
Figure 1. Overview of the Study Design and the Germline Variant Detection Methods Used.
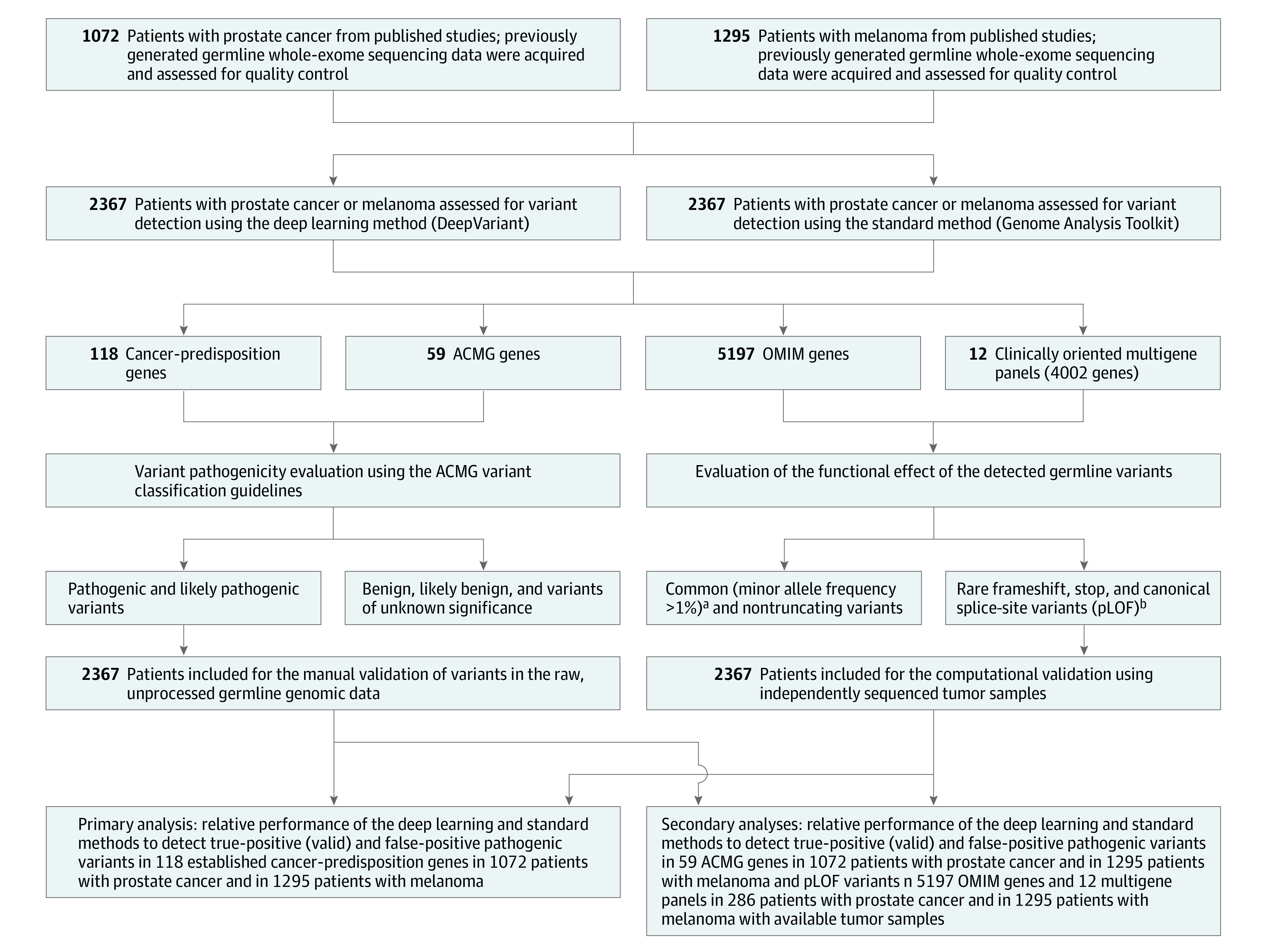
Germline whole-exome sequencing data of 2 independent cohorts were analyzed using a deep learning variant detection approach and the current standard germline variant detection method to assess the sensitivity, specificity, positive predictive value, and negative predictive value of detecting pathogenic variants in 118 cancer-predisposition genes as well as in 59 American College of Medical Genetics and Genomics (ACMG) genes. In addition, an exome-wide analysis of putative loss-of-function (pLOF) variants in 5197 Online Mendelian Inheritance in Man (OMIM) genes and in 12 clinically oriented multigene panels was conducted in 286 patients with prostate cancer and in 1295 patients with melanoma whose tumors were available for independent variant validation. For each analysis, the performance of the deep learning method and the standard method was compared with the performance obtained when both methods are concurrently used (additional information appears in the Methods section). The performance of the standard and deep learning methods to detect pathogenic variants in the cancer-predisposition and ACMG gene sets was tested on all patients with prostate cancer and melanoma (n = 2367). However, the performance of the 2 methods in the OMIM gene set and the multigene panels was tested on 286 patients with prostate cancer and 1295 patients with melanoma (n = 1581) whose tumor sequencing data were available for computational validation of the identified pLOF variants.
aRepresents the frequency of the allele (also called variant) in the general population.
bGenetic variants that are likely to severely disrupt the protein function by truncating the gene transcript. Examples of pLOF variants include frameshift, stop-codon, and canonical splice-site variants.