


Abstract
DNA methyltransferases interact with their CpG target sites in the context of variable flanking sequences. We investigated DNA methylation by the human DNMT3B catalytic domain using substrate pools containing CpX target sites in randomized flanking context and identified combined effects of CpG recognition and flanking sequence interaction together with complex contact networks involved in balancing the interaction with different flanking sites. DNA methylation rates were more affected by flanking sequences at non-CpG than at CpG sites. We show that T775 has an essential dynamic role in the catalytic mechanism of DNMT3B. Moreover, we identify six amino acid residues in the DNA-binding interface of DNMT3B (N652, N656, N658, K777, N779, and R823), which are involved in the equalization of methylation rates of CpG sites in favored and disfavored sequence contexts by forming compensatory interactions to the flanking residues including a CpG specific contact to an A at the +1 flanking site. Non-CpG flanking preferences of DNMT3B are highly correlated with non-CpG methylation patterns in human cells. Comparison of the flanking sequence preferences of human and mouse DNMT3B revealed subtle differences suggesting a co-evolution of flanking sequence preferences and cellular DNMT targets.
INTRODUCTION
DNA methylation is an essential epigenetic mechanism that has important roles in the regulation of gene expression, genomic stability, cell differentiation and mammalian development (1–3). Mammalian DNA methylation predominantly occurs at the C-5 position of cytosine residues within CpG dinucleotides, affecting about 70–80% of all CpG sites in the genome (4). The DNMT3A and DNMT3B paralogs (5,6) are de novo DNA methyltransferases which generate DNA methylation during gametogenesis (mainly DNMT3A) and post-implantation development (mainly DNMT3B) (3,7), but they also contribute to the preservation of DNA methylation at repetitive elements (8,9). In this process DNMT3B is particularly relevant, as it has been shown to be essential for the methylation of Satellite II repeats in human cells (10–12).
The structure of the heterotetrameric complex comprising the catalytic domain of DNMT3B and the C-terminal domain of DNMT3L has been solved with four different DNA substrates (Supplementary Table S1) (13,14). The interaction between DNMT3B and the DNA involves three regions, the so-called TRD loop (residues 772–791, colored red in Figure 1A), catalytic loop (residues 648–672, colored green in Figure 1A), and the RD homodimeric interface region (residues 822–828, colored yellow in Figure 1A). The catalytic loop and TRD loop approach the DNA at the CpG target region from the minor and major groove side, respectively, generating a clamp holding the DNA. The recognition of the CpG guanine residue is based on a hydrogen bond of its O6 atom to the side chain of N779 and a water mediated H-bond of its N7 atom to the side chain of T775. However, the CpG specificity of DNMT3A and DNMT3B is not absolute, and both enzymes also introduce methylation at lower level at non-CpG sites in vitro (15–17). DNMT3B catalyzed non-CpG methylation was found in ES and neural cells, mainly at CpA sites followed by a G (CAG context) (4,18,19). Non-CpG methylation has been connected with gene regulatory and chromatin modulating functions and to X-chromosome inactivation mainly in the nervous system so far (20,21).
Figure 1.
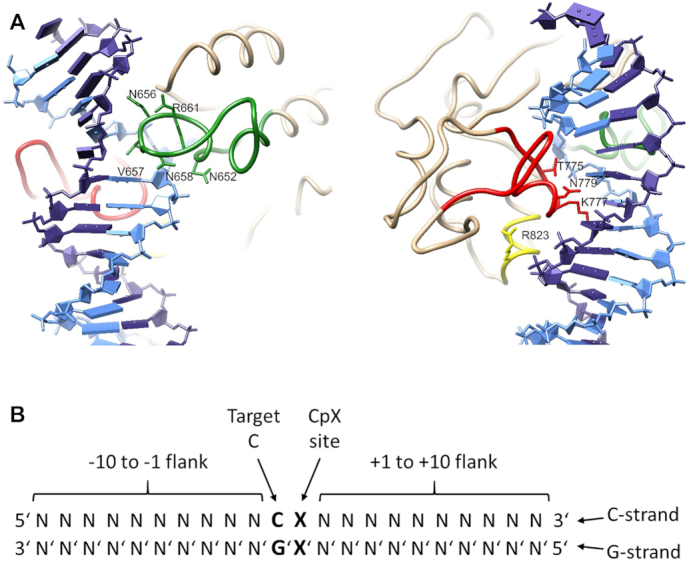
Design of the enzyme mutants and substrates used in this study. (A) Structure of the CGA complex of DNMT3B (6U8P) in two views focusing on the catalytic loop (green), TRD loop (red) and RD interface loop (yellow). The target strand for DNA methylation (C-strand) is shown in light blue, the non-target strand (G-strand) in dark blue. The amino acid residues mutated in this study are indicated. (B) The terminology used in this study to describe the DNA substrate.
Previous work has shown that DNMT3B does not methylate all CpG sites equally. Instead, its catalytic activity is modulated by the adjacent base pairs, called flanking sequence here (17,22). These flanking sequence preferences differ between DNMT3B and DNMT3A (14,23) and they determine the biological activities of DNMT3A and DNMT3B in the methylation of repetitive elements, for example the essential role of DNMT3B for the methylation of SatII repeats (14). Moreover, we showed that genomic CpG methylation observed after reintroduction of DNMT3A or DNMT3B into TKO hESC correlates with the in vitro flanking sequence preferences of both enzymes (14). However, from a biological perspective, extensive flanking sequence preferences of DNMTs are not beneficial, because they would reduce the capacity of the system to introduce arbitrary methylation states at any CpG site and thereby limit the regulatory power of DNA methylation. Therefore, DNMTs need to balance the methylation rates of CpG sites in different sequence context but the molecular basis of such flanking context equalization processes in DNMTs is not known. For example in our previous work, we showed that CpG sites embedded into the SatII flanking sequence (ATTCGATG) are among the preferred substrates of DNMT3B but the detailed effects of individual residues on the recognition are not known. Finally, the question appeared if flanking sequence preferences are subject to an evolutionary optimization.
To gain better understanding of these important questions, we performed a detailed substrate preference analysis of humanDNMT3B and selected DNMT3B mutants in the context of the isolated catalytic domain using a deep enzymology method and determined CpG and non-CpG methylation in libraries of substrates containing CpX target sites embedded into a context of 10 randomized base pairs on either side. In order to study the role of individual amino acid residues in the DNA interaction process, we selected nine DNA-interacting residues of DNMT3B for mutagenesis and studied their detailed effects on the methylation of CpG and non-CpG sites in different flanking contexts. Five of the selected residues are located in the catalytic loop (N652, N656, V657, N658 and R661) (Figure 1A). N652 forms a backbone phosphate contact to the +1 flank base pair (see Figure 1B for the terminology that is used). N656 contacts the -1 base and DNA backbone at the -1 and -2 flank. V657 partially occupies the space left by the rotated target cytosine and it contacts the G’ together with the -1 flank and the CpX site. N658 contacts the CpX site G/A residue and the DNA backbone at the CpX site and +1 flank. R661 forms a side-chain hydrogen bond with N656, a structural feature distinct from that of DNMT3A (14). Three of the mutated amino acid residues are located in the TRD loop (T775, K777 and N779). T775 approaches the sugar residue of the target cytosine and it forms a water mediated H-bond to the N7 atom of the CpX site G/A residue. K777 forms complex specific interactions at the CpX site, the +1 site (where it hydrogen bonds to the N7 of the G), and contacts to +2 and +3 flank sites. N779 forms complex specific interactions to the CpG site (including a hydrogen bond to the O6 atom of G) and +1 flank. In the CAG complex, it is rotated by 120 degrees and these contacts are not formed. R823 is in the RD interface and it forms a phosphate contact at the +3 flank site. This residue corresponds to R882 in DNMT3A, which is a hotspot of DNMT3A mutations in AML (24) and its mutation has been shown to lead to strong changes of flanking sequence preferences of DNMT3A (23,25) and structural adjustments of this region (26). Moreover, the residues investigated here are connected by two hydrogen bonding networks with each other, one established between catalytic loop residues N652, N656, N658 and R661 and the other between the TRD loop residues T775, K777 and N779. Therefore, conformational changes of one residue may be transmitted to others potentially leading to cooperative effects. A detailed list of the DNA contacts formed by these residues in the different structures is provided in Supplementary Tables S2 and S3 and structural views are shown in Supplementary Figure S1. All residues were mutated to alanine, N656 and N779 to alanine and aspartate, and T775 to alanine, asparagine and glutamine.
Our data illustrate several examples of the combined readout of the CpX site and the base pairs in the flanking regions. The mutational analysis allowed us to connect some of the complex sequence-readout effects with specific protein-DNA contacts mediated by the mutated amino acid residues. Our study discovered several important new findings including (i) the novel strategy of DNMT3B to minimize flanking sequence effects by the establishment of alternative interaction networks with different flanking contexts, (ii) the finding that the SatII sequence is recognized by the combined effects of several residues, (iii) a strong correlation of in vitro non-CpG methylation preferences of DNMT3B with its cellular activity and genomic non-CpG methlyation patterns and (iv) the observation that human and mouse DNMT3B have optimized flanking sequence preferences for human and mouse repeats, suggesting that flanking sequence preferences are under evolutionary pressure.
MATERIALS AND METHODS
Cloning and mutagenesis
The catalytic, C-terminal domain of human DNMT3B (DNMT3B-C) (amino acids 553–853 of Q9UBC3) was cloned as His-tagged fusion protein into a pET28 expression vector. Mutagenesis was performed by inverse PCR amplifying the entire pET28(+) vector with Q5® High-Fidelity DNA Polymerase (NEB) using primers with up to three mismatches at the 5′ end to introduce the desired mutation. The PCR products were loaded on a 0.5% agarose TPE gel and purified using NucleoSpin Gel and PCR Clean-up Mini kit (MACHEREY-NAGEL). The purified linearized vectors were then phosphorylated using T4 Polynucleotide Kinase (NEB) followed by circularization of the plasmids using T4 DNA Ligase (NEB). The ligated plasmids were then transformed into One Shot® Stbl3™ Chemically Competent Escherichia coli (ThermoFischer Scientific). After transformation, the cells were recovered in 1.5 ml centrifugation tubes in SOC medium at 37°C with horizontal shaking at 200 rpm for 1 h. Cell suspensions were then plated on 2% LB-agar containing kanamycin and incubated overnight at 37°C. Single colonies were used for inoculating liquid LB cultures containing kanamycin, which were grown overnight at 37°C with shaking at 200 rpm. The plasmid DNA was then isolated using the NucleoSpin Plasmid Mini kit for plasmid DNA (MACHEREY NAGEL) and sequences were confirmed by Sanger DNA sequencing.
Protein expression and purification
The His-tagged DNMT3B-C and the N652A, N656A, N656D, V657A, N658A, R661A, T775A, T775N, T775Q, K777A, N779A, N779D and R823A mutants were overexpressed in BL21 (DE3) Codon+ RIL E. coli cells (Stratagene) and purified as described for DNMT3A-C (27). The purity of the preparations was estimated to be >95% from Coomassie stained SDS gels. The concentrations of the proteins were determined by UV spectrophotometry and confirmed by densitometric analysis of Coomassie stained SDS–polyacrylamide gels.
Radioactive DNA methylation kinetics
Methylation activities of DNMT3B and DNMT3B mutants were determined using an avidin-biotin methylation plate assay (28). For this, a biotinylated double-stranded 30-mer oligonucleotide with a single CpG site was used (GAA GCT GGG ACT TCCGGG AGG AGA GTG CAA). Methylation with DNMT3B-C was conducted at 37°C with 2 μM enzyme in 1× methylation buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 50 mM KCl, 0.25 mg/ml bovine serum albumin) in the presence of 1 μM of the biotinylated substrate, the reactions were started by adding 0.76 μM radioactively labeled AdoMet (Perkin Elmer). The initial slope of the enzymatic reaction was determined by linear regression.
Flanking sequence preference analysis with a randomized substrate and bioinformatics analysis
The preparation of the substrate with CpX site in a randomized sequence context, methylation, bisulfite conversion and library preparation was conducted as described (14,29). Two sets of barcodes were introduced in the PCR steps to distinguish different samples and experiments. NGS data sets were bioinformatically analyzed using a local instance of the Galaxy server (30). Sequence reads were trimmed with Trim Galore! (the tool was developed by Felix Krueger at the Babraham Institute), paired using Pear (31), and filtered according to the expected DNA size using the Filter FASTQ tool (32). Duplicates were removed. They were observed at very low levels showing the absence of clonal PCR. The original DNA sequence was then reconstituted based on the bisulfite converted upper and lower strands to investigate the average methylation state of both CpX sites. As preparations of mutants with very low activity showed some detectable methylation at CCWGG sequences, which could be attributed to the E. coli dcm methyltransferase, all CCWGG sequences were removed from the analysis. Average methylation levels for each base at different flanking positions were determined as well as average methylation levels of all NNCGNN flanks. Pearson correlation factors were calculated with Excel using the correl function. P-values were determined using the distribution of r-values from >200 correlation analyses with one data set shuffled. Box plots were generated usnig MS Excel Professional Plus 2016.
For a global analysis, methylation data for the different CpX sites were separated and average methylation levels of all substrates containing a particular base at one of the flanking positions were determined. For a more detailed analysis, data were separated into 256 bins for the 256 NNCXNN flanking sites and average methylation levels of each NNCXNN site was determined. In both analyses, results of independent experimental repeats correlated closely (Supplementary Figure S3 and Supplementary Figure S4A) and both repeats were merged for downstream analysis. The sequencing depth of the combined data sets exceeds 55000 reads in all cases corresponding to an average coverage of >350× for CpG and >230× for CpA, CpT and CpC. Among all data sets, 98.7% showed a coverage >100× and the smallest number of sequence reads in one of the analysis bins was 36. These numbers well exceed established standards of whole genome bisulfite analysis, for which the NIH Roadmap Epigenomics Project currently recommends the use of two replicates with a combined total coverage of 30× (http://www.roadmapepigenomics.org/protocols, retrieved in June 2020). Moreover, the sequencing depth was not correlated with the average methylation levels of bins (Supplementary Figure S4B).
Analysis of genomic methylation data
DNA methylation in human H1 ES cells was investigated using whole genome bisulfite data from Encode (data set: GSM2138820, CHG and CHH data were merged). Data from DNMT3B KO H1 hESC were from GSM3122510 (33). CpG sites were filtered for coverage >9 and only methylation data of the upper DNA strand were used. DNA sequences surrounding the CpG sites were retrieved using BEDTools GetFastaBed (34) using a local instance of the Galaxy server (30). Average methylation levels in all NNCGNN flanks were determined with a home written program. The 11 CpG sites in mouse minor satellite repeats were derived from Genebank entry Z22168.1 and the 67 CpG sites of the human SatII sequence were taken from X72623.1.
RESULTS
It was the aim of this study to investigate details of the DNA recognition and interaction mechanisms of the catalytic domain of the human DNA methyltransferase DNMT3B. We specifically addressed the combined interaction and readout of the CpG target region together with flanking sequence base pairs. To this end, CpG and non-CpG methylation was tested in libraries of substrates containing CpX target sites embedded into a context of 10 randomized base pairs on either side and the methylation levels were determined by bisulfite conversion coupled to ultra-deep next generation sequencing (NGS) readout. In order to study the role of individual amino acid residues in the DNA interaction process, 13 DNMT3B mutants with amino acid exchanges of DNA-interacting residues were generated (N652A, N656A, N656D, V657A, N658A, R661A, T775A, T775N, T775Q, K777A, N779A, N779D, R823A) and the mutant proteins were purified in the context of the catalytic domain of DNMT3B (Supplementary Figure S2). Afterwards, the effects of the mutations on the methylation of CpG and non-CpG sites in different flanking contexts were studied in detail.
CpG methylation activity of wildtype DNMT3B and DNMT3B mutants
The catalytic activities of the wildtype human DNMT3B catalytic domain (WT) enzyme and the 13 selected mutants were determined with a radioactive DNA methylation assay using a 30mer oligonucleotide substrate containing a single CpG site in a TTCCGGGA sequence context (Figure 2A). In addition, we conducted deep enzymology experiments and investigated methylation of a pool of DNA substrates, in which the target CpX site was flanked by 10 random nucleotides on either side. The substrate pool was methylated by WT DNMT3B and DNMT3B mutants, the reaction products were subjected to hairpin ligation, bisulfite conversion, PCR amplification and NGS analysis as described previously (14,23,29). As the T775 mutants showed very weak activity in the radioactive kinetics (<0.5% of WT DNMT3B), only T775A was used for the deep enzymology experiments. Data were generated in two independent repeats and sequenced at great depth (Supplementary Table S4).
Figure 2.

CpG and non-CpG catalytic activities of WT DNMT3B and DNMT3B mutants. (A) Relative CpG methylation activities determined by radioactive kinetics. Numbers represent averages of three experiments, error bars show the SD. (B) Relative CpG methylation activities determined in the deep enzymology experiments. Numbers represent averages of two experiments, error bars show the SD. (C) Relative non-CpG methylation activities determined in the deep enzymology experiments. Numbers represent averages of two experiments, error bars show the SD.
In the initial analysis, methylation levels of CpG sequences were analyzed. As shown in Figure 2A and B, the results of both activity assays agree closely with each other. When considering both assays, the activities of N652A, N656A, N779A and N779D were similar to WT (<25% reduction when compared to WT). V657A, R661A, K777A and R823A showed a moderate reduction in activity (residual activities between 25 and 75% of WT). N656D and N658A showed a strong reduction of activity with residual activities between 5 and 15% of WT. The activity of the T775 mutants was even weaker (around 0.5% of WT activity). Next, the global non-CpG methylation activity of WT DNMT3B and the mutants was determined (Figure 2C). The CpA methylation activity of the WT enzyme was 17% of the activity observed at CpG sites, CpT activity was 7% and CpC 6%. The relative overall non-CpG activities of most mutants were similar to WT, with a few interesting exceptions: K777A showed an increase in non-CpG methylation in all three sequence contexts. Similarly, T775A showed an increase in non-CpG methylation, but error margins between the repeats were larger due to the very low overall methylation levels. N658A showed increased relative activity at CpA sites, and V657A showed an increase in the relative activity at CpT sites. Moreover, N656D and R823A showed globally reduced non-CpG methylation.
Flanking sequence preferences of CpG and non-CpG methylation by WT DNMT3B
To investigate if and to which extend flanking sequence interactions modulate the CpG recognition, two approaches for data analysis of the deep enzymology data were applied. First in a global analysis, for the different types of CpX methylation by human WT DNMT3B, the average methylation levels of all substrates containing a particular base at one of the -8 to +8 flank sites were determined to identify bases favorable or unfavorable for activity. The data were expressed in observed/expected values of the methylation levels. We first compared the results of both independent experimental repeats showing low error levels (Supplementary Figure S3A and S3B) and high correlation of the derived profiles (Supplementary Figure S3C). Therefore, the two data sets were merged and the magnitude of the position specific enrichments and depletions was calculated showing that the –2 to +3 flanks were most important for catalytic activity (Supplementary Figure S3D). In the merged data set, the position specific enrichment of bases was calculated for the different types of CpX methylation for the -4 to +4 flanking region (Figure 3A). For WT CpG methylation, the profile was very similar to previous results obtained with murine DNMT3B showing strongest preferences for T at the –2 site, and G/A at +1 (14). Even weaker trends of the previous data set were confirmed like a preference for A and disfavor for G at –1 and favor for C and disfavor for A at +2. In this global analysis, all four types of CpX methylation showed similar trends in flanking sequence preferences indicating that at a global level, a flanking sequence favorable for CpG methylation is also favorable for non-CpG methylation (Figure 3B). Correlation analysis showed that the flanking sequence preferences of CpA and CpT methylation were most similar at the overall level, followed by CpG and CpA. CpC methylation flanking sequence preferences were most distinct from the other profiles (Figure 3C).
Figure 3.
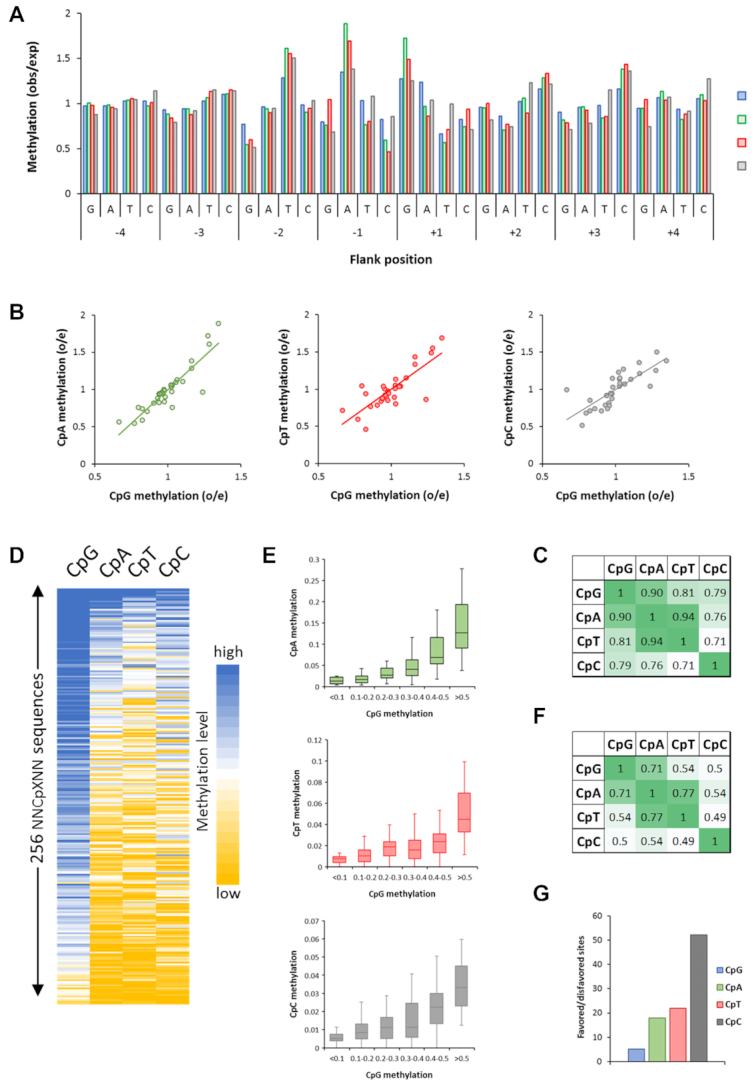
Global analysis of flanking sequence effects on CpG and non-CpG methylation by wildtype DNMT3B showing overall correlations of CpX methylation. (A) Average CpX methylation levels of all substrates containing specific bases at –4 to +4 flank positions. Methylation levels are given as observed/expected (obs/exp) values. (B) Correlation of non-CpG and CpG –4 to +4 flanking sequence preferences. (C) Pearson correlation factors of the different CpX methylation flanking sequence profiles. (D) Global correlation of CpX methylation in 256 NNCXNN sequences. (E) Boxplot showing the correlation of CpG and non-CpG methylation levels of NNCXNN sequences. The boxes display the first and third quartiles with medians indicated by vertical lines. Whiskers display the data range. (F) Pearson correlation factors of the flanking preferences of all types of CpG and non-CpG methylation. (G) Ratio of the average methylation rates of the 15% most preferred and most disfavored flanking sites for CpG and CpN methylation.
Next, a more specific analysis was performed. For this, the sequencing reads for each CpX specificity were separated and average methylation levels of all 256 NNCXNN hexanucleotides were determined. Due to the separate treatment of each NNCXNN sequence, this analysis is optimized to detect the combined effect of more than one flanking position on the methylation activity. The methylation levels of both repeats showed high correlation (Supplementary Figure S4A) and therefore corresponding data sets were merged. A correlation analyses of the CpG, CpA, CpT and CpC methylation levels in the different NNCXNN flanking sequence contexts (Figure 3D–F) revealed a good correlation and similar trends as observed in the –4 to +4 flanking sequence enrichment analysis (Figure 3A–C). However, comparison of the average methylation rates of the 15% most favored and 15% most disfavored sites clearly indicates that the overall flanking sequence preferences were enhanced for non-CpG methylation (Figure 3G).
Detailed analysis of non-CpG flanking sequence preferences
A more detailed comparison of the CpG profiles with the profiles observed for CpA, CpT and CpC methylation revealed few but striking differences in flanking sequence effects in the –4 to +4 flank base preferences (Figure 4A). For CpA methylation, many trends observed for CpG methylation were enhanced, T was more favored and G more disfavored at the –2 site, A was more preferred at the –1 site where G was more disfavored. In contrast, G was more favored at +1, +2 and +3. Strikingly, the preference for A at +1 observed with CpG methylation was specific for the CpG context and lost with CpA and CpT. For CpT methylation, the profile shows high similarity with CpA. For CpC, the pattern looks different with T favored at many places, most prominently at –2 and at +2, but it is also tolerated at +1 where T is highly disfavored in CpG methylation. It is also noteworthy that G is highly disfavored at –2 in all types of non-CpG methylation.
Figure 4.

Detailed differences of WT DNMT3B flanking sequence preferences in CpG and non-CpG methylation and comparison with DNMT3B dependent genomic non-CpG methylation patterns. (A) Global methylation analysis showing average methylation levels of all substrates which carry a specific base at each –4 to +4 flank site. Methylation levels are given as observed/expected (obs/exp) values. (B) Analysis based on correlation of the hexanucleotide sequences. The 10 NNCXNN flanks with highest CpX methylation were used to illustrate the enrichment of bases with a Weblogo. (C) Enrichment and depletion of bases at –4 to +4 flank positions for CpX methylation in the genomic DNMT3B dependent methylation (Genome) and the DNMT3B in vitro preferences (DNMT3B). Data are shown as observed/expected values (obs/exp). (D) Correlation of genomic (Genome) and enzymatic (DNMT3B) observed/expected methylation profiles. The P-values for the correlations of CpA, CpT and CpC methylation are 2.8 × 10-7, 7.6 × 10-6 and 9.2 × 10-5, respectively.
To explore the flanking sequence effects in CpG and non-CpG methylation in more details, we inspected the average methylation levels in NNCXNN sequence contexts. This analysis illustrated the strong effects of the –2 to +2 flanks mainly on the non-CpG methylation activity of DNMT3B, because the ratio of the average methylation rates of the 15% best and worst flanking sites was 5.1 for CpG, but 18, 21 and 52 for CpA, CpT and CpC, respectively (Figure 3G and Supplementary Figure S5A). In case of CpT and CpC, many of the worst sites showed no detectable methylation. Next, the flanks with highest CpX methylation levels were used to calculate Weblogos (Figure 4B). Overall, this analysis reproduced most of the conclusions drawn from the global analysis shown in Figure 4A, including the preference for T, A, G/A and C at the –2, –1 and +2 sites in CpG methylation. It is interesting to note that the human SatII sequence context (TTCGAT) although it does not reflect the most preferred residues for CpG methylation at the –1 and +2 positions has rank of 19 among all 256 NNCGNN sites (where a low rank indicates high activity). This result illustrates that the combined interaction with all positions leads to a high preference for this sequence. For non-CpG methylation, key findings of the global analysis were reproduced as well, including the strong preference for A(–1) in CpA methylation and reduction of A preference at +1 and the strong preference for T(–2) in CpC methylation. We observed another striking result for CpA methylation, because the four sequences with highest CpA methylation all were TACAGN sequences. As shown in Supplementary Figure S5B, the preference of DNMT3B for the TACXG sequence was much higher in CpA and CpT methylation than in CpG methylation providing an impressive example of the coupling of flanking sequence contacts with CpG recognition and the stronger effects of flanking sequences on non-CpG methylation.
Comparison of genomic non-CpG methylation patterns with in vitro preferences
To determine the in vivo flanking preferences of DNMT3B, public whole genome bisulfite data of CpG and non-CpG methylation from human ES cells (hESC) were compared with data from the same cell line after KO of DNMT3B (33). The difference of both data sets was used as indication of the activity of DNMT3B in the native cells. As already reported (33), the data show a major contribution of DNMT3B to CpA and CpT methylation in this cell line (Table 1). The contribution of DNMT3B to CpG methylation was minor (Table 1), which is in agreement with the general observation that CpG methylation is most strongly determined by DNMT1 (29). CpC methylation levels were lowest and close to background and the measurable contribution of DNMT3B was moderate. Next, the flanking sequence dependent methylation levels of the DNMT3B associated genomic methylation were determined and compared with the in vitro flanking sequence preferences of DNMT3B (Figure 4C and D). For CpG methylation, no significant correlation was observed in agreement with the notion that DNMT3B does not strongly contribute to CpG methylation in this cell line. However, the flanking profiles of genomic CpA and CpT methylation were very strongly and highly significantly correlated with the activity profiles of DNMT3B with Pearson correlation r-values of 0.96 and 0.9 and P-values below 1 × 10-5 (Figure 4C and D, Table 1). For CpC methylation, a weaker but still highly significant correlation was observed. These findings indicate that the cellular non-CpG methylation activity of DNMT3B is strongly influenced by its flanking sequence preferences, in particular at CpA and CpT sites.
Table 1.
Methylation levels in hESC and DNMT3B KO hESC
| Methylation level (%) | Correlation genomic vs. enzymatic obs/exp methylation profiles | |||||
|---|---|---|---|---|---|---|
| WT -H1 hESC | DNMT3B KO H1 cells | WT-KO | DNMT3B contribution to WT methylation (%) | r-value | P-value | |
| CpG | 84.34 | 79.16 | 5.18 | 6.1 | 0.05 | n.s. |
| CpA | 2.47 | 0.85 | 1.62 | 65.5 | 0.96 | 2.8E-07 |
| CpT | 0.81 | 0.46 | 0.35 | 43.5 | 0.90 | 7.6E-06 |
| CpC | 0.57 | 0.50 | 0.07 | 12.1 | 0.68 | 9.2E-05 |
Detailed flanking sequence preferences of CpG and non-CpG methylation by DNMT3B mutants
Next, detailed deep enzymology based flanking sequence preferences of all DNMT3B mutants (except T775A) were determined. In case of all CpG and most non-CpG data sets, the individual repeats showed a strong correlation (Supplementary Figure S6) indicating a good quality of the data sets, which were then merged for further analysis. In support of the reliable data quality, most of the profiles reproduced the key preferences of WT DNMT3B including the preferences for T(-2), A(–1) and G(+1) (Figure 5). However, there were interesting global and mutant specific effects observed. K777A showed the strongest local changes in flanking sequence preferences at the +1 site where G was strongly disfavored in CpG methylation and T was strongly favored in non-CpG methylation. N656D showed a massively altered profile at the –2 to +1 sites indicating that larger perturbation of the catalytic loop conformation was caused by the negative charge of the aspartate. Another striking effect was observed at the +1 flanking site, where G and A are preferred by WT in CpG methylation. As described above, the A preference was lost in non-CpG methylation, and the same effect was also observed for many mutants even in CpG methylation, most strongly in the case of N779D, but also for N779A, N658A and N656A.
Figure 5.

Enrichment and depletion of bases in the –4 to +4 flank region of CpG and non-CpG substrates methylated by DNMT3B mutants. On the x-axis the flank positions are shown. On the y-axis the relative methylation levels are shown as observed/expected.
To investigate if the mutations affected the equalization of methylation rates in different flanking contexts, we compared the degree of the flanking sequence preferences in WT DNMT3B and its mutants. Visual inspection of the data revealed that GGCGGG and GGCGTG sites often were only weakly methylated by the mutants. We, therefore, determined the relative methylation rates of these two target sites for WT and all mutants showing that WT was able to methylate them with good activity, but four of the mutants (N652A, V657A, N658A and K777A) had no activity at one of these sites or even at both (Figure 6A). We also compared the ratios of average methylation rates of the 15% most preferred and most disfavored sites, which is around 5.1 in the case of WT. Strikingly, this value increased to 15 in the case of N656D, 20 for N658A and 17 for N779D (Figure 6B) indicating that these mutants showed increased sensitivity towards flanking sequence effects. For example, N656D had almost zero activity at A(C/T)CGTG sites, N779D was not active on GGCGA sites, and N658A had zero activity at 6 out of the 256 flanking contexts indicating that this residue has a very important role in the adaptation of DNMT3B to different flanking sequences.
Figure 6.

Effects of amino acid mutations on flanking sequence preferences of DNMT3B. (A) Mutations in DNMT3B increase the flanking sequence sensitivity of CpG methylation leading to a drop in the ability of some DNMT3B mutants to methylate GGCGGG and GGCGTG sequences, here expressed as the ratio of their methylation and the average of all NNCGNN methylation rates. Note that four of the mutations led to a complete loss of GGCGGG and/or GGCGTG methylation (N652A, V657A, N658A and K777A). (B) Ratio of the methylation rates of the 15% best and 15% worst NNCGNN sites. Note, the elevated effects of flanking sequences in N658A, N779D and N656D. (C) Rank of the SatII sequence (TTCGAT) methylation preference by WT and mutant DNMT3B in all 256 NNCGNN flanks. A low rank indicates high methylation activity. (D) Heatmap of the activity of DNMT3B and R823A at the 4096 different NNNCGNNN sequences sorted by the difference in preferences. (E) Occurrence of bases at the +1 to +3 flank sites in the 5% of sequences most preferred and disfavored by R823A. (F) DNA shape effect on the CpG methylation activity of WT DNMT3B and the K777A and N779D mutants. Differences in the minor groove width were determined between the methylated and unmethylated sequences. In both mutants, an increased minor groove width at the +2 to +4 flanks is associated with activity. Data are shown as average of two repeats, error bars indicate the standard deviation.
Next, we were interested to investigate the role of the mutated amino acid residues in the preferred interaction of DNMT3B with the SatII sequence. As mentioned above this sequence is at rank 19 in WT DNMT3B. Interestingly, the preference was reduced moderately in the case of N656A, V657A, N658A, T775A and N779A and more strongly for N656D, R661A, K777A and N779D. Actually, for N779D the SatII sequence was already in the disfavored fraction of flanking sequences (Figure 6C). In case of N779A/D the reduced preference for the SatII sequence is correlated with the loss of the preference for A(+1). The strong effects of R661A and K777A can be connected to their loss of the T(–1) and T(–2) preferences, which are unique among all investigated mutants. Overall, these results show that the SatII interaction depends on the positioning of both the catalytic loop and TRD loop based on several protein-DNA contacts.
Finally, we investigated the CpG flanking sequence preference of R823A more closely. R823 forms a phosphate contact at the +3 flank site (14). The corresponding residue in DNMT3A (R882) is a hotspot of DNMT3A mutations in AML (24) and its mutation has been shown to lead to strong changes of flanking sequence preferences of DNMT3A (23,25). To investigate potential changes in preferences at the +3 site, CpG methylation datasets of WT DNMT3B and R823A determined with a substrate pool containing a hemimethylated CpG target site were sequenced at greater depth (Supplementary Table S4). Initially, the data were analyzed with respect to the average methylation levels of all substrates containing a particular base at one of the –4 to +4 flank sites (Supplementary Figure S7). This analysis revealed high similarity between the profiles determined with the substrate libraries containing CN and hemimethylated CpG target sites, despite the much higher sequencing depth of the latter. Moreover, the profiles of WT DNMT3B and R823A were very similar to each other. Next, the more sensitive analysis of the NNNCGNNN flanking sequence preferences was conducted using the hemimethylated CpG data. As shown in Figure 6D, both WT DNMT3B and R823A showed similar activities at many sites, which is in agreement with the previous analysis. However, sorting of the data by the ratio of the activities of R823A and WT revealed a subset of sites that was methylated better by R823A than by wildtype and vice versa. An analysis of sequences present in the 5% sequences most favored and disfavored by R823A (Figure 6E) discovered that sites preferred by R823A show strong enrichment of GGG sequences at flank position +1 to +3. The enrichment of T at +1 in favored and disfavored sequences appeared because T at the +1 site is very disfavored, leading to low catalytic rates which amplifies ratios of rates. Hence, one role of the R823 residue appears to be in balancing a strong interaction with triple-G sequences located in the +1 to +3 flank region. More generally, our data show that DNA phosphate contact of R823 plays a role in the interaction with the 3′ flank and as observed with its R882 counterpart in DNMT3A, mutation of R823 leads to changes in the flanking sequence preferences.
DNA shape readout
In addition to contacts to the edges of the bases in the major and minor groove (called direct readout), DNA sequence can be determined by DNA binding proteins through probing sequence specific conformational preferences of the DNA (called DNA shape readout or indirect readout). To investigate if the sequence preferences determined here may affect the DNA conformation, the DNA shape server was used (http://rohslab.cmb.usc.edu) (35), which provides parameters for the roll, helical twist, minor groove width and propeller twist based on penta- or hexanucleotide sequences centered at one base pair (minor groove width, propeller twist) or base pair step (roll, helical twist). The occurrence and methylation of all base or base pair centered penta- and hexanucleotides was determined and the corresponding changes of the DNA shape parameters comparing methylated sequences with all sequences were calculated. While no significant changes were observed in most cases, an increased minor groove width at flank position +2 to +4 was correlated with the activity of the K777A and N779D mutants (Figure 6F). The TRD loop approaches the DNA from the major groove at the plus flank side and in the K777A mutant structure a compression of the major groove has been observed (14). According to our activity data, the corresponding enlargement of the minor groove is favorable for methylation activity of this mutant. Similar effects may occur with N779D. The correlation of an increased minor groove width at flanks sites +2 to +4 with activity of the K777A and N779D mutants illustrates the cooperative effects of DNA contacts from the major and minor groove side.
Comparison of the flanking sequence preferences of human and mouse DNMT3B
The catalytic domains of human and mouse DNMT3B enzymes differ at 17 out of 285 amino acid residues. Among these, a K-to-R substitution is observed at the residue corresponding to human DNMT3B K782, which is in the TRD loop and contacts the backbone of +2 and +3 flank of the non-target strand. In contrast, the catalytic domains of human and mouse DNMT3A are identical in amino acid sequence. To find out if flanking sequence preferences of the human and mouse DNMT3B enzymes differ, CpG methylation preferences of human and mouse DNMT3B (mouse data were taken from our previous publication (14)) were analyzed regarding the average methylation of NNNCGNNN sites, because our previous paper has shown that the +3 site is very important for SatII preferences (14). As shown in Figure 7A, flanking sequence preferences of human and mouse DNMT3B were highly correlated, but preferences at some flanking sequences differ significantly (Figure 7B), suggesting that some minor changes of flanking sequence preferences occurred in the evolution of mouse and human DNMT3B. Genetic studies have shown that human SatII repeats are a major methylation target of DNMT3B in human cells (10–12) while minor satellite repeats are major targets of mouse DNMT3B (36). Therefore, we investigated the preferences of human and mouse DNMT3B for the CpG sites in these repeat elements. For this analysis, DNMT3B methylation preferences were taken for the preferred DNA strand, assuming that DNMT1 will convert hemimethylated sites generated by DNMT3B into the fully methylation state. Our data revealed that human DNMT3B shows an increased preference for SatII sequences while murine DNMT3B prefers minor satellite repeats (Figure 7C and D). Although these changes are only moderate, they suggest an evolutionary adaptation of the flanking sequence preferences of DNMT3B enzymes for their most important physiological targets. In contrast and as reported before, DNMT3A strongly disfavors both sequences (Supplementary Figure S8).
Figure 7.

Comparison of the flanking sequence preferences of human and mouse DNMT3B. (A) Heatmap of the average methylation activities of mouse and human DNMT3B and DNMT3A at NNNCGNNN sites sorted by the average activity of DNMT3B. The amino acid sequence of catalytic domain of human and mouse DNMT3A is identical. DNMT3A data were taken from (14). (B) Heatmap of the average methylation activities of mouse and human DNMT3B at NNNCGNNN sites sorted by the difference between human and mouse enzymes. (C) Boxplot of the ranks of the CpG sites in the human SatII repeat (n = 67) and murine minor satellite repeat (n = 11) in the NNNCGNNN flanking preferences of human and mouse DNMT3B. A low rank indicates high preference. The boxes display the first and third quartiles with medians indicated by vertical lines. Whiskers display the data range. (D) Ratio of the median ranks of human and mouse DNMT3B for the methylation of CpG sites in human SatII and mouse MinSat repeats.
DISCUSSION
Similar to other DNA interacting enzymes and proteins, DNMTs interact with their CpG target sites in the context of flanking base pairs. This leads to flanking sequence preferences in the methylation rates of CpG sites, because the shape, conformations and dynamics of DNA vary strongly depending on the sequence context (37–39) and some DNA sequences fit better or worse into the DNA binding site of the enzyme. Three interesting questions stem from this situation: (i) How do DNMTs manage to recognize their short CpG target site in the context of different flanking sequences imposing very different structural constraints on the DNA? (ii) How do DNMTs balance flanking sequence preferences and allow methylation of all CpG sites with comparable methylation rate constants? (iii) Do flanking sequence preferences co-evolve with the biological targets of DNMTs?
In our previous work, we reported the structure of DNMT3B catalytic domain and compared the flanking sequence preferences of murine DNMT3B catalytic domain and human/murine DNMT3A catalytic domain (which are identical in sequence) at the level of ±3 base pair flanking sequences (14). Based on this we could rationalize differences in flanking sequence preferences of both enzymes on the basis of the enzyme structures (14,40) and showed that the flanking sequence preferences are relevant in the context of the ICF syndrome. In the current study, we expanded the structure/function and flanking sequence analysis of human DNMT3B catalytic domain by analysis of 12 mutants and deepening the data basis in case of two more mutants (K777A and N779A), for which the +1 flanking data were provided for the murine enzyme in the previous work. Using these new data, we investigated details of the CpX target site recognition by DNMT3B and its interaction with flanking base pairs. Our first aims were to examine the connection of CpX site and flanking sequence readout, which occur next to each other in the protein-DNA complex. Moreover, we aimed to unravel the strategies DNMT3B uses to balance its interaction with different flanking contexts and thereby equalize the methylation rates of CpG sites in different flanking contexts. Our data reveal a preference of DNMT3B for CpG methylation in a TACG(G/A) context. CpA and CpT flanking sequence preferences are similar. CpG site and flanking sequence interaction are correlated as indicated by the observations that in non-CpG methylation the preference for A(+1) is lost and the preference for a TACXG context is much stronger than in CpG methylation. These data illustrate that the equalization of the interaction with different flanking sites is less efficient in the non-CpG context, where the interaction with the guanine at the CpG site is missing. Moreover, CpC flanking preferences are different, perhaps because of the presence of a GG dinucleotide on the non-target strand. We showed that the cellular non-CpG methylation activity of DNMT3B in hESC is highly correlated with the corresponding in vitro flanking sequence preferences of DNMT3B mainly in between the flanking sites –3 and +3. The correlation of genomic CpC methylation with in vitro preferences was weaker but still highly significant. These findings document the high biological relevance of the processes investigated in our work.
Several DNMT3B mutants were investigated allowing to draw detailed conclusions regarding the DNA recognition process of DNMT3B. Among all the studied DNA contacting residues, T775 was most important for DNA methylation by DNMT3B as indicated by the exceptional loss in activity for all three mutants of this residue studied here (T775A/N/Q). Structural studies showed that T775 approaches the purine of the CpX site where it forms a water mediated H-bond to the N7 atom of the purine base (13,14), but most likely this contact alone cannot explain the very strong loss of activity after mutation of T775 in any other residue. However, T775 also contacts the ribose of the target cytosine and it occupies parts of the intrahelical space emptied by the flipped cytosine. By this, it blocks the back-rotation of the flipped cytosine into the DNA, where the target base would not be accessible for methylation. The correct and stable positioning of the side chain of T775 may, therefore, be needed to keep the target cytosine in a flipped state, suggesting that T775 has a dynamic role in coupling the sequence readout and complex formation with stable target base flipping.
The second strongest effects were observed with K777A. Previously, we already observed very specific changes of the preferences mainly at the +1 flanking site, most strongly a disfavor for G and gain of preference for T which was also observed in cellular methylation data (14). The loss of the G(+1) preference of DNMT3B in K777A can be explained by the specific H-bond of K777 to the N7 of a G at this site (14). Mutational loss of K777 leads to a strong disfavor of G(+1) causing a complete inability of the K777A mutant for methylation of CGG sites. The preference of K777A for a T(+1) may be related to a Van der Waals contact (3.5 Å) of the K777-Cγ atom to the methyl group of T(+1). This contact apparently leads to a rearrangement of K777, which weakens the contact to the CpX site and leads to the loss of the H-bonds formed between K777-NZ and the DNA in the different complexes. These data indicate that K777 has an important role in equalizing of flanking sequence context: it compensates the T(+1) preference and G(+1) disfavor inherent in WT DNMT3B by forming a contact to G(+1) that increases DNA methylation and by forming a contact to T(+1) that is unfavorable and reduces DNA methylation. The altered readout of the +1 flank of K777A is combined with a pronounced loss of CpG specificity indicating towards a coupling of CpG and flank interactions. This effect may be explained by the approach of the side chain methylene groups of K777 and C8 of the CpG guanine. Thereby, K777 creates one wall of the binding pocket for the CpG guanine base and it is well established for DNA polymerases that accurate base recognition requires a confined binding pocket (41). Loss of the K777 side chain may increase the conformational freedom of the base at the CpX position allowing DNMT3B to tolerate bases different than guanine more easily. In a similar way, R823 has a role to compensate an intrinsic preference for triple-G sequences in the 3′ flank possibly by interaction with a triple-G associated DNA conformation.
The N779A and D mutations showed a similar change in sequence preferences, most strikingly a strong loss of the preference for an AT’ base pair at the +1 flanking site in CpG methylation. This effect can be explained by the H-bond between N779 and the T’(+1) O4 atom, which is specifically formed in the CpG complex. The same CpG specific contact can explain the loss of A(+1) preference of WT DNMT3B in non-CpG context. Hence, N779 appears to be particularly involved in the readout of a CGA trinucleotide sequence indicating that it has a special role in the methylation of SatII repeats which contain an A(+1). Moreover, the N779 mediated gain in CGA methylation partially compensates the CGG preference mediated by K777 avoiding a too strong preference for G at the +1 site.
Among the residues in the catalytic loop, V657A and N658A showed the largest overall changes in CpG specificity, because V657 showed an increased CpT methylation and N658A showed increased CpA methylation. Moreover, N658 was most important for the equalization of flanking sequence interaction, in particular for the efficient methylation of CpG sites in G-rich flanking contexts. N658 contacts the ribose at the CpX site and +1 flank in all complexes, and our data suggest that this contact is relevant for CpG recognition. Interestingly, the changes in the +1 preferences observed with N658A are opposite to K777A, suggesting that N658 also has a role in dampening of the effects of K777. Regarding the increased CpT methylation of V657A, no structural data are available for the CpT site interaction of DNMT3B, but the effect of V657A may be mutation specific. The V657-Cγ atom forms a close contact to the CpG and CpA guanine N7 and modeling suggests that an alanine in the V657A mutant might engage in a productive interaction with the methyl group of a T at a CpT site. Interestingly, N656A from the catalytic loop and to a lesser degree N658A show a similar reduction of A(+1) preference in CpG methylation as N779A/D suggesting that the double sided grip of the DNA from the major and minor groove side is required for the specific formation of the N779-AT’(+1) contact. Strikingly, N652, N658 and K777 are all needed to allow methylation of GGCGGG sites suggesting that the close grip of the DNA is particularly important for binding of substrates with G-rich flanks. Moreover, the catalytic loop residues cooperate in the balancing of flanking sequence effects, which is likely mediated by the H-bond network connecting all of them. This effect is most clearly illustrated with R661A, which showed a pronounced reduction of SatII sequence preferences caused by a disfavor for T(-1) that was unique among all mutants. In the currently available structures, R661 does not engage in DNA contacts but it has an important role in stabilizing the conformation of the catalytic loop by forming H-bonds with N656 and N658.
CONCLUSIONS AND OUTLOOK
Our data reveal a complex balancing of flanking sequence effects and their connection to CpG readout by DNMT3B. We show striking correlations of the non-CpG flanking sequence preferences of DNMT3B and non-CpG methylation patterns in human cells. Moreover, our data illustrate a co-evolution of DNMT3B flanking sequence preferences with its key targets, SatII repeats in the human genome and minor satellite repeats in mouse. DNMT3B executes a sandwiched approach of the DNA by the catalytic loop and TRD loop from the minor and major groove sides, respectively, which is necessary for tight DNA contacts and methylation of CpG sites in G-rich flanks. WT DNMT3B has an inherent preference for methylation in a TACG(A/G) context that is elevated and confined to TACXG in CpA and CpT methylation. T775 is very important for catalysis suggesting that this residue has an essential dynamic role in coupling the DNA recognition and target base flipping. A complex network of balancing contacts of amino acid side chains with different flanking base pairs is involved in the equalization of methylation rates of CpG sites in different flanking contexts. K777 compensates a T(+1) preference inherent in WT DNMT3B and supports methylation of G(+1) flanks. A too large preference for G(+1) is counteracted by N779 and N658 which support CGA methylation. Finally, R823 compensates an intrinsic preference of DNMT3B for GG dinucleotides at the +2 and +3 sites. Unfortunately, the currently available structural data are insufficient to explain the detailed structural basis for many of the biochemical effects observed here. However, a more comprehensive structural analysis of the DNA interaction of DNMT3B would be a formidable task, because if one considers the ±2 flanks of the four different CpX sites, 1024 different crystal structures would be required. This number contrasts sharply with the four DNMT3B structures in different DNA sequence context, which are currently available. Therefore, a full structural interpretation of these effects would require a large effort that may not be possible until more automated structural analysis methods are established perhaps using the Cryo-EM technology. Moreover, the experiments reported here were conducted in the context of the catalytic domain of DNMT3B. To explain the specificity of DNMT3B in vivo these data must be connected with the targeting and regulation effects of the N-terminal part of DNMT3B (5,6).
DATA AVAILABILITY
NGS kinetic raw data have been uploaded at DaRUS, the Data Repository of the University of Stuttgart (https://doi.org/10.18419/darus-815). All scripts used in this study for data analysis of the NGS kinetics are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: A.J. devised the study. M.D. performed the experimental work with support from S.A. A.J. and P.B. supervised the work. M.D., S.A., P.B. and A.J. were involved in NGS data analysis and bioinformatic analyses. M.B. and J.S. provided structural data essential for the design of the project. M.D. and A.J. prepared the manuscript draft. All authors were involved in final data analysis and interpretation and writing of the manuscript.
Contributor Information
Michael Dukatz, Institute of Biochemistry and Technical Biochemistry, University of Stuttgart, Allmandring 31, 70569 Stuttgart, Germany.
Sabrina Adam, Institute of Biochemistry and Technical Biochemistry, University of Stuttgart, Allmandring 31, 70569 Stuttgart, Germany.
Mahamaya Biswal, Department of Biochemistry, University of California, Riverside, CA 92521, USA.
Jikui Song, Department of Biochemistry, University of California, Riverside, CA 92521, USA.
Pavel Bashtrykov, Institute of Biochemistry and Technical Biochemistry, University of Stuttgart, Allmandring 31, 70569 Stuttgart, Germany.
Albert Jeltsch, Institute of Biochemistry and Technical Biochemistry, University of Stuttgart, Allmandring 31, 70569 Stuttgart, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
DFG [JE 252/10, JE 252/36 to A.J.], NIH [R35GM119721 to J.S.]. Funding for open access charge: The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Law J.A., Jacobsen S.E.. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010; 11:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schubeler D. Function and information content of DNA methylation. Nature. 2015; 517:321–326. [DOI] [PubMed] [Google Scholar]
- 3. Chen Z., Zhang Y.. Role of mammalian DNA methyltransferases in development. Annu. Rev. Biochem. 2019; 89:135–158. [DOI] [PubMed] [Google Scholar]
- 4. Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M. et al.. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jeltsch A., Jurkowska R.Z.. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 2016; 44:8556–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gowher H., Jeltsch A.. Mammalian DNA methyltransferases: new discoveries and open questions. Biochem. Soc. Trans. 2018; 46:1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeng Y., Chen T.. DNA methylation reprogramming during mammalian development. Genes. 2019; 10:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dodge J.E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E.. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005; 280:17986–17991. [DOI] [PubMed] [Google Scholar]
- 9. Jeltsch A., Jurkowska R.Z.. New concepts in DNA methylation. Trends Biochem. Sci. 2014; 39:310–318. [DOI] [PubMed] [Google Scholar]
- 10. Okano M., Bell D.W., Haber D.A., Li E.. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. [DOI] [PubMed] [Google Scholar]
- 11. Xu G.L., Bestor T.H., Bourc’his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E.. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999; 402:187–191. [DOI] [PubMed] [Google Scholar]
- 12. Hansen R.S., Wijmenga C., Luo P., Stanek A.M., Canfield T.K., Weemaes C.M., Gartler S.M.. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. PNAS. 1999; 96:14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin C.C., Chen Y.P., Yang W.Z., Shen J.C.K., Yuan H.S.. Structural insights into CpG-specific DNA methylation by human DNA methyltransferase 3B. Nucleic Acids Res. 2020; 48:3949–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao L., Emperle M., Guo Y., Grimm S.A., Ren W., Adam S., Uryu H., Zhang Z.M., Chen D., Yin J. et al.. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 2020; 11:3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramsahoye B.H., Biniszkiewicz D., Lyko F., Clark V., Bird A.P., Jaenisch R.. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. PNAS. 2000; 97:5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gowher H., Jeltsch A.. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001; 309:1201–1208. [DOI] [PubMed] [Google Scholar]
- 17. Suetake I., Miyazaki J., Murakami C., Takeshima H., Tajima S.. Distinct enzymatic properties of recombinant mouse DNA methyltransferases Dnmt3a and Dnmt3b. J. Biochem. (Tokyo). 2003; 133:737–744. [DOI] [PubMed] [Google Scholar]
- 18. Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C.T., Low H.M., Kin Sung K.W., Rigoutsos I., Loring J. et al.. Dynamic changes in the human methylome during differentiation. Genome Res. 2010; 20:320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J.H., Park S.J., Nakai K.. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017; 7:11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He Y., Ecker J.R.. Non-CG methylation in the human genome. Annu. Rev. Genomics Hum. Genet. 2015; 16:55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jeltsch A., Broche J., Bashtrykov P.. Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes. 2019; 10:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Handa V., Jeltsch A.. Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J. Mol. Biol. 2005; 348:1103–1112. [DOI] [PubMed] [Google Scholar]
- 23. Emperle M., Adam S., Kunert S., Dukatz M., Baude A., Plass C., Rathert P., Bashtrykov P., Jeltsch A.. Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res. 2019; 47:11355–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamidi T., Singh A.K., Chen T.. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015; 7:247–265. [DOI] [PubMed] [Google Scholar]
- 25. Emperle M., Rajavelu A., Kunert S., Arimondo P.B., Reinhardt R., Jurkowska R.Z., Jeltsch A.. The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res. 2018; 46:3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anteneh H., Fang J., Song J.. Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 2020; 11:2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emperle M., Rajavelu A., Reinhardt R., Jurkowska R.Z., Jeltsch A.. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 2014; 289:29602–29613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bashtrykov P., Jankevicius G., Smarandache A., Jurkowska R.Z., Ragozin S., Jeltsch A.. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 2012; 19:572–578. [DOI] [PubMed] [Google Scholar]
- 29. Adam S., Anteneh H., Hornisch M., Wagner V., Lu J., Radde N.E., Bashtrykov P., Song J., Jeltsch A.. DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat. Commun. 2020; 11:3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Gruning B.A. et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46:W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang J., Kobert K., Flouri T., Stamatakis A.. PEAR: a fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2014; 30:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blankenberg D., Gordon A., Von Kuster G., Coraor N., Taylor J., Nekrutenko A.. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010; 26:1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan H.K., Wu C.S., Li J., Tan Z.H., Hoffman J.R., Fry C.J., Yang H., Di Ruscio A., Tenen D.G.. DNMT3B shapes the mCA landscape and regulates mCG for promoter bivalency in human embryonic stem cells. Nucleic Acids Res. 2019; 47:7460–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou T., Yang L., Lu Y., Dror I., Dantas Machado A.C., Ghane T., Di Felice R., Rohs R.. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013; 41:W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen T., Ueda Y., Dodge J.E., Wang Z., Li E.. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003; 23:5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garvie C.W., Wolberger C.. Recognition of specific DNA sequences. Mol. Cell. 2001; 8:937–946. [DOI] [PubMed] [Google Scholar]
- 38. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abe N., Dror I., Yang L., Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S.. Deconvolving the recognition of DNA shape from sequence. Cell. 2015; 161:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Z.M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S.B., Wang Y. et al.. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018; 554:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kool E.T. Active site tightness and substrate fit in DNA replication. Annu. Rev. Biochem. 2002; 71:191–219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS kinetic raw data have been uploaded at DaRUS, the Data Repository of the University of Stuttgart (https://doi.org/10.18419/darus-815). All scripts used in this study for data analysis of the NGS kinetics are available upon request.