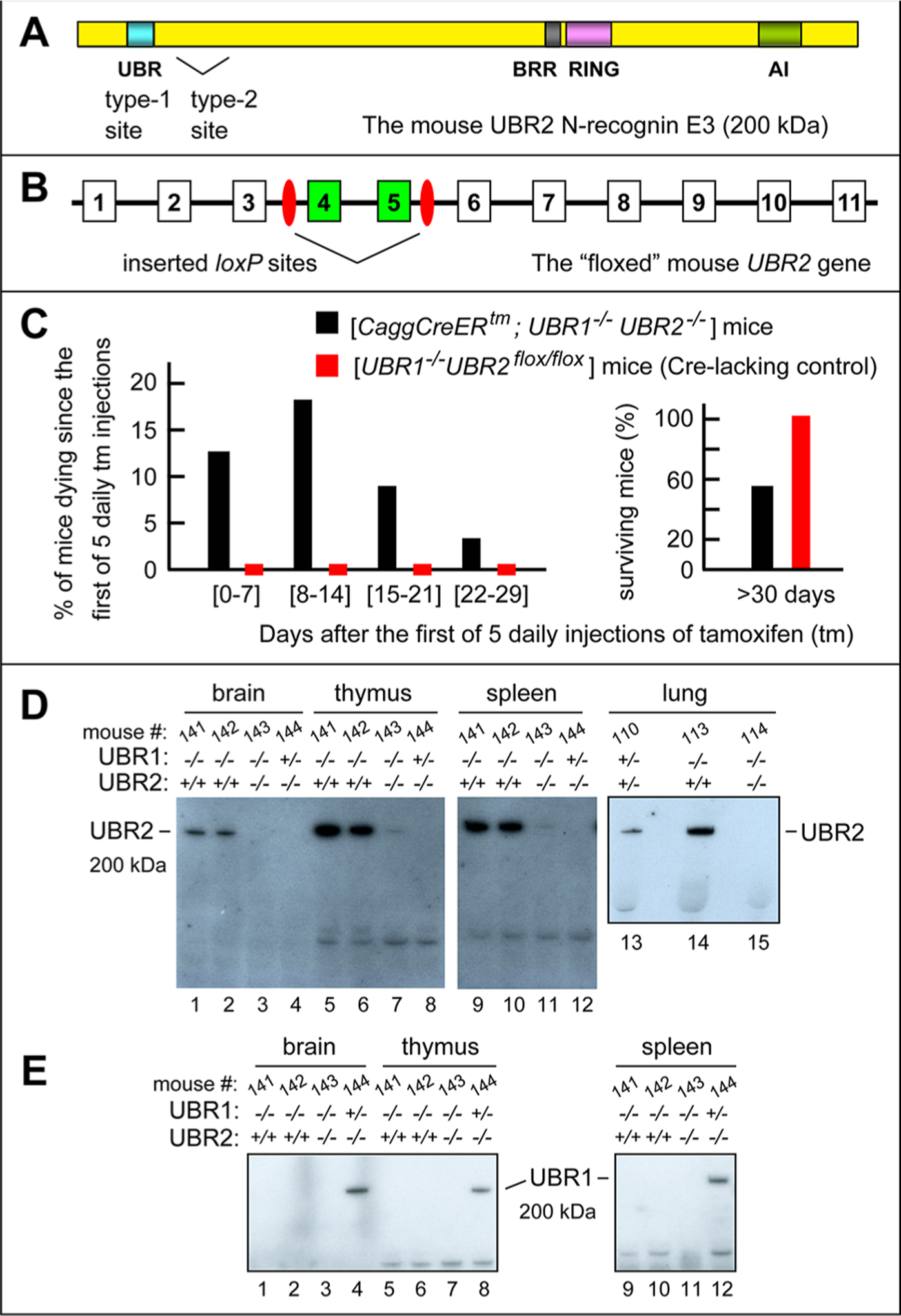
Mouse strains lacking both mmUBR1 and mmUBR2 E3 ubiquitin ligases. (A) The 200 kDa mmUBR2 and its domains.2,24,26 BRR is the basic residue-rich domain. RING is a Cys-His-rich domain that is present in a large class of E3 Ub ligases. (B) The first 11 exons (of 48 exons total) of the mmUBR2 gene, with exons and introns not to scale.72 Inserted 34 bp loxP sites, recognized by Cre recombinase, are colored red. “Floxed” exons 4 and 5, which are deleted upon activation of Cre by tamoxifen (TM), are colored green. (C) Percentages of [CaggCreER; mmUBR1−/− mmUBR2flox/flox] mice (black bars) vs Cre-lacking [mmUBR1−/− mmUBR2flox/flox] mice (red bars) that died by the indicated days after five daily injections of TM. On the right, these percentages are summed to tabulate survival of these mice more than 30 days after TM injections, with 100% survival of Cre-lacking [mmUBR1−/− mmUBR2flox/flox] mice. See also Materials and Methods. (D) Immunoblotting (IB) analyses of specific tissues of mice (with the indicated mmUBR1/mmUBR2 genotypes), using the anti-UBR2 antibody. In UBR2 notations, a plus sign denotes the UBR2flox allele. Note undetectable (lanes 3, 4, 8, 12, and 15) or nearly undetectable (lanes 7 and 11) levels of mmUBR2 in TM-generated [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice, in contrast to readily detectable mmUBR2 in identically treated Cre-lacking [mmUBR1−/− mmUBR2flox/flox] mice (lanes 1, 2, 5, 6, 9, 10, and 14). (E) Same as panel D but IB with the anti-UBR1 antibody. Note the absence of detectable mmUBR1 in unconditionally mmUBR1-lacking mice (lanes 1–3, 5–7, and 9–11) vs its presence in the mmUBR1+/− heterozygous genetic background (lanes 4, 8, and 12).
Conditional double-mutant (2-KO) [CaggCreER; mmUBR1−/− mmUBR2flox/flox] mice were generated through a series of heterozygous matings of mmUBR1−/− mice,73 mmUBR2flox/flox mice [created in this study (Figures S2 and S3)], and CaggCreER mice.103 The latter mouse strain expressed a conditional (activatable by TM) Cre recombinase from the ubiquitously active chimeric PCagg promoter.103 An otherwise identical (and similarly produced) control strain, [mmUBR1−/− mmUBR2flox/flox], contained mmUBR2flox/flox but lacked the CreER recombinase.
[CaggCreERtm; mmUBR1−/− mmUBR2flox/flox] mice that were aged between 4 and 16 weeks (and control [mmUBR1−/− mmUBR2flox/flox] mice, as well) were treated by daily intraperitoneal injections, over 5 days, of TM [Sigma, T5648; 0.2 mL injections of TM, at 10 mg/mL in sesame oil (MP Biomedicals)]. Cre-mediated excision of the “floxed” DNA segment that encompassed exons 4 and 5 (Figure 2A,B and Figure S2) from both copies of the mmUBR2 gene was verified using genomic DNA, primers TV227 and TV228 (Table S2), and PCR, which yielded a 269 bp DNA fragment that signified the presence of the functionally inactivated mmUBR2− allele versus a 3340 bp fragment in the absence of excision of the DNA segment mentioned above (Figure S3).
The excision was further verified using primers TV507 and TV510 (Table S2), cDNA produced from RNA that had been isolated from several mouse tissues, and RT-PCR, which yielded a 565 bp amplified DNA fragment for the excision-inactivated mmUBR2− allele and a 810 bp fragment for the initial mmUBR2flox allele. Figure S4 shows examples of the resulting data, for the brain and kidney tissues of a [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mouse that was produced upon TM treatment, and for its identically treated but Cre-lacking (and mmUBR2flox/flox-retaining) [mmUBR1−/− mmUBR2flox/flox] counterpart. TM-induced mmUBR2flox/flox → mmUBR2−/−conversions in examined tissues were usually at least 90% complete (Figure S4), were always higher than 60%, and varied, within that interval, among specific tissues of a TM-treated mouse and among different TM-treated mice. Furthermore, and most importantly, we also carried out IBs with anti-UBR2 and anti-UBR1 antibodies to verify the ablation of the mmUBR2 protein in several tissues of [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice, in addition to the unconditional absence of the mmUBR1 protein in these mice (Figure 2D,E).
Mouse Tissue Extracts.
Mouse tissues were processed as previously described.40,41 Briefly, specific tissues were lysed in RIPA buffer104 containing “protease inhibitor cocktail” (Roche, 11697498001), using an MP FastPrep-24 instrument and Lysing Matrix D (MP Biomedicals), with three runs at 6.5 m/s for 20 s each and 5 min incubations on ice between the runs. Extracts were centrifuged at 10000g for 10 min at 4 °C. The total protein concentration in supernatants was measured by the bicinchoninic acid (BCA) assay (ThermoFisher, 23225). The resulting samples [50 μg of total protein in 45 μL of lithium dodecyl sulfate (LDS) sample buffer] were heated at 70 °C for 10 min, followed by SDS-PAGE and IB analyses.
Construction of [hsUBR1−/− hsUBR2−/−] Human HEK29T Cell Lines.
Double-mutant [hsUBR1−/− hsUBR2−/−] HEK29T cell lines were constructed through sequential knockouts, using the CRISPR-Cas9 technique.85–87 Plasmid PX459 [pSpCas9(BB)-2A-Puro V2.0]85 (Table S1) was from Addgene (plasmid 62988; http://n2t.net/addgene:62988; RRID: Addgene, 62988). Guide sequences targeting hsUBR1 and hsUBR2 at their exon 5 regions were cloned into PX459 using oligonucleotides TV766–TV769 (Table S2). The human HEK293T cell line105 (American Type Culture Collection, https://www.atcc.org/products/all/crl-3216.aspx) was transfected, using GeneJuice (Sigma, 70967), with pTV463 (Table S1), which targeted exon 5 of hsUBR2, and transformants were selected with puromycin at 1 μg/mL (Sigma, P8833) for 1 day. Surviving cells were transfected with pTV462 (Table S1), which targeted exon 5 of hsUBR1, and transformants were again selected with puromycin at 1 μg/mL for 1 day. Monoclonal cell populations were obtained, then, through limiting dilution into 96-well plates at a density of ∼0.5 cell/well. The resulting cell clones were expanded and assayed for hsUBR1 and hsUBR2 using IBs with anti-hsUBR1 and anti-hsUBR2 antibodies (Figure 3A,B).
Figure 3.
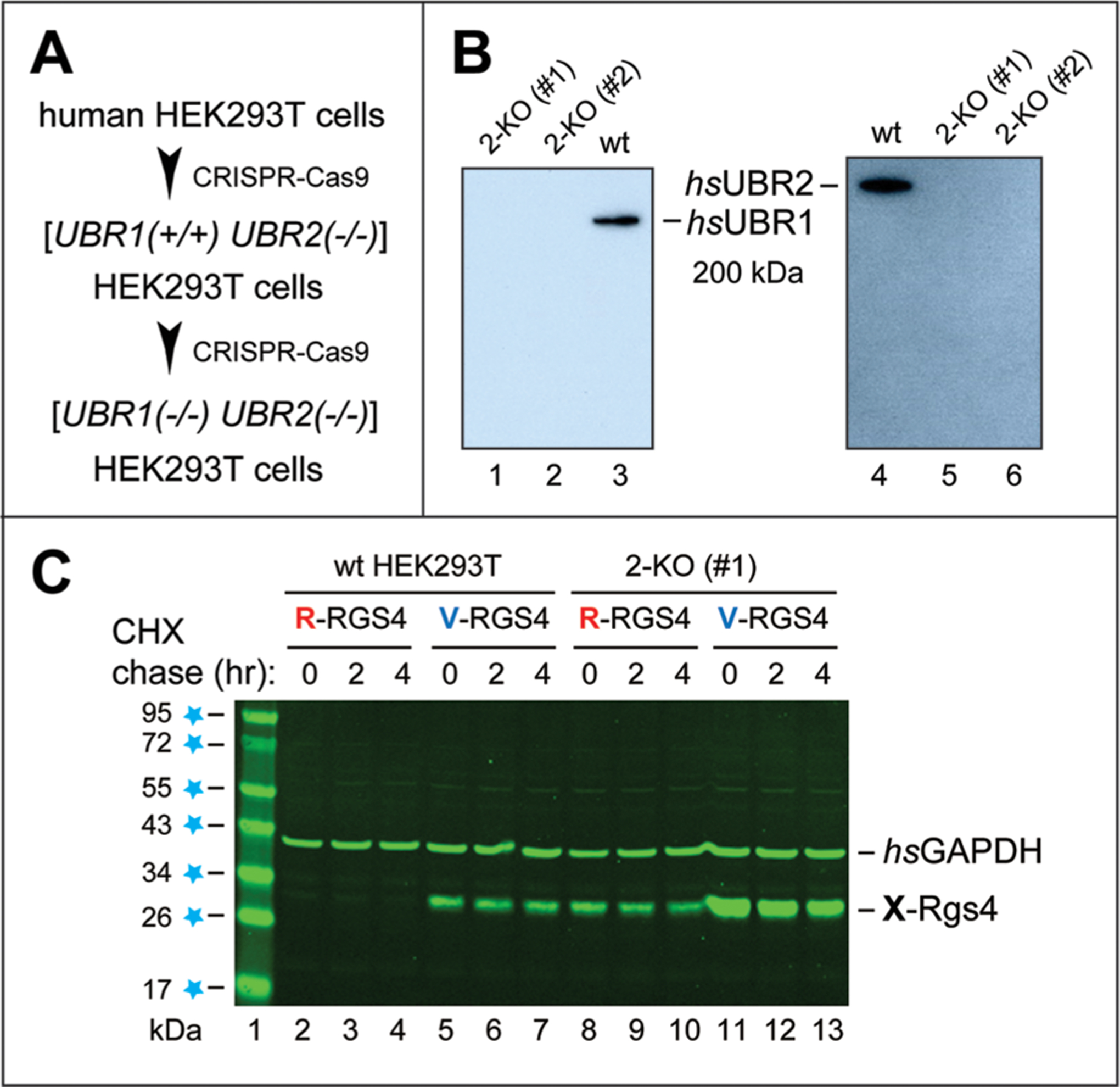
Human HEK293T cell lines lacking both hsUBR1 and hsUBR2 E3s. (A) Sequential, CRISPR-based85–87 construction of double-mutant [hsUBR1−/− hsUBR2−/−] human HEK293T cell lines (see Materials and Methods). (B) IB analyses of the wild-type (parental) HEK293T cell line and two independently produced [hsUBR1−/− hsUBR2−/−] HEK293T cell lines [denoted as 2-KO (knockout) (#1) and 2-KO (#2)], using anti-hsUBR1 and anti-hsUBR2 antibodies. (C) Metabolic stabilization of an Arg/N-degron protein substrate in 2-KO (#1) HEK293T cells. Lane 1, molecular mass markers, with values in kilodaltons indicated on the left. Lanes 2–4, cycloheximide (CHX) chase, for 0, 2, and 4 h, respectively, of wild-type HEK293T cells transiently transfected by a plasmid expressing C-terminally flag-tagged Arg-RGS4f (R-RGS4) (see Materials and Methods and Results). Lanes 5–7, same as lanes 2–4, respectively, but with Val-RGS4f (V-RGS4), bearing an Nt-residue (Val) that is not recognized by the Arg/N-degron pathway (Figure 1). Lanes 8–10, same as lanes 2–4, respectively, but with 2-KO (#1) double-mutant HEK293T cells. Lanes 11–13, same as lanes 5–7, respectively, but with 2-KO (#1) HEK293T cells. The bands of X-RGS4 and glyceraldehyde-3-phosphate dehydrogenase (hsGAPDH, a loading control, detected by the anti-GAPDH antibody) are indicated on the right.
Chase-Degradation Assays.
They were carried out largely as previously described.40,106 Briefly, wild-type and mutant HEK293T cells were seeded at a density of 400000 cells/well in six-well plates in a DMEM-based medium described above and grown at 37 °C. For degradation assays that involved transient transfections, seeded cells were transfected, a day after, with 0.2 μg of indicated plasmids per plate, followed by overnight incubation. For degradation assays involving endogenous untagged proteins, cells were grown for 2 days. Thereafter, cell monolayers were gently washed with phosphate-buffered saline (PBS) at 37 °C, followed by addition of fresh DMEM-based medium at 37 °C. At that point, cells in “zero-time” plates were removed and processed as described below. At the same time, cycloheximide (CHX) (Sigma, C7698) was added to other wells (to a final concentration of 0.25 mg/mL), initiating the chase. At indicated times, cells in individual wells were briefly and gently washed with ice-cold PBS, harvested into 1.5 mL tubes, and pelleted by centrifugation at 10000g for 1 min. Cells were then lysed by sonicating a suspension for 10 s in 0.2 mL of RIPA buffer104 containing “protease inhibitor cocktail” (Roche, 11697498001), using a microtip (Branson sonicator, 101-148-062) at 10% duty cycle and output 2, followed by centrifugation at 10000g for 10 min. The total protein concentration in supernatants was measured by the bicinchoninic acid (BCA) assay (ThermoFisher, 23225). The resulting samples (30 μg of total protein in 45 μL of LDS sample buffer) were heated at 70 °C for 10 min, followed by SDS–PAGE and IB assays. Protein degradation in these assays was quantified using ImageJ.107 Briefly, ImageJ quantified the brightness of specific IB-detected protein bands versus the background brightness.
Immunoblotting.
IB analyses were carried out largely as described previously.40,44,104,106 Briefly, following SDS–PAGE, fractionated proteins in a polyacrylamide gel were electroblotted onto a nitrocellulose or PVDF membrane using iBlot (Invitrogen, 25-0912; Program 3; 7 min transfer for regular detection or 8 min transfer for detection of high-molecular mass proteins). Membranes with electroblotted proteins were blocked104 and thereafter incubated with a relevant primary antibody, followed by either LI-COR IRDye-conjugated secondary antibodies or HRP-conjugated secondary antibodies. Protein bands were detected and quantified using an Odyssey-9120 instrument and its software, or using chemilumenescence and ImageJ.107
Quantitative Reverse Transcription PCR (RT-qPCR).
RT-qPCR was carried out as described previously.108 Briefly, samples of a mouse tissue or cells grown in culture were lysed using QIAshredder (Qiagen, 79654), and RNA was purified using the RNeasy Mini Kit (Qiagen, 74104), with an additional treatment by RNase-Free DNase (Qiagen, 79254) to preclude DNA contamination. RNA integrity and purity were assessed in part by spectrophotometric measurements (with A260/A280 > 1.8, and A260/A230 > 1.5), using NanoPhotometer (Implen), and also by agarose gel electrophoresis. Reverse transcription, to synthesize cDNA, was carried at 42 °C for 1 h in a 20 μL reaction sample containing RNA (25 ng/μL), 400 units of M-MulV reverse transcriptase (Lucigen, 97065-184), 20 units of RNase Inhibitor (Lucigen, 97065-224), 6 μM Random Primer Mix (NEB, S1330S), and 2 mM Deoxynucleotide (dNTP) Solution Mix (NEB, N00447S). cDNA synthesis was terminated by heating samples at 85 °C for 1 min. qPCR was carried out with 0.5 ng/μL cDNA, 1× SYBR Green I-based qPCR Master Mix (Bioland, QP01), and 250 nM mmUBR2 cDNA-specific oligonucleotides, TV282 (5′-TTTCCCTACCAACCAACCTC-3′) and TV283 (5′-AGCTTATCGCTCCTCTCTCG-3′), in a 20 μL reaction sample using Mastercycler RealPlex2 (Eppendorf) and the following qPCR program: one cycle at 95 °C for 10 min, 45 cycles at 95 °C for 15 s and at 60 °C for 30 s, and one cycle at 95 °C for 15 s and at 60 °C for 15 s. PCR products were then subjected to increases in temperature from 60 to 95 °C at a rate of 1.75 °C/min and thereafter incubated at 95 °C for 15 s (for melting-curve analyses). All RT-qPCR assays were carried out at least in duplicate. Analyses of relative levels of gene expression were carried out using the 2-Δ/ΔCt method for a gene of interest versus the GAPDH gene, as previously described.109,110
Yeast Strains, Media, and Genetic Techniques.
S. cerevisiae media included YPD (1% yeast extract, 2% peptone, and 2% glucose; only most relevant components are cited), SD medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, and 2% glucose), and synthetic complete (SC) medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, and 2% glucose) with a drop-out mixture of compounds required by specific auxotrophic strains. S. cerevisiae strains used in this work are cited in the next section. Standard techniques were used for the construction of yeast strains and transformation by DNA.104,111
Split-Ubiquitin Binding Assay.
The assay’s concept98,99 is described in Results. A version of the split-Ub assay was carried out in S. cerevisiae largely as described previously.44,112 S. cerevisiae NMY51 (MATa trp1 leu2 his3 ade2 LYS2::lexA-HIS3 ade2::lexA-ADE2 URA3::lexA-lacZ) (Dualsystems Biotech AG, Schlieren, Switzerland) was cotransformed with split-Ub-based bait and prey plasmids (Table S1) using the lithium acetate method.111 Transformants were selected for the presence of both bait and prey plasmids during ∼3 days of growth at 30 °C on SC(−Trp, −Leu) medium (minimal medium containing 2% glucose, 0.67% yeast nitrogen base, 2% bacto-agar, and a complete amino acid mixture that lacked Leu and Trp). Single colonies of the resulting cotransformants were grown in SC(-Trp, -Leu) liquid medium to an A600 of ∼1.0, a near-stationary phase. The cultures were thereafter serially diluted by 3-fold, and 10 μL samples of cell suspensions were spotted onto either double-dropout SC(-Trp, -Leu), triple-dropout SC(-Trp, -Leu, -His), or quadruple-dropout SC(-Trp, -Leu, -His, -Ade) plates, which were incubated at 30 °C for 2–4 days.
Bacterial Adenylate Cyclase-Based (BACTH) Protein Binding Assay.
BACTH was carried out as previously described,100 using E. coli strain BTH101 that lacked the CyaA adenylate cyclase [F−, cya-99, araD139, galE15, galK16, rpsL1 (Strr), hsdR2, mcrA1, mcrB1]. Briefly, the ORFs encoding the Nt-fragment of mmUBR1 (mmUBR11–1032), the Nt-fragment of mmUBR2 (mmUBR21–1041), and the full-length 24 kDa mmATF3 (see Results) were cloned into the pKT25 and pUT18 plasmids, yielding fusions of mmUBR1/2 fragments with the T25 Nt-fragment of the Bordetella pertussis CyaA cyclase and fusions of mmATF3 with the cyclase’s T18 Ct-fragment. The resulting bait and prey plasmids were cotransformed into BTH101 E. coli,108 followed by selection for growth at 30 °C on plates containing 4% MacConkey Agar (Difco, 212123), 1% maltose (Difco, 216830), 50 μg/mL ampicillin (VWR, 0039), and 25 μg/mL kanamycin (VWR, 0408). Single colonies of the resulting cotransformants were grown to an A600 of ∼1.0 (a near-stationary phase) in LB liquid media containing 2 mM IPTG (VWR, 97063–282), 50 μg/mL ampicillin, and 25 μg/mL kanamycin. The resulting cultures were serially diluted by 2-fold, and 10 μL samples of cell suspensions were spotted onto M63/maltose plates containing 1× M63 minimal medium at pH 7.0 [76 mM ammonium sulfate, 0.5 M monopotassium phosphate, 9 μM iron(II) sulfate heptahydrate, 1.5% agar, 0.2% maltose, and 2 mM IPTG]. Plates were incubated at 30 °C for 2–7 days.
RESULTS
Construction of “Floxed” [CaggCreERtm; mmUBR1−/− mmUBR2flox/flox] Mouse Strains.
Previous studies by this laboratory produced and characterized single-mutant null mmNTAN1−/−, mmNTAQ1−/−, mmATE1−/−, mmUBR1−/−, and mmUBR2−/− mouse strains that lacked specific components of the Arg/N-degron pathway41,64,66,73,74,113 (Figure 1). mmUBR1−/− mice, which retain all other Arg/N-recognins and thus a significant fraction of the pathway’s activity, are counterparts of hsUBR1−/− humans with JBS (see the introduction).80–82 mmUBR1−/− mice have a milder version of human JBS. Other abnormal phenotypes of mmUBR1−/− mice include a perturbed regulation of fatty acid synthase in the skeletal muscle during starvation.73
The 200 kDa mmUBR1 and 200 kDa mmUBR2 E3s are expressed in all or most mouse tissues, and their sequences are 47% identical.2,72 One phenotype of adult mmUBR2−/− mice in the 129/B6 strain background is infertility of males, owing to the near absence of synaptonemal complexes and apoptosis of mmUBR2−/− spermatocytes.74 Embryonic fibroblasts (EFs) derived from mmUBR2−/− mice exhibit genomic instability.114 In contrast to the viability of mmUBR1−/− and mmUBR2−/− mice, double-mutant [mmUBR1−/− mmUBR2−/−] mice die as midgestation embryos, with severe neural and cardiovascular defects.73,74,83 We asked whether it might be possible to generate viable adult [mmUBR1−/− mmUBR2−/−] mice, despite the 100% embryonic lethality of the [mmUBR1−/− mmUBR2−/−] genotype.
To produce, conditionally, a [mmUBR1−/− mmUBR2−/−] mouse strain, we used the previously generated mmUBR1−/− mice73 (C57BL/6N strain background), and also Gt(ROSA)-26Sortm1(FLP1)Dym, mmUBR2tm1a(KOMP)Mbp/+, and CaggCreERtm mouse strains (Figure 2A,B and Figures S2–S4). Our aim was to produce a double mutant [mmUBR1−/− mmUBR2flox/flox] strain that was poised to convert both copies of the mmUBR2flox gene (which encoded the wild-type mmUBR2 protein) into their inactive counterparts through a conditional expression of Cre recombinase. Active Cre would excise an essential mmUBR2 genomic DNA fragment (Figure 2A,B and Figure S2). Using procedures described in Materials and Methods, we constructed, initially, a homozygous mmUBR2flox/flox mouse strain, in which both copies of mmUBR2 had two inserted Cre-recognized loxP sites that flanked exons 4 and 5 (Figures S2–S4).
In the next step, we produced conditional [CaggCreER; mmUBR1−/− mmUBR2flox/flox] mice, using a series of heterozygous matings among mmUBR1−/− mice,73 mmUBR2flox/flox mice, and CaggCreER mice.103 The latter mouse strain contained the CaggCreER gene, expressed from the ubiquitously active chimeric Pcagg promoter.103 CaggCreER encodes CreER, a fusion between Cre and the mouse estrogen receptor ligand-binding domain that can interact with tamoxifen (TM), a synthetic estrogen-like compound that binds to CreER. When CreER is expressed in a mouse, it is inactive as a Cre recombinase but can be activated by injecting mice intraperitoneally with TM.103
A control strain was [mmUBR1−/− mmUBR2flox/flox], which contained both copies of mmUBR2 as mmUBR2flox but lacked the CreER recombinase. The functional intactness of the mmUBR2flox allele was inferred from the fact that both [CaggCreER; mmUBR1−/− mmUBR2flox/flox] and [mmUBR1−/− mmUBR2flox/flox] mice survived embryogenesis at frequencies indistinguishable from those for parental mmUBR1−/− mice, and that the resulting adult “floxed” mice were phenotypically similar to single-mutant mmUBR1−/− mice. In addition, tissues of these mice contained mmUBR2 E3, as determined by IB (Figure 2D).
In summary, [CaggCreER; mmUBR1−/− mmUBR2flox/flox], the desired conditional mouse strain, unconditionally lacked mmUBR1 and contained apparently normal levels of mmUBR2 (Figure 2D,E). Injections of TM into adult (∼2 months old) [CaggCreER; mmUBR1−/− mmUBR2flox/flox] mice activated Cre, which inactivated both copies of mmUBR2flox in these mice, yielding the [CaggCreER; mmUBR1−/− mmUBR2−/−] genotype (Figure 2D,E and Figures S2–S4).
Death of ∼40% of [CaggCreERtm; mmUBR1−/− mmUBR2−/−] Mice within a Month from Induction of the Double-Mutant Genotype.
In [mmUBR1−/− mmUBR2flox/flox] control mice, which lacked Cre recombinase, the mmUBR2flox allele would not be inactivated by TM (Figure S2). As expected, a 5-day regimen of intraperitoneal TM injections was reproducibly (without exceptions) nonlethal to [mmUBR1−/− mmUBR2flox/flox] mice over subsequent weeks (Figure 2C, left panel, red rectangles). In striking contrast, the same TM injection regimen (carried out, so far, with 33 [CaggCreER; mmUBR1−/− mmUBR2−/−] mice) resulted in the death of 4 (∼12%), 6 (∼18%), 3 (∼9%), and 1 (∼3%) of these adult mice during the first, second, third, and fourth weeks, respectively, counting from the beginning of the 5-day TM injection period (Figure 2C). The remainder (∼58%) of this 33-mouse cohort survived for significantly more than 30 days from the start of injections (Figure 2C, right panel), with some of them surviving for at least 300 days.
Given the uniformly nonlethal TM injection results with the control (Cre-lacking) [mmUBR1−/− mmUBR2flox/flox] strain, it was the onset of the [CaggCreER; mmUBR1−/− mmUBR2−/−] genotype (starting from the initial and robustly viable [CaggCreER; mmUBR1−/− mmUBR2flox/flox] genotype) that killed, over the first month, ~42% of [CaggCreER; mmUBR1−/− mmUBR2−/−] double-mutant mice (Figure 2C). All data, below, about [CaggCreER; mmUBR1−/− mmUBR2−/−] mice were obtained with the surviving cohort of these mice. We do not know, at present, specific causes of death of tamoxifen-induced [CaggCreER; mmUBR1−/− mmUBR2−/−] mice that died within a month of the start of the 5-day tamoxifen regimen (Figure 2C).
Mouse strains were produced and maintained at a pathogen-free (barrier) facility (see Materials and Methods). However, both TM injections and post-TM work with these mice were performed in conventional (nonbarrier) facilities. Thus, one verifiable possibility, which can be addressed by carrying out all procedures with [CaggCreER; mmUBR1−/− mmUBR2−/−] mice inside a barrier facility, is that the absence of both mmUBR1 and mmUBR2 may compromise specific aspects of the immune system (given the emerging immunity/inflammation functions of the Arg/N-degron pathway29,67,115) and therefore would make newly generated double-mutant mice at least transiently more susceptible to infections that would be lethal in these mice but nonlethal in single-mutant mmUBR1−/− mice.
IB assays for hsUBR1 and hsUBR2 involved SDS–PAGE of extracts from mouse tissues and antibodies to the E3s mentioned above. As measured by levels of the hsUBR2 protein, the penetrance of cre-lox-mediated inactivation of both copies of mmUBR2flox in TM-treated (and surviving) [CaggCreER; mmUBR1−/− mmUBR2−/−] mice was nearly 100% in the lung and at least 90% in the brain, thymus, and spleen (Figure 2D). In addition, IBs with the anti-UBR1 antibody confirmed the absence of mmUBR1 in the unconditional mmUBR1−/− background (Figure 2E). In agreement with IB results, RT-PCRs with the brain and kidney RNA preparations for RNA derived from the intact (mmUBR2flox) versus exon 4/5-deleted (mmUBR2−) alleles (see Materials and Methods) showed nearly undetectable (<5%) levels of “flox”-derived mmUBR2 RNA sequences (Figure 2A,B,D and Figures S2–S4).
Unconditional Double-Mutant [hsUBR1−/− hsUBR2−/−] Human HEK293T Cells.
In addition to conditional adult double-mutant [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice (Figure 2 and Figures S2–S4), we used the CRISPR-Cas9 technique85–87 to construct, sequentially (at first hsUBR2−/− cells and thereafter [hsUBR1−/− hsUBR2−/−] cells), double-mutant human HEK293T cell lines that lacked both hsUBR1 and hsUBR2 E3s (Figure 3A,B; see Materials and Methods). The absence of both hsUBR1 and hsUBR2 E3s in two independently produced [hsUBR1−/− hsUBR2−/−] HEK293T cell lines was verified by IBs with antibodies to these E3s (Figure 3A,B). [hsUBR1−/− hsUBR2−/−] HEK293T cells did not seem to differ significantly from parental HEK293T cells in either light-microscopic appearance or growth rates.
The mammalian Arg/N-degron pathway is mediated by at least four E3s: UBR1, UBR2, UBR4, and UBR5 (Figure 1 and the introduction). In previously characterized cell culture settings, UBR1 and UBR2 appeared to mediate the bulk of degradation of model substrates bearing Arg/N-degrons.2,24 To address this question with [hsUBR1−/− hsUBR2−/−] HEK293T cells, we used plasmids that expressed, from the PCMV promoter, a set of previously constructed C-terminally flag-tagged X-RGS4f proteins.116 In these 25 kDa proteins, X was either Arg, a destabilizing Nt-residue, or Val, an Nt-residue that is not recognized as destabilizing (Figure 1). To produce, in vivo, X-RGS4f proteins bearing a desired Nt-X residue, they were expressed as constructs of the Ub reference technique (URT), a variant of the Ub fusion technique.22,116,117
Cycloheximide (CHX) chase-degradation assays,40,106 with chases for 0, 2, and 4 h, used wild-type versus [hsUBR1−/− hsUBR2−/−] HEK293T cells that had been transiently transfected with plasmids expressing X-RGS4f proteins (X = Arg or Val) (Figure 3C). Extracts of these cells were fractionated by SDS–PAGE, followed by IBs with anti-flag to detect X-RGS4f, and with an antibody to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (as well as Coomassie staining of total protein patterns) to verify the uniformity of protein loads (Figure 3C).
Arg-RGS4f, known to be rapidly degraded by the Arg/N-degron pathway,22,116 was so short-lived in wild-type HEK293T cells that it could be detected but barely so even at the start of the CHX chase (at the level of IB sensitivity in these assays) (Figure 3C, lanes 2–4). In contrast, Arg-RGS4f was readily detectable and stable in [hsUBR1−/− hsUBR2−/−] double-mutant cells (Figure 3C, lanes 2–4; compare with lanes 8–10). However, the otherwise identical Val-RGS4f was a long-lived protein in both wild-type and [hsUBR1−/− hsUBR2−/−] cells (Figure 3C, lanes 5–7 and 11–13; compare with lanes 2–4). These chase-degradation results (Figure 3C) indicated that hsUBR1 and hsUBR2 are the major contributors, in human HEK293T cells, to the targeting of protein substrates that bear Arg/N-degrons, as distinguished from substrates bearing other degrons recognized by the Arg/N-degron pathway (see the introduction).
Increased Levels of the Endogenous, Untagged mmATF3 Transcription Factor in [mmUBR1−/− mmUBR2−/−] Mice.
[hsUBR1−/− hsUBR2−/−] human cell lines and [CaggCreER; mmUBR1−/− mmUBR2−/−] adult mice that have been generated in this study (Figures 2 and 3 and Figures S2–S4) became complementary tools in our work. The data about ATF3 in this section and the following sections stem from the use of these tools. We concentrated, initially, on ATF3 in preference to other proteins because our split-Ub binding assays have revealed, at early stages of this study, a physical binding of ATF3 to UBR1/UBR2 (see below), and also because a readily available anti-ATF3 antibody was of uncommonly high quality (see Materials and Methods).
Mammalian ATF3 is a 24 kDa bZIP TF. It contains a positively charged DNA-binding region next to a leucine zipper coiled coil motif that mediates the formation of a homodimer ATF3 or its complexes with other bZIP TFs, other TFs, or non-TF proteins.88–97 ATF3 is a member of a family of bZIP TFs that includes the JUN, FOS, ATF/CREB, and JDP subfamilies and consists of nearly 60 distinct bZIP-coding genes in a mouse or human.97 Mammalian ATF3 is induced by many different stresses, controls hundreds of genes, regulates its own gene, as well, and can act as either a repressor or an activator of transcription.88–96 ATF3 is short-lived in vivo. The MDM2 E3 Ub ligase has been shown to mediate a fraction of ATF3 degradation.94 Other E3s that can target ATF3 for degradation were unknown until this study.
The levels of endogenous, untagged mmATF3 were assayed by IB in tissues of either double-mutant [CaggCreER; mmUBR1−/− mmUBR2−/−] or single-mutant (mmUBR1-lacking, mmUBR2-containing) [mmUBR1−/− mmUBR2flox/flox] adult mice. We found that the level of mmATF3 was reproducibly and significantly increased in (at least) kidneys and thymi of [CaggCreER; mmUBR1−/− mmUBR2−/−] mice, relative to levels of mmATF3 in these tissues of [mmUBR1−/− mmUBR2flox/flox] mice (Figure 4A, lane 1, compared to lane 2, and lane 3, compared to lane 4). A parsimonious interpretation of these results was that the increases in the steady-state levels of mmATF3 in tissues of double-mutant mice (Figure 4A) were caused, at least in part, by metabolic stabilization of mmATF3 in the absence of both mmUBR1 and mmUBR2 E3s. This interpretation is in agreement with the results of chase-degradation assays in the setting of HEK293T human cells that lack both hsUBR1 and hsUBR2 (see below and Figure 4C–E).
Figure 4.
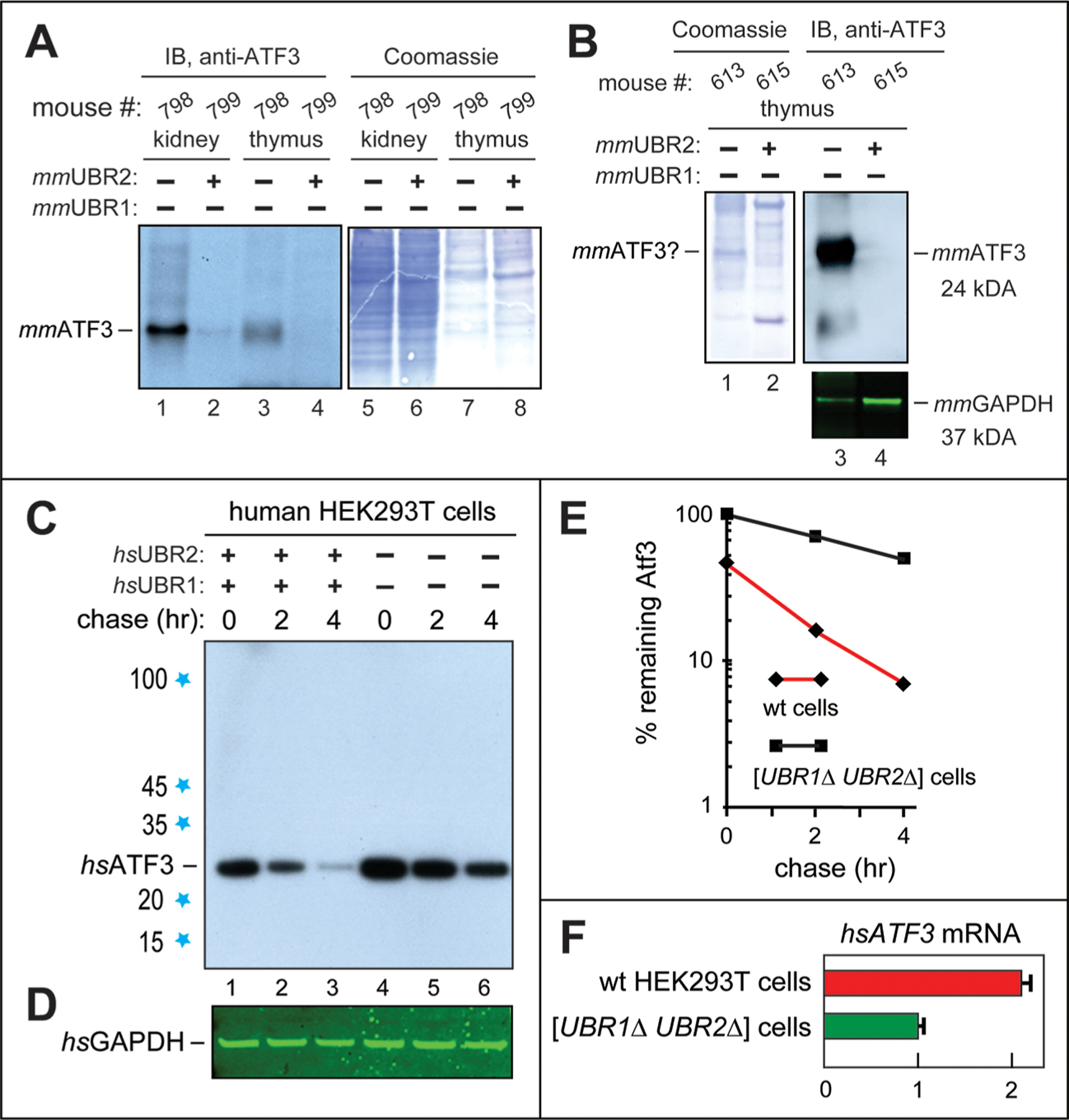
Increases in the levels of the ATF3 transcription factor in [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice and [hsUBR1−/− hsUBR2−/−] HEK293T human cells, and the degradation of hsATF3 by the human Arg/N-degron pathway. (A) Lanes 1 and 2, IB comparisons, using the anti-ATF3 antibody, of the levels of mmATF3 in extracts from kidneys of a [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mouse (lane 1) and a mmUBR2-containing [mmUBR1−/− mmUBR2flox/flox] mouse (lane 2) (see Materials and Methods). Lanes 3 and 4, same as lanes 1 and 2, respectively, but in extracts from thymi of the same mice. Lanes 5–8, same as lanes 1–4, respectively, Coomassie-stained total protein patterns. (B) Lanes 1 and 2, Coomassie-stained total protein patterns of extracts from thymi of another (“exceptional”) [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mouse vs a [mmUBR1−/− mmUBR2flox/flox] mouse. Lanes 3 and 4, same as lanes 1 and 2, respectively, but IB with the anti-ATF3 antibody. The bottom panel shows the corresponding IB-determined levels of GAPDH. Whereas the level of mmATF3 was undetectably low in the thymus of the [mmUBR1−/− mmUBR2flox/flox] mouse (lane 4; similar to results in lane 4 of panel A), the level of mmATF3 in the thymus of the exceptional [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mouse (lane 3) was so high that the band of mmATF3 may have become detectable as a distinct Coomassie-stained band in the total protein pattern (this remains to be verified; lane 1, denoted with a question mark on the left). Only two (of six) examined [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice, and only in the thymus, exhibited this “runaway” increase in the level of mmATF3, as distinguished from significant but much lower increases in the level of mmATF3 in other [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice (panel A, lanes 1 and 3). See Results for additional discussion. (C) CHX-based chase-degradation analysis of the endogenous, untagged hsATF3 in wild-type vs [hsUBR1−/− hsUBR2−/−] human HEK293T cells. Lanes 1–3, CHX chase with hsATF3 for 0, 2, and 4 h, respectively, in wild-type HEK293T cells. Lanes 4–6, same as lanes 1–3, respectively, but with [hsUBR1−/− hsUBR2−/−] HEK293T cells. (D) Levels of GAPDH (loading control) in the same experiment, determined by IB with the anti-GAPDH antibody. (E) Quantification (a semilog graph) of the results in panel C. These curves are a direct quantification of the patterns in panel C, hence the absence of standard deviations. All degradation assays were performed at least twice, yielding results that differed by <10%. (F) RT-qPCR analysis of the levels of hsATF3 mRNA in wild-type (wt) HEK293T cells vs [hsUBR1−/− hsUBR2−/−] cells (relative to the levels of GAPDH mRNA, a calibration control), with the level of hsATF3 mRNA in [hsUBR1−/− hsUBR2−/−] HEK293T cells taken as 1.0.
Remarkably, in the thymi of two [CaggCreER; mmUBR1−/− mmUBR2−/−] mice (but not in four other mice of the double-mutant genotype that had been examined by IB), the significant increases in the levels of mmATF3 mentioned above (Figure 4A) were dwarfed by strikingly high levels of the mmATF3 protein in these mice (Figure 4B). Specifically, those levels of mmATF3, assayed by IB, were so high (vs undetectably low levels of mmATF3 in the thymi of control [mmUBR1−/− mmUBR2flox/flox] mice) that the normally scarce mmATF3 protein apparently became a significant band in the Coomassie-stained total protein pattern (Figure 4B, lanes 1 and 3 vs lanes 2 and 4; see the legend of Figure 4B for additional details).
Depending on the nucleotide sequences of specific transcriptional promoters and on interactions of ATF3 with itself and other TFs, ATF3 can act as either a repressor or an activator of transcription of many genes, including its own.88−96 ATF3 can be upregulated by many stresses, including inflammation.89,91,92 Significant (but not runaway) increases in the level of mmATF3 were observed in (at least) the kidneys and thymi of all other examined mice of the [mmUBR1−/− mmUBR2−/−] genotype (Figure 4A). It is possible that the two exceptional cases of double-mutant mice with particularly high levels of the mmATF3 protein in their thymi (Figure 4B) had occurred when a metabolic stabilization of mmATF3 in the absence of both mmUBR1 and mmUBR2 Ub ligases was accompanied, in addition, by a transcriptional/translational upregulation (mediated by inflammation?) of the mmATF3 protein that led to a runaway positive feedback. Molecular circuits that underlie this phenomenon in the thymus (the only tissue, among examined ones, in which the effect was observed) remain to be addressed.
Interestingly, ATF3 apparently represses its own gene,90 making the cases of runaway levels of the mmATF3 protein in the thymi of two exceptional double-mutant mice (Figure 4B) more difficult to explain and therefore particularly worthy of reproduction and understanding. In summary, the hypothesis presented above ascribes strikingly high increases in the level of the mmATF3 protein in two [CaggCreERtm; mmUBR1−/− mmUBR2−/−] mice (of six examined double-mutant mice) (Figure 4B) to a combination of two inputs: (i) the absence, in all mice of the double-mutant genotype, of the mmUBR1 and mmUBR2 E3s that normally target mmATF3 for degradation and (ii) a sporadic (in only two such mice so far) infection/inflammation process that unleashed a transcription/translation positive feedback (apparently confined to the thymus) that increased the level of metabolically stabilized mmATF3 protein to strikingly high levels in these thymi (Figure 4B; compare to Figure 4A). One approach to verifying this conjecture would be to determine mmATF3 levels in [CaggCreER; mmUBR1−/− mmUBR2−/−] mice, in comparison to both [mmUBR1−/− mmUBR2flox/flox] and wild-type mice, under inflammation-inducing conditions, for example upon treatments with bacterial lipopolysaccharide (LPS).
Increased Levels of Endogenous hsATF3 in [hsUBR1−/− hsUBR2−/−] Human Cells.
The results of comparing steady-state levels of endogenous, untagged human hsATF3 (its amino acid sequence is >95% identical to that of mmATF3) between exponentially growing wild-type HEK293T cells and their [hsUBR1−/− hsUBR2−/−] counter-parts were similar to findings with [CaggCreER; mmUBR1−/− mmUBR2−/−] double-mutant mice versus mmUBR1-lacking, mmUBR2-containing single-mutant mice (Figure 4A,B and Figure S5). Specifically, whereas the endogenous hsATF3 was barely detectable in wild-type HEK293T cells, its levels, while variable, were always significantly higher in cells lacking both hsUBR1 and hsUBR2 E3s, e.g., ∼5-fold higher in the example shown in Figure S5.
The relative level of hsATF3 mRNA (measured by RT-qPCR) in wild-type HEK293T cells versus their [hsUBR1−/− hsUBR2−/−] counterparts was 2.1-fold higher in wild-type cells (Figure 4F). Thus, a significant increase in the level of the hsATF3 protein in [hsUBR1−/− hsUBR2−/−] HEK293T cells took place despite a >2-fold decrease in the level of hsATF3 mRNA in these cells, in comparison to wild-type HEK293T cells (Figure 4F and Figure S5). A decrease in the level of hsATF3 mRNA in cells that overproduce the hsATF3 protein (Figure 4F and Figure S5) is in agreement with evidence that ATF3 represses transcription of its own gene.90 It remains to be determined whether the observed increase in the level of hsATF3 in [hsUBR1−/− hsUBR2−/−] HEK293T cells (Figure 4C–E and Figure S5) stems entirely from the metabolic stabilization of hsATF3 in hsUBR1/hsUBR2-lacking cells or whether, for example, a translational upregulation of hsATF3 is involved, as well.
hsATF3 as a Short-Lived Substrate of the Arg/N-Degron Pathway.
We also carried out CHX-based chase-degradation assays with the endogenous, untagged hsATF3 in [hsUBR1−/− hsUBR2−/−] HEK293T cells versus their wild-type counterparts. The rate of degradation of hsATF3 in wild-type HEK293T cells (t1/2 < 1 h) was found to be much higher than in [hsUBR1−/− hsUBR2−/−] double-mutant cells (t1/2 ∼ 4 h), thereby identifying hsUBR1 and hsUBR2 as functionally overlapping Ub ligases that mediate the degradation of hsATF3 (Figure 4C,E).
In addition, chase-degradation assays with endogenous hsATF3 and wild-type versus [hsUBR1−/− hsUBR2−/−] HEK292T cells have also revealed a significant “time-zero” effect of ablating both hsUBR1 and hsUBR2 (Figure 4C–E). Specifically, the level of hsATF3 in wild-type cells at the start of the chase, i.e., the steady-state level of the hsATF3 protein, was ∼2-fold lower than the analogous time-zero level of hsATF3 in [hsUBR1−/− hsUBR2−/−] cells (Figure 4C–E). This effect was qualitatively similar to the results of independent steady-state IB analyses described in Figure S5. The increases of the steady-state levels of hsATF3 in [hsUBR1−/− hsUBR2−/−] HEK293T cells described above varied between ∼2- and ∼5-fold in different experiments. The reason for this variability remains to be understood. In summary, the significantly slower degradation of hsATF3 during the chase in [hsUBR1−/− hsUBR2−/−] human cells was accompanied, in addition, by a significant increase in the time-zero (start of chase) level of hsATF3 in these cells (Figure 4C–E).
An incomplete in vivo stabilization of hsATF3 in the absence of hsUBR1/hsUBR2 (Figure 4C–E) was expected, as many TFs contain more than one degron.97,118 In addition, the degradation of hsATF3 was previously shown to be mediated, in part, by the hsMDM2 Ub ligase.94 Together, our analyses of the endogenous, untagged ATF3 in adult [CaggCreER; mmUBR1−/− mmUBR2−/−] mice and in [hsUBR1−/− hsUBR2−/−] HEK293T human cells (in the latter case, using chase-degradation assays) indicated that mammalian ATF3 is a short-lived substrate of the Arg/N-degron pathway (Figure 4). This conclusion is supported, independently, by the findings, described below, that human and mouse ATF3 proteins specifically interact with fragments of the 200 kDa UBR1 and UBR2 E3 Ub ligases.
Protein Binding Assays.
A majority of TFs are autoactivating in the classic two-hybrid protein interaction assay.119 In part for this reason, our analyses of mammalian TFs employed a version of the split-Ub technique44,98,99,112 (Figure 5A). In this method, two test proteins are expressed, in S. cerevisiae, as fusions to a Ct-half of Ub (CUb) and to its mutant Nt-half (NUb). A mutation in the Nt-half of Ub serves to weaken the constitutive binding of NUb to CUb. An interaction between test proteins that contain linked Ub halves would reconstitute a quasinative Ub moiety from CUb and mutant NUb, causing the in vivo cleavage of a CUb-containing test fusion by (constitutively present) deubiquitylases immediately downstream from the reconstituted Ub moiety. This cleavage acts, through additional steps, as readout of split-Ub assays98,99 (Figure 5A).
Figure 5.
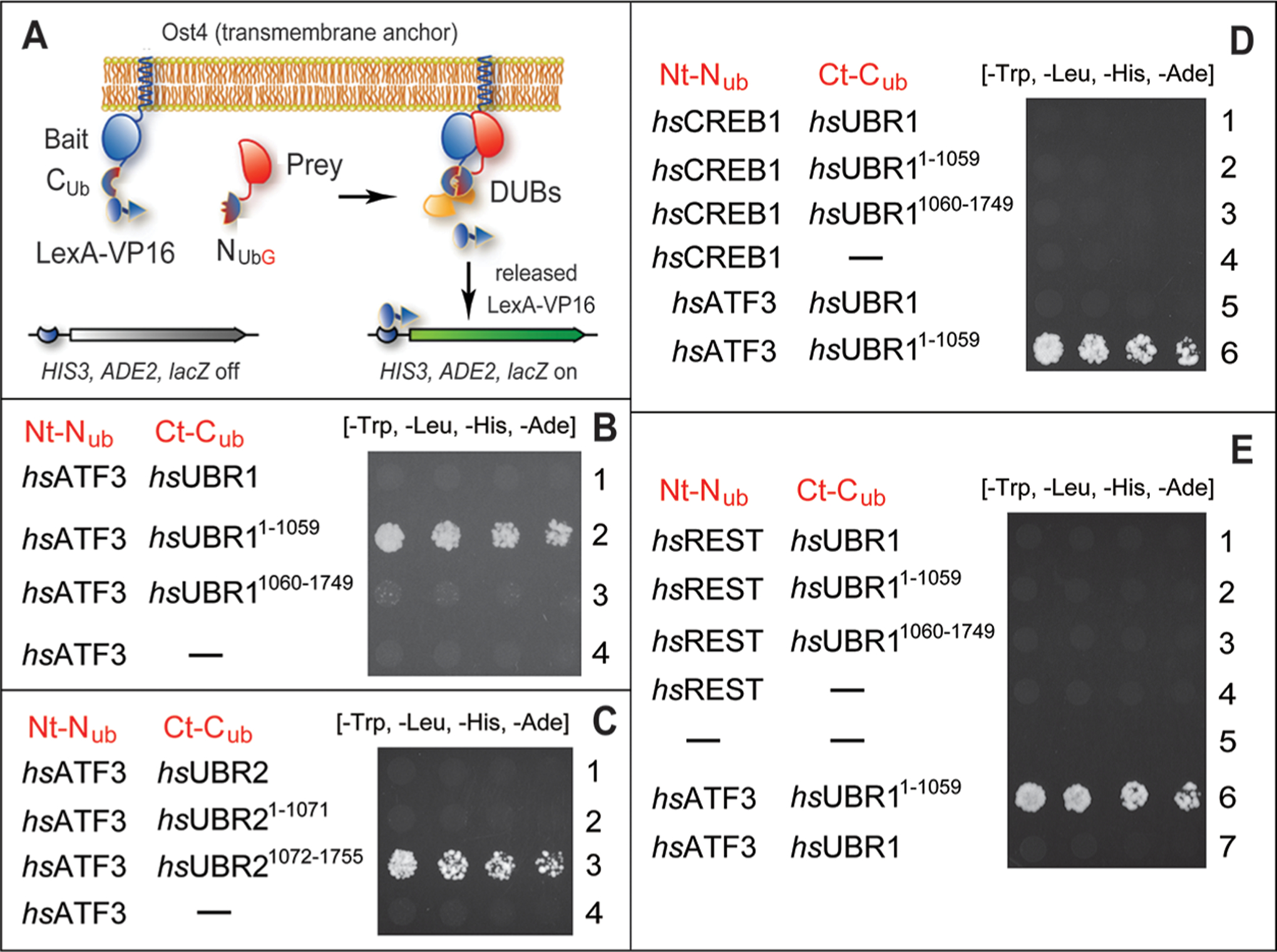
Split-ubiquitin protein interaction assays with human transcription factors ATF3, CREB1, and REST vs human UBR1 and UBR2 E3 ubiquitin ligases. (A) Design of split-Ub assays44,98,99,112 (see Materials and Methods). (B) Row 1, hsATF3 vs full-length hsUBR1. Row 2, hsATF3 vs the Nt-fragment of hsUBR1 (hsUBR11–1059). Row 3, hsATF3 vs the Ct-fragment of hsUBR1 (hsUBR11060–1749). Row 4, hsATF3 vs the vector alone. (C) Row 1, hsATF3 vs full-length hsUBR2. Row 2, hsATF3 vs the Nt-fragment of hsUBR2 (hsUBR21–1071). Row 3, hsATF3 vs the Ct-fragment of hsUBR2 (hsUBR11072–1755). Row 4, hsATF3 vs the vector alone. (D) Row 1, hsCREB1 vs full-length hsUBR1. Row 2, hsCREB1 vs the Nt-fragment of hsUBR1 (hsUBR11–1059). Row 3, hsCREB1 vs the Ct-fragment of hsUBR1 (hsUBR11060–1749). Row 4, hsCREB1 vs the vector alone. Row 5, hsATF3 vs full-length hsUBR1 (the same as in row 1 of panel A but an independent split-Ub assay). Row 6, hsATF3 vs the Nt-fragment of hsUBR1 (hsUBR11–1059) (the same as in row 2 of panel B but an independent split-Ub assay). (E) Row 1, hsREST vs full-length hsUBR1. Row 2, hsREST vs the Nt-fragment of hsUBR1 (hsUBR11–1059). Row 3, hsREST vs the Ct-fragment of hsUBR1 (hsUBR11060–1749). Row 4, hsREST vs the vector alone. Row 5, vectors alone. Row 6, hsATF3 vs the Nt-fragment of hsUBR1 (hsUBR11–1059) (the same as in row 2 of panel B but an independent split-Ub assay). Row 7, hsATF3 vs full-length hsUBR1 (the same as in row 1 of panel A but an independent split-Ub assay).
Specific readouts, in the S. cerevisiae strain for split-Ub assays (Figure 5A and Materials and Methods), comprise, independently, an induction of the scHIS3 gene, the scADE2 gene, and the E. coli lacZ gene (Figure 5A). The activity of scHIS3 and scADE2 makes possible yeast growth assays on minimal medium plates that lack either histidine (His) or both His and adenine (Ade) (Figure 5A). Controls included IBs to examine the in vivo levels of split-Ub fusions, and also verifying that binding positive fusions were not autoactivating, i.e., that none of them were positive in split-Ub assays that contained just one of two fusions. The results summarized below passed all of these controls. As described below, this study also employed a different, E. coli-based, and mechanistically distinct BACTH protein interaction assay120 (Figure S7).
The ATF3 Transcription Factor Binds to the N-Terminal Fragment of UBR1 but Not to Full-Length UBR1.
In agreement with chase-degradation results, indicating the hsUBR1/hsUBR2-mediated degradation of hsATF3 in HEK293T cells (Figure 4C–E), split-Ub protein interaction assays revealed the binding of the 123 kDa Nt-fragment of the 200 kDa human hsUBR1 (hsUBR11–1059) to full-length 24 kDa hsATF3 (Figure 5B, row 2, Figure 5D, row 6, and Figure 5E, row 6). Similar split-Ub results were obtained with mouse mmATF3 versus the 119 kDa Nt-fragment of mouse mmUBR1 (mmUBR11–1032) (Figure S6A,B).
In contrast, hsATF3 did not bind to either full-length hsUBR1 or its 77 kDa Ct-fragment (hsUBR11060–1749) (Figure 5B, rows 1 and 3, compared with row 2; Figure 5D, row 5, and Figure 5E, row 7). Similar results were obtained with mmATF3 versus mmUBR1 (Figure S6A). The absence of hsATF3 binding to full-length UBR1 was unexpected, given the robust binding of, for example, hsATF3 to the hsUBR11–1059 Nt-fragment (Figure 5B, rows 1 and 3, compared with row 2; Figure 5D, row 5, and Figure 5E, row 7).
The binding of a physiological substrate of UBR1 to its Nt-fragment but not to full-length UBR1 has been encountered earlier, in this lab’s studies of the conditional degradation of S. cerevisiae CUP9, a homeodomain transcriptional repressor, by the yeast Arg/N-degron pathway.61,62,121 Specifically, scCUP9 binds unconditionally to an Nt-fragment of scUBR1 but would bind to full-length scUBR1 only in the presence of dipeptides bearing destabilizing (type 1/type 2) Nt-residues. As shown previously, such peptides can interact with the substrate-binding sites of scUBR1, thereby altering its conformation and enabling the binding of the scCUP9 repressor.62 The resulting circuit, regulated by short peptides, mediates the control of peptide import in yeast.61,62,121,122 Thus far, our binding assays did not indicate any influence of short peptides on interactions between hsATF3 and hsUBR1 or its fragments.
Our initial attempts to identify a UBR1-binding segment of ATF3 (i.e., to map ATF3 degron) by carrying out split-Ub assays with hsUBR11–1059 versus hsATF3 fragments were unsuccessful, owing to autoactivation by ATF3 fragments (but not by full-length ATF3). Specifically, positive split-Ub readouts were observed with ATF3 fragments in the absence of a partner ligand. We are addressing this problem by exploring other protein interaction assays. To assess the specificity of UBR1-ATF3 interactions in a different way, split-Ub assays were also performed with full-length hsUBR1, its Nt-fragment (hsUBR11–1059), and its Ct-fragment (hsUBR11060–1749) versus other, non-ATF3 TFs, specifically hsCREB1 (another bZIP TF)97 and hsREST (a Krüppel-type zinc finger TF).97 In contrast to results with hsATF3 (Figure 5B,C), split-Ub assays did not detect a binding of either hsCREB1 or hsREST to any one of the hsUBR1-based proteins mentioned above (Figure 5D,E). Similar results (no binding to hsCREB1) were also obtained with hsUBR2 and its fragments versus hsCREB1 (Figure S6C).
BACTH, an E. coli-Based Protein Interaction Assay, Detects the Binding of ATF3 to the N-Terminal Fragment of UBR1.
To verify the discovered interaction between ATF3 and the Nt-fragment of UBR1 by changing a binding-detection method, we used the BACTH (“bacterial two-hybrid”) system, an in vivo interaction assay that is carried out in E. coli and is based on conditional reconstitution of a split bacterial (Bordetella pertussis) CyaA adenylate cyclase that forms cAMP from AMP (Figure S7).120 In this assay, a pair of potentially interacting proteins is expressed, in E. coli, as fusions to the T25 Nt-part and the T18 Ct-part of CyaA cyclase (Figure S7A–C). Interactions between two test proteins that contain the linked T25 and T18 parts of CyaA would reconstitute the ability of CyaA to produce cAMP. An increase in the level of cAMP would trigger cAMP-dependent transcriptional activation of E. coli catabolic circuits, including the lac and mal operons. Thus, an interaction between test proteins can be detected, for example, through the ability of a BACTH E. coli strain to grow on a minimal medium with maltose as the sole carbon source120 (Figure S7A–C).
The results of BACTH assays (ability to grow on maltose) with mouse mmATF3 versus Nt- and Ct-fragments of mmUBR1 and mmUBR2 are shown in Figure S7D. First, both mmATF3 alone and mmUBR11–1032 alone are not autoactivating in the BACTH assay (Figure S7D, rows 3 and 4). Second, a robust E. coli growth with mmATF3 versus mmUBR11–1032 indicates their interaction (Figure S7D, row 1, compared with rows 3 and 4), in agreement with the results by split-Ub assays (Figure 5B, row 2, Figure 5D, row 6, and Figure 5E, row 6). In summary, the binding of human or mouse ATF3 to the Nt-fragment mentioned above of human or mouse UBR1 could be detected using both the S. cerevisiae-based split-Ub assay and the mechanistically distinct E. coli-based BACTH assay.
ATF3 Binds to the C-Terminal Fragment of UBR2 but Not to Full-Length UBR2 or Its N-Terminal Fragment.
Split-Ub assays were also carried out with hsATF3 versus hsUBR2. The latter is of the same size (200 kDa) as hsUBR1, and the sequences of two E3s are 47% identical. Similarly to the observed absence of binding of hsATF3 to full-length hsUBR1 (Figure 5B, rows 1 and 3; compare with row 2; Figure 5D, row 5, and Figure 5E, row 7), the split-Ub assay detected no binding of hsATF3 to full-length hsUBR2 (Figure 5C, row 1). Surprisingly, however, hsATF3 also did not bind to the Nt-fragment of hsUBR2 (hsUBR21–1071), in contrast to the binding of the same hsATF3 to the Nt-fragment of hsUBR1 (hsUBR11–1059) (Figure 5C, row 2, compared with Figure 5A, row 2).
Even more unexpectedly, hsATF3 was found to bind to the Ct-fragment of hsUBR2 (hsUBR21072–1755) (Figure 5C, row 3, compared with rows 1, 3, and 4). This pattern of hsATF3 binding vis-à-vis the Nt- and Ct-fragments of hsUBR2 (it is reproducible among independent split-Ub assays) is the “opposite” of that observed with hsUBR1 Nt- and Ct-fragments. Specifically, hsATF3 binds to the Nt-fragment of hsUBR1 (hsUBR11–1059), but not to its Ct-fragment (hsUBR11–1059) (Figure 5B; compare with Figure 5C). The absence of binding of hsATF3 to the Nt-fragment of hsUBR2 that was observed using the S. cerevisiae-based split-Ub assay (Figure 5C, row 2) was reproduced using BACTH, the E. coli-based and mechanistically distinct interaction assay (Figure S7B, row 2, compared with row 1).
On the likely assumption that these surprising but robust and reproducible in vivo interaction results by split-Ub and BACTH assays (Figure 5B,C and Figure S7) would be confirmed through further verifications by other methods, the current set of findings suggests that a functional overlap between sequelogous UBR1 and UBR2 is far from complete, in that the two Ub ligases may play distinct roles in their (possibly conditional) targeting of ATF3 for degradation. A major priority in our ongoing studies of the ATF3-UBR1/UBR2 circuit is to map and understand a degron(s) of ATF3 that is recognized by UBR1 and/or UBR2.
DISCUSSION
This paper describes the following main results.
We constructed conditionally double-mutant [CaggCreER; mmUBR1−/− mmUBR2flox/flox] mouse strains that lacked the mmUBR1 E3 Ub ligase and lost another, sequelogous mmUBR2 E3 in adulthood, upon TM-mediated induction of Cre recombinase, thus bypassing the 100% embryonic lethality of the unconditional [mmUBR1−/− mmUBR2−/−] genotype (Figure 2). The induced [mmUBR1−/− mmUBR2−/−] genotype led to the death of nearly 50% of adult [mmUBR1−/− mmUBR2−/−] mice within a month of the genotype’s onset (Figure 2C).
The CRISPR-Cas9 technique85–87 was used to produce double-mutant [hsUBR1−/− hsUBR2−/−] human HEK293T cell lines that lacked both hsUBR1 and hsUBR2 (Figure 3A,B). A normally short-lived test protein bearing an Arg/N-degron became long-lived in [hsUBR1−/− hsUBR2−/−] human cells (Figure 3C).
The levels of the endogenous, untagged mmATF3 bZIP TF protein in examined tissues of mmUBR1/mmUBR2-lacking [CaggCreER; mmUBR1−/− mmUBR2−/−] mice were significantly higher than in mmUBR1-lacking single-mutant mice (Figure 4A). In the thymi of two (of six examined) [CaggCreER; mmUBR1−/− mmUBR2−/−] mice, the levels of the normally scarce mmATF3 protein were strikingly high, indicating a runaway positive feedback (Figure 4B). One verifiable possibility is that a major increase in the level of the mmATF3 protein (Figure 4B) stemmed from both a metabolic stabilization of mmATF3 (in the absence of the mmUBR1/mmUBR2 Ub ligases) and a strong upregulation of mmATF3 expression that may have been caused by infection and inflammation in these double-mutant mice.
Chase-degradation assays with the endogenous, untagged hsATF3 in human [hsUBR1−/− hsUBR2−/−] HEK293T cells and their wild-type counterparts showed that the Arg/N-degron pathway mediates a large fraction of hsATF3 degradation in wild-type cells (Figure 4C–E). To the best of our knowledge, ATF3 is the first physiological substrate of the mammalian Arg/N-degron pathway that is targeted for degradation through an internal (remaining to be mapped) degron, as distinguished from an Arg/N-degron.
Split-Ub interaction assays revealed the binding of the 123 kDa Nt-fragment of the 200 kDa human hsUBR1 to full-length 24 kDa hsATF3 (Figure 5B,D,E). In contrast, hsATF3 did not bind to either full-length hsUBR1 or its 77 kDa Ct-fragment (Figure 5B). The binding of hsATF3 to the Nt-fragment of hsUBR1 was in agreement with the identification of hsATF3 as a short-lived substrate of the Arg/N-degron pathway (Figure 4C–E). These findings, obtained through the S. cerevisiae-based split-Ub assay (Figure 5), were reproduced using the E. coli-based and mechanistically distinct BACTH protein interaction assay (Figure S7).
Two other human TFs, hsCREB1 and hsREST, did not bind to either hsUBR1, hsUBR2, or their examined fragments (Figure 5D,E and Figure S6C), supporting the specificity of hsATF3-hsUBR1 interaction.
hsUBR1 and hsUBR2 of the Arg/N-degron pathway are both 200 kDa in size and highly sequelogous (47% identical), and there is a significant (though not complete) functional overlap among these E3s (Figure 1).2,72 Therefore, it was particularly unexpected to find that while hsATF3 interacts with the 123 kDa Nt-fragment of hsUBR1, the same hsATF3 exhibits an “opposite” binding pattern with hsUBR2, in that hsATF3 binds to the 81 kDa Ct-fragment of hsUBR2 (Figure 5B,C).
Broad spans of mammalian DNA occupied by transcriptional promoters (if their definition includes the often remote enhancer and silencer sites); a number of different TF-binding sites in a complete promoter; the ∼1600 distinct TFs encoded by the human genome;97 the tendency of TFs to form not only homo-oligomers but also hetero-oligomeric complexes with other proteins, including other TFs; the functionally relevant shuttling of TFs between the nucleus and cytosol; and the conditional degradation of most TFs123–126 make transcriptional regulation an uncommonly complex setting, particularly vis-à-vis TFs that regulate hundreds of genes.
The ATF3 bZIP TF is a previously studied TF of the latter kind.88–97 It can form a homodimer and also heterodimers with other bZIP TFs (of which there are –60 in a mouse or human97), and with other proteins, as well. Mammalian ATF3 is induced by many different stresses (including infection and inflammation), controls hundreds of functionally diverse genes, regulates its own gene, as well, and can act as either a repressor or an activator of transcription.88–96 To this complexity of ATF3 roles and functional interactions is now added its degradation, as ATF3 is shown, here, to be targeted for destruction by the Arg/N-degron pathway (Figure 4C–E) and has also been shown to be targeted by the MDM2 E3 Ub ligase.94
Rape, Mena, and colleagues recently discovered the first example of dimerization quality control.123,124 It involves specific folded domains called BTBs, which are present in TFs and other proteins. Although the functionally relevant configuration of a BTB domain is usually a homodimer, a BTB domain can also heterodimerize with other BTB domains. It was found that while a homodimeric BTB-containing protein does not expose its conditional BTB-localized degron, such a degron becomes active (exposed) in a heterodimeric (incorrectly assembled) protein, leading to its selective degradation.123,124
Homodimeric versus heterodimeric alternatives are also relevant to a number of other protein domains, including the leucine zipper domain of ATF3, a bZIP TF. Specific DNA-binding properties and transcriptional functions of ATF3 depend on whether it acts, in vivo, as a homodimer or as a heterodimer with other bZIP TFs or other proteins, including other TFs.88–97 One verifiable possibility is that distinct dimeric configurations of ATF3 may have different in vivo half-lives, because a degron(s) recognized by UBR1/UBR2 in ATF3 may be either active (exposed) or sterically shielded, depending on a particular homodimeric/heterodimeric state of ATF3 in its complexes with itself or other proteins. One possibility is that the observed binding of ATF3 to specific fragments of UBR1 and UBR2 (but not to full-length UBR1/UBR2) in split-Ub and BACTH interaction assays (Figure 5B,C and Figures S6A and S7B) may signify a conditionality of the ATF3 degron vis-à-vis its recognition by the Arg/N-degron pathway.
Our recent analyses of in vivo interactions, by the UBR1 and UBR2 E3 Ub ligases, with TFs other than ATF3 showed that UBR1/UBR2 can specifically bind to a number of different TF proteins, including TFs of structural classes that are distinct from bZIP. We work to determine a set of human TFs that can be recognized and targeted for degradation by UBR1/UBR2. We also work to map and understand a specific degron(s) of ATF3 that mediates the UBR1/UBR2-dependent degradation of ATF3, a process identified in this study.
These results about mammalian bZIP ATF3 and the UBR1/UBR2 E3 Ub ligases (Figures 2–5) complement the earlier discovery that the Arg/N-degron pathway mediates the degradation of S. cerevisiae scCUP9, a homeodomain transcriptional repressor that downregulates, in particular, the expression of the transmembrane peptide transporter scPTR2. The resulting circuit, which involves the conditional (modulated by short peptides) scUBR1-mediated degradation of scCUP9 (a specific TF), controls the import of di/tripeptides in yeast.61,62,121,122 Mammalian cells contain sequelogs (and thus potential functional counterparts) of scCUP9, suggesting that the mammalian Arg/N-degron pathway may also regulate the transport of peptides through a conditional degradation of a cognate transcriptional repressor(s). Together, the present and earlier advances identify the Arg/N-degron pathway as a regulator of gene expression. A significant aspect of this regulation in mammals includes the UBR1/UBR2-mediated degradation of ATF3 TF, and possibly of other TFs, as well.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to J. Sheng, a former member of the Varshavsky laboratory, for contributing to an early stage of this project. The authors also thank the former and current lab members for their advice and assistance throughout this study.
Funding
This work was supported by National Institutes of Health Grants R01DK039520 and R01GM031530 (A.V.).
ABBREVIATIONS
- BACTH
bacterial two-hybrid
- bZIP
basic leucine zipper
- CHX
cycloheximide
- DUB
deubiquitylase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IB
immunoblotting
- ORF
open reading frame
- RT-qPCR
reverse transcription/quantitative polymerase chain reaction
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TM
tamoxifen
- Ub
ubiquitin
- UPS
ubiquitin–proteasome system
- wt
wild-type
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00514.
N-degron pathways (Figure S1), construction of the mmUBR2flox allele (Figure S2), genomic DNA sequences, recombination sites, and PCR probes (Figure S3), RT-qPCR analyses (Figure S4), immunoblotting analyses with the anti-ATF3 antibody (Figure S5), split-Ub interaction assays (Figure S6), BACTH interaction assays (Figure S7), plasmids used in this study (Table S1), oligonucleotide primers used in this study (Table S2), and cited references (PDF)
The authors declare no competing financial interest.
“Sequelog” denotes a sequence that is similar, to a specified extent, to another sequence.75 Derivatives of “sequelog” include “sequelogy” (sequence similarity) and “sequelogous” (similar in sequence). The usefulness of “sequelog” and derivative notations stems from the rigor and clarity of their evolutionary neutrality. By contrast, in settings that use “homolog”, “ortholog”, and “paralog” (they denote common descent and functional similarity and dissimilarity, respectively), these terms are often interpretation-laden and imprecise. Homologue, ortholog, and paralog are compatible with the sequelog terminology. The former terms can be used to convey understanding about common descent and biological functions if this additional information, distinct from sequelogy per se, is actually present.75
REFERENCES
- (1).Hershko A, Ciechanover A, and Varshavsky A (2000) The ubiquitin system. Nat. Med 6, 1073–1081. [DOI] [PubMed] [Google Scholar]
- (2).Varshavsky A (2019) N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. U. S. A 116, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Finley D, Ulrich HD, Sommer T, and Kaiser P (2012) The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vittal V, Stewart MD, Brzovic PS, and Klevit RE (2015) Regulating the regulators: recent revelations in the control of E3 ubiquitin ligases. J. Biol. Chem 290, 21244–21251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pohl C, and Dikic I (2019) Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822. [DOI] [PubMed] [Google Scholar]
- (6).Ji CH, and Kwon YT (2017) Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol. Cells 40, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Balchin D, Hayer-Hartl M, and Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- (8).Sun Z, and Brodsky JL (2019) Protein quality control in the secretory pathway. J. Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zheng N, and Shabek N (2017) Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem 86, 129–157. [DOI] [PubMed] [Google Scholar]
- (10).Watson ER, Brown NG, Peters JM, Stark H, and Schulman BA (2019) Posing the APC/C E3 ubiquitin ligase to orchestrate cell division. Trends Cell Biol 29, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schapira M, Calabrese MF, Bullock AN, and Crews CM (2019) Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (12).Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, and Martin A (2018) Structure and function of the 26S proteasome. Annu. Rev. Biochem 87, 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Forster F, and Baumeister W (2016) Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U. S. A 113, 7816–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Finley D, and Prado MA (2020) The Proteasome and its network: engineering for adaptability. Cold Spring Harbor Perspect. Biol 12, a033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Collins GA, and Goldberg AL (2017) The logic of the 26S proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Budenholzer L, Cheng CL, Li Y, and Hochstrasser M (2017) Proteasome structure and assembly. J. Mol. Biol 429, 3500–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yu H, and Matouschek A (2017) Recognition of client proteins by the proteasome. Annu. Rev. Biophys 46, 149–173. [DOI] [PubMed] [Google Scholar]
- (18).Bachmair A, Finley D, and Varshavsky A (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186. [DOI] [PubMed] [Google Scholar]
- (19).Bachmair A, and Varshavsky A (1989) The degradation signal in a short-lived protein. Cell 56, 1019–1032. [DOI] [PubMed] [Google Scholar]
- (20).Gonda DK, Bachmair A, Wünning I, Tobias JW, Lane WS, and Varshavsky A (1989) Universality and structure of the N-end rule. J. Biol. Chem 264, 16700–16712. [PubMed] [Google Scholar]
- (21).Rao H, Uhlmann F, Nasmyth K, and Varshavsky A (2001) Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 410, 955–960. [DOI] [PubMed] [Google Scholar]
- (22).Hu R-G, Sheng J, Qi X, Xu Z, Takahashi TT, and Varshavsky A (2005) The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 437, 981–986. [DOI] [PubMed] [Google Scholar]
- (23).Hu R-G, Wang H, Xia Z, and Varshavsky A (2008) The N-end rule pathway is a sensor of heme. Proc. Natl. Acad. Sci. U. S. A 105, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Varshavsky A (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci 20, 1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dougan DA, Micevski D, and Truscott KN (2012) The N-end rule pathway: from recognition by N-recognins to destruction by AAA+ proteases. Biochim. Biophys. Acta, Mol. Cell Res 1823, 83–91. [DOI] [PubMed] [Google Scholar]
- (26).Tasaki TS, Sriram SM, Park KS, and Kwon YT (2012) The N-end rule pathway. Annu. Rev. Biochem 81, 261–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ree R, Varland S, and Arnesen T (2018) Spotlight on protein N-terminal acetylation. Exp. Mol. Med 50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gibbs DJ, Bacardit J, Bachmair A, and Holdsworth MJ (2014) The eukaryotic N-end rule pathway: conserved mechanisms and diverse functions. Trends Cell Biol 24, 603–611. [DOI] [PubMed] [Google Scholar]
- (29).Holdsworth MJ, Vicente J, Sharma G, Abbas M, and Zubrycka A (2020) The plant N-degron pathways of ubiquitin-mediated proteolysis. J. Integr. Plant Biol 62, 70–89. [DOI] [PubMed] [Google Scholar]
- (30).Dissmeyer N, Rivas S, and Graciet E (2018) Life and death of proteins after protease cleavage: protein degradation by the N-end rule pathway. New Phytol 218, 929–935. [DOI] [PubMed] [Google Scholar]
- (31).Hwang CS, Shemorry A, and Varshavsky A (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Shemorry A, Hwang CS, and Varshavsky A (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 50, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kim HK, Kim RR, Oh JH, Cho H, Varshavsky A, and Hwang CS (2014) The N-terminal methionine of cellular proteins as a degradation signal. Cell 156, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nguyen KT, Lee CS, Mun SH, Truong NT, Park SK, and Hwang CS (2019) N-terminal acetylation and N-end rule pathway control degradation of the lipid droplet protein PLIN2. J. Biol. Chem 294, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen SJ, Wu X, Wadas B, Oh J-H, and Varshavsky A (2017) An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355, eaal3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dong C, Zhang H, Li L, Tempel W, Loppnau P, and Min J (2018) Molecular basis of GID-mediated recognition of degrons for the Pro/N-end rule pathway. Nat. Chem. Biol 14, 466–473. [DOI] [PubMed] [Google Scholar]
- (37).Dougan DA, and Varshavsky A (2018) Understanding the Pro/N-end rule pathway. Nat. Chem. Biol 14, 415–416. [DOI] [PubMed] [Google Scholar]
- (38).Qiao S, Langlois CR, Chrustowicz J, Sherpa D, Karayel O, Hansen FM, Beier V, von Gronau S, Bollschweiler D, Schafer T, Alpi AF, Mann M, Prabu JR, and Schulman BA (2020) Interconversion between anticipatory and active GID E3 ubiquitin ligase conformations via metabolically driven substrate receptor assembly. Mol. Cell 77, 150–163. [DOI] [PubMed] [Google Scholar]
- (39).Kim JM, Seok OH, Ju S, Heo JE, Yeom J, Kim DS, Yoo JY, Varshavsky A, Lee C, and Hwang CS (2018) Formylmethionine as an N-degron of a eukaryotic N-end rule pathway. Science 362, eaat0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Piatkov KI, Brower CS, and Varshavsky A (2012) The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. U. S. A 109, E1839–E1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brower CS, and Varshavsky A (2009) Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One 4, e7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Brower CS, Piatkov KI, and Varshavsky A (2013) Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol. Cell 50, 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shearer RF, Iconomou M, Watts CK, and Saunders DN (2015) Functional roles of the E3 ubiquitin ligase UBR5 in cancer. Mol. Cancer Res 13, 1523–1532. [DOI] [PubMed] [Google Scholar]
- (44).Oh JH, Hyun JY, and Varshavsky A (2017) Control of Hsp90 chaperone and its clients by N-terminal acetylation and the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 114, E4370–E4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rivera-Rivera I, Román-Hernández G, Sauer RT, and Baker TA (2014) Remodeling of a delivery complex allows ClpS-mediated degradation of N-degron substrates. Proc. Natl. Acad. Sci. U. S. A 111, E3853–E3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yoo YD, Mun SR, Ji CH, Sung KW, Kang KY, Heo AJ, Lee SH, An JY, Hwang J, Xie XQ, Ciechanover A, Kim BY, and Kwon YT (2018) N-terminal arginylation generates a bimodal degron that modulates autophagic proteolysis. Proc. Natl. Acad. Sci. U. S. A 115, E2716–E2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vicente J, Mendiondo GM, Movahedi M, Peirats-Llobet M, Juan YT, Shen YY, Dambire C, Smart K, Rodriguez PL, Charng YY, Gray JE, and Holdsworth MJ (2017) The Cys-Arg/N-end rule pathway is a general sensor of abiotic stress in flowering plants. Curr. Biol 27, 3183–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Masson N, Keeley TP, Giuntoli B, White MD, Puerta ML, Perata P, Hopkinson RJ, Flashman E, Licausi F, and Ratcliffe PJ (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 365, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gao X, Yeom J, and Groisman EA (2019) The expanded specificity and physiological role of a widespread N-degron recognin. Proc. Natl. Acad. Sci. U. S. A 116, 18629–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, and Elledge SJ (2019) A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Park JS, Lee JY, Nguyen YTK, Kang NW, Oh EK, Jang DM, Kim HJ, Kim DD, and Han BW (2020) Structural analyses on the deamidation of N-terminal Asn in the human N-degron pathway. Biomolecules 10, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kim MK, Oh SJ, Lee BG, and Song HK (2016) Structural basis for dual specificity of yeast N-terminal amidase in the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 113, 12438–12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Weaver BP, Weaver YM, Mitani S, and Han M (2017) Coupled caspase and N-end rule ligase activities allow recognition and degradation of pluripotency factor LIN-28 during non-apoptotic development. Dev. Cell 41, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Suzuki T, and Varshavsky A (1999) Degradation signals in the lysine-asparagine sequence space. EMBO J 18, 6017–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tomita T, and Matouschek A (2019) Substrate selection by the proteasome through initiation regions. Protein Sci 28, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Oh JH, Hyun JY, Chen SJ, and Varshavsky A (2020) Five enzymes of the Arg/N-degron pathway form a targeting complex: the concept of superchanneling. Proc. Natl. Acad. Sci. U. S. A 117, 10778–10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Piatkov KI, Oh J-H, Liu Y, and Varshavsky A (2014) Calpain-generated natural protein fragments as short-lived substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 111, E817–E826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Eldeeb MA, and Fahlman RP (2014) The anti-apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget 5, 2714–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kim ST, Lee YJ, Tasaki T, Hwang J, Kang MJ, Yi EC, Kim BY, and Kwon YT (2018) The N-recognin UBR4 of the N-end rule pathway is required for neurogenesis and homeostasis of cell surface proteins. PLoS One 13, e0202260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grofte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen S, Mailand N, Neumann B, Heriche JK, Shearer R, Saunders D, Bartek J, Lukas J, and Lukas C (2012) TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709. [DOI] [PubMed] [Google Scholar]
- (61).Turner GC, Du F, and Varshavsky A (2000) Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 405, 579–583. [DOI] [PubMed] [Google Scholar]
- (62).Du F, Navarro-Garcia F, Xia Z, Tasaki T, and Varshavsky A (2002) Pairs of dipeptides synergistically activate the binding of substrate by ubiquitin ligase through dissociation of its autoinhibitory domain. Proc. Natl. Acad. Sci. U. S. A 99, 14110–14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Graciet E, Hu RG, Piatkov K, Rhee JH, Schwarz EM, and Varshavsky A (2006) Aminoacyl-transferases and the N-end rule pathway in a human pathogen. Proc. Natl. Acad. Sci. U. S. A 103, 3078–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Piatkov KI, Colnaghi L, Bekes M, Varshavsky A, and Huang TT (2012) The auto-generated fragment of the Usp1 deubiquitylase is a physiological substrate of the N-end rule pathway. Mol. Cell 48, 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Gibbs DJ, and Holdsworth MJ (2020) Every breath you take: new insights into plant and animal oxygen sensing. Cell 180, 22–24. [DOI] [PubMed] [Google Scholar]
- (66).Kwon YT, Balogh SA, Davydov IV, Kashina AS, Yoon JK, Xie Y, Gaur A, Hyde L, Denenberg VH, and Varshavsky A (2000) Altered activity, social behavior, and spatial memory in mice lacking the NTAN1 amidase and the asparagine branch of the N-end rule pathway. Mol. Cell. Biol 20, 4135–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, and Bachovchin DA (2019) N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Choi WS, Jeong B-C, Joo YJ, Lee M-R, Kim J, Eck MJ, and Song HK (2010) Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat. Struct. Mol. Biol 17, 1175–1181. [DOI] [PubMed] [Google Scholar]
- (69).Matta-Camacho E, Kozlov G, Li FF, and Gehring K (2010) Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat. Struct. Mol. Biol 17, 1182–1188. [DOI] [PubMed] [Google Scholar]
- (70).Kwon YT, Kashina AS, and Varshavsky A (1999) Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol. Cell. Biol 19, 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Baker RT, and Varshavsky A (1995) Yeast N-terminal amidase: a new enzyme and component of the N-end rule pathway. J. Biol. Chem 270, 12065–12074. [DOI] [PubMed] [Google Scholar]
- (72).Kwon YT, Reiss Y, Fried VA, Hershko A, Yoon JK, Gonda DK, Sangan P, Copeland NG, Jenkins NA, and Varshavsky A (1998) The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 95, 7898–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kwon YT, Xia Z, Davydov IV, Lecker SH, and Varshavsky A (2001) Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 (E3-alpha) of the N-end rule pathway. Mol. Cell. Biol 21, 8007–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kwon YT, Xia ZX, An JY, Tasaki T, Davydov IV, Seo JW, Sheng J, Xie Y, and Varshavsky A (2003) Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol 23, 8255–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Varshavsky A (2004) Spalog and sequelog: neutral terms for spatial and sequence similarity. Curr. Biol 14, R181–R183. [DOI] [PubMed] [Google Scholar]
- (76).Wang H, Piatkov KI, Brower CS, and Varshavsky A (2009) Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol. Cell 34, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Park MS, Bitto E, Kim KR, Bingman CA, Miller MD, Kim HJ, Han BW, and Phillips GN Jr. (2014) Crystal structure of human protein N-terminal glutamine amidohydrolase, an initial component of the N-end rule pathway. PLoS One 9, e111142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, and Kwon YT (2005) RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 102, 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Gibbs DJ, Md Isa N, Movahedi M, Lozano-Juste J, Mendiondo GM, Berckhan S, Marín-de la Rosa N, Vicente Conde J, Sousa Correia C, Pearce SP, Bassel GW, Hamali B, Talloji P, Tomé DF, Coego A, Beynon J, Alabadí D, Bachmair A, León J, Gray JE, Theodoulou FL, and Holdsworth MJ (2014) Nitric oxide sensing in plants is mediated by proteolytic control of group VII ERF transcription factors. Mol. Cell 53, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, Beemer FA, Hamel B, Vanlieferinghen P, Gershoni-Baruch R, Vieira MW, Dumic M, Auslender R, Gil-da-Silva-Lopes VL, Steinlicht S, Rauh R, Shalev SA, Thiel C, Winterpacht A, Kwon YT, Varshavsky A, and Reis A (2005) Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat. Genet 37, 1345–1350. [DOI] [PubMed] [Google Scholar]
- (81).Hwang CS, Sukalo M, Batygin O, Addor MC, Brunner H, Aytes AP, Mayerle J, Song HK, Varshavsky A, and Zenker M (2011) Ubiquitin ligases of the N-end rule pathway: assessment of mutations in UBR1 that cause the Johanson-Blizzard syndrome. PLoS One 6, e24925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Sukalo M, Fiedler A, Guzman C, Spranger S, Addor MC, McHeik JN, Oltra Benavent M, Cobben JM, Gillis LA, Shealy AG, Deshpande C, Bozorgmehr B, Everman DB, Stattin EL, Liebelt J, Keller KM, Bertola DR, van Karnebeek CD, Bergmann C, Liu Z, Duker G, Rezaei N, Alkuraya FS, Ogur G, Alrajoudi A, Venegas-Vega CA, Verbeek NE, Richmond EJ, Kirbiyik O, Ranganath P, Singh A, Godbole K, Ali FA, Alves C, Mayerle J, Lerch MM, Witt H, and Zenker M (2014) Mutations in the human UBR1 gene and the associated phenotypic spectrum. Hum. Mutat 35, 521–531. [DOI] [PubMed] [Google Scholar]
- (83).An JY, Seo JW, Tasaki T, Lee MJ, Varshavsky A, and Kwon YT (2006) Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 103, 6212–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Rajewsky K, Gu H, Kühn R, Betz UA, Müller W, Roes J, and Schwenk F (1996) Conditional gene targeting. J. Clin. Invest 98, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Zheng Q, Cai X, Tan MH, Schaffert S, Arnold CP, Gong X, Chen CZ, and Huang S (2014) Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. BioTechniques 57, 115–124. [DOI] [PubMed] [Google Scholar]
- (87).Knott GJ, and Doudna JA (2018) CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Hai T, Wolfgang CD, Marsee DK, Allen AE, and Sivaprasad U (1999) ATF3 and stress responses. Gene Expression 7, 321–335. [PMC free article] [PubMed] [Google Scholar]
- (89).Hai T, Wolford CC, and Chang YS (2010) ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expression 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Wolfgang CD, Liang G, Okamoto Y, Allen AE, and Hai T (2000) Transcriptional autorepression of the stress-inducible gene ATF3. J. Biol. Chem 275, 16865–16870. [DOI] [PubMed] [Google Scholar]
- (91).Chang YS, Jalgaonkar SP, Middleton JD, and Hai T (2017) Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. U. S. A 114, e7159–e7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Fazio EN, Young CC, Toma J, Levy M, Berger KR, Johnson CL, Mehmood R, Swan P, Chu A, Cregan SP, Dilworth FJ, Howlett CJ, and Pin CL (2017) Activating transcription factor 3 promotes loss of the acinar cell phenotype in response to cerulein-induced pancreatitis in mice. Mol. Biol. Cell 28, 2347–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Labzin LI, Schmidt SV, Masters SL, Beyer M, Krebs W, Klee K, Stahl R, Lutjohann D, Schultze JL, Latz E, and De Nardo D (2015) ATF3 is a key regulator of macrophage IFN responses. J. Immunol 195, 4446–4455. [DOI] [PubMed] [Google Scholar]
- (94).Mo P, Wang H, Lu H, Boyd DD, and Yan C (2010) MDM2 mediates ubiquitination and degradation of activating transcription factor 3. J. Biol. Chem 285, 26908–26915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Lu D, Wolfgang CD, and Hai T (2006) Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J. Biol. Chem 281, 10473–10481. [DOI] [PubMed] [Google Scholar]
- (96).Wang Z, and Yan C (2016) Emerging roles of ATF3 in the suppression of prostate cancer. Mol. Cell. Oncol 3, e1010948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018) The human transcription factors. Cell 172, 650–665. [DOI] [PubMed] [Google Scholar]
- (98).Johnsson N, and Varshavsky A (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U. S. A 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Dunkler A, Muller J, and Johnsson N (2012) Detecting protein-protein interactions with the split-ubiquitin sensor. Methods Mol. Biol 786, 115–130. [DOI] [PubMed] [Google Scholar]
- (100).Ouellette SP, Karimova G, Davi M, and Ladant D (2017) Analysis of membrane protein interactions with a bacterial adenylate cyclase-based two-hybrid (BACTH) technique. Curr. Protoc. Mol. Biol 118, 20.12.21–20.12.24. [DOI] [PubMed] [Google Scholar]
- (101).Mortensen R (2006) Overview of gene targeting by homologous recombination Current Protocols in Molecular Biology, Chapter 23, Unit 23.21, Wiley-Interscience, New York. [DOI] [PubMed] [Google Scholar]
- (102).Bouabe H, and Okkenhaug K (2013) Gene Targeting in Mice. Methods Mol. Biol 1064, 315–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Hayashi S, and McMahon A (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol 244, 305–318. [DOI] [PubMed] [Google Scholar]
- (104).Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, and Struhl K (2017) Current Protocols in Molecular Biology, Wiley-Interscience, New York. [Google Scholar]
- (105).Stepanenko AA, and Dmitrenko VV (2015) HEK293 in cell biology and cancer research: phenotype, karyotype, tumor-igenicity, and stress-induced genome-phenotype evolution. Gene 569, 182–190. [DOI] [PubMed] [Google Scholar]
- (106).Wadas B, Borjigin J, Huang Z, Oh J-H, Hwang C-S, and Varshavsky A (2016) Degradation of serotonin N-acetyltransferase, a circadian regulator, by the N-end rule pathway. J. Biol. Chem 291, 17178–17196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Green MR, and Sambrook J (2012) Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- (109).Pfaffl MW (2001) A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Livak KJ, and Schmittgen TD (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- (111).Andrews B, Boone C, Davis TN, and Fields S (2016) Budding Yeast (a laboratory manual), Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- (112).Gisler SM, Kittanakom S, Fuster D, Wong V, Bertic M, Radanovic T, Hall RA, Murer H, Biber J, Markovich D, Moe OW, and Stagljar I (2008) Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol. Cell. Proteomics 7, 1362–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Kwon YT, Kashina AS, Davydov IV, Hu R-G, An JY, Seo JW, Du F, and Varshavsky A (2002) An essential role of N-terminal arginylation in cardiovascular development. Science 297, 96–99. [DOI] [PubMed] [Google Scholar]
- (114).Ouyang Y, Kwon YT, An JY, Eller D, Tsai S-C, Diaz-Perez S, Troke J, Teitell MA, and Marahrens Y (2006) Loss of Ubr2, an E3 ubiquitin ligase, leads to chromosome fragility and impaired homologous recombinational repair. Mutat. Res., Fundam. Mol. Mech. Mutagen 596, 64–75. [DOI] [PubMed] [Google Scholar]
- (115).de Marchi R, Sorel M, Mooney B, Fudal I, Goslin K, Kwasniewska K, Ryan PT, Pfalz M, Kroymann J, Pollmann S, Feechan A, Wellmer F, Rivas S, and Graciet E (2016) The N-end rule pathway regulates pathogen responses in plants. Sci. Rep 6, 26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Wadas B, Piatkov KI, Brower CS, and Varshavsky A (2016) Analyzing N-terminal arginylation through the use of peptide arrays and degradation assays. J. Biol. Chem 291, 20976–20992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Varshavsky A (2005) Ubiquitin fusion technique and related methods. Methods Enzymol 399, 777–799. [DOI] [PubMed] [Google Scholar]
- (118).Chao CCK (2015) Mechanisms of p53 degradation. Clin. Chim. Acta 438, 139–147. [DOI] [PubMed] [Google Scholar]
- (119).Vidal M, and Fields S (2014) The yeast two-hybrid assay: still finding connections after 25 years. Nat. Methods 11, 1203–1206. [DOI] [PubMed] [Google Scholar]
- (120).Karimova G, Pidoux J, Ullmann A, and Ladant D (1998) A bacterial two-hybrid system based on reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A 95, 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Byrd C, Turner GC, and Varshavsky A (1998) The N-end rule pathway controls the import of peptides through degradation of a transcriptional repressor. EMBO J 17, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Kitamura K, and Fujiwara H (2013) The type-2 N-end rule peptide recognition activity of Ubr11 ubiquitin ligase is required for the expression of peptide transporters. FEBS Lett 587, 214–219. [DOI] [PubMed] [Google Scholar]
- (123).Mena EL, Kjolby RAS, Saxton RA, Werner A, Lew BG, Boyle JM, Harland R, and Rape M (2018) Dimerization quality control ensures neuronal development and survival. Science 362, eaap8236. [DOI] [PubMed] [Google Scholar]
- (124).Herhaus L, and Dikic I (2018) Dimerization quality control via ubiquitylation. Science 362, 151–152. [DOI] [PubMed] [Google Scholar]
- (125).Rubenstein EM, and Hochstrasser M (2010) Redundancy and variation in the ubiquitin-mediated proteolytic targeting of a transcription factor. Cell Cycle 9, 4282–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Hickey CM, Xie Y, and Hochstrasser M (2018) DNA binding by the MATα2 transcription factor controls its access to alternative ubiquitin-modification pathways. Mol. Biol. Cell 29, 542–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.