

Abstract
Mutations in the human cystathionine beta synthase (CBS) gene are known to cause endothelial dysfunction responsible for cardiovascular and neurovascular diseases. CBS is the predominant hydrogen sulfide (H2S)-producing enzyme in endothelial cells (ECs). Recently, H2S was shown to attenuate ROS and improve mitochondrial function. Mitochondria are metabolic organelles that actively transform their ultrastructure to mediate their function. Therefore, we questioned whether perturbation of CBS/H2S activity could drive mitochondrial dysfunction via mitochondrial dynamics in ECs. Here we demonstrate that silencing CBS induces mitochondria fragmentation, attenuates efficient oxidative phosphorylation, and decreases EC function. Mechanistically, CBS silencing significantly elevates ROS production, thereby leading to reduced mitofusin 2 (MFN2) expression, decouple endoplasmic reticulum-mitochondria contacts, increased mitochondria fission, enhanced receptor-mediated mitophagy, and increased EC death. These defects were significantly rescued by the treatment of H2S donors. Taken together our data highlights a novel signaling axis that mechanistically links CBS with mitochondrial function and ER-mitochondrial tethering and could be considered as a new therapeutic approach for the intervention of EC dysfunction-related pathologies.
Keywords: endothelial cells, mitochondrial dynamics, mitophagy, mitochondrial fusion, mitochondrial fission
1 |. INTRODUCTION
Cystathionine beta synthase (CBS) is one of three enzymes that generates hydrogen sulfide (H2S), the third gasotransmitter, in all mammalian tissues and is the predominant H2S-producing enzyme in endothelial cells (ECs).1 Loss of CBS function in humans is linked with a wide variety of pathologies associated with significant endothelial dysfunction and impaired angiogenesis—including cardiovascular and neurovascular disorders.2–4 Additionally, studies in CBS knockout mice have shown poor response to vasodilators and impaired angiogenesis that have been largely attributed to redox imbalance and high levels of plasma homocysteine.5,6
Hydrogen sulfide is a direct scavenger of reactive oxygen species (ROS) and peroxynitrite.7 Moreover, it attenuates mitochondrial ROS production and prevents mitochondrial membrane depolarization in the cardiovascular and nervous system and has proangiogenic role.8 A recent report demonstrates that treatment with the H2S donor, NaHS, can significantly increase collateral vessel growth, capillary density, and regional tissue blood flow in a rat model of hind limb ischemia.9 Additionally, Papapetropoulos et al. established that endogenous and exogenous H2S stimulates EC-related angiogenic properties through a KATP channel/MAPK pathway.10 Interestingly, recent studies have demonstrated that the CBS protein is also present in mitochondrial fractions and that H2S produced by mitochondrial cystathionine γ-lyase (CSE) or CBS exerts protective effects on mitochondrial function.11–15
Mitochondria are dynamic organelles that continually undergo morphological changes through the process of mitochondrial fusion and fission. Mitochondria dynamics maintain the shape, size, and number of mitochondria, as well as influence their physiological functions—like mitochondrial DNA stability, respiratory capacity, apoptosis, response to cellular stress, and mitophagy.16 Moreover, recent reports suggested that metabolism and mitochondrial morphology dynamics are highly linked and regulate one another.17 Mechanistically, mitochondria fusion is mediated by mitofusins 1 and 2 (MFN1, MFN2) and optic atrophy 1 (OPA1),13 and is known to maximize OXPHOS activity18,19 and enhance cell longevity.20–22 Moreover, mitochondria fission is mediated by dynamin-related protein 1 (DRP1) and fission-related proteins, like fission 1 (FIS1),7 mitochondria fission factor (MFF), MiD51, and MiD4913). It is known that fission generates fragmented mitochondria that can enhance ROS production,23 induce mitophagy,24,25 and mediate apoptosis.26 Because of these important functions, mitochondrial dynamics are essential for proper cellular function, resilience, and metabolism; even mild defects in these machinery can disturb oxidative phosphorylation (OXPHOS) and glycolysis27 and are associated with disease,16 such as endothelial dysfunction.28,29
Endothelial cells are motors for angiogenesis—the process by which blood vessels are formed. They switch from a quiescent state to a migratory/proliferative phenotype in order to support angiogenesis, which is a dynamic and energy-consuming process. ECs utilize glycolysis as an energy source to meet this demand.30 However, there are recent reports that proliferating and lymphatic ECs can utilize OXPHOS for fatty acid oxidation (FAO) to sustain DNA synthesis31 and epigenetic regulation of lymphatic gene expression,32 respectively. Moreover, it has also been recently reported that quiescent ECs can utilize FAO for redox homeostasis.31–35 In this context, endothelial mitochondria—by coordinating ROS and calcium signaling, metabolism, and apoptosis—have emerged as signaling hubs that can modulate a wide range of endothelial functions, including angiogenesis.
Autophagy is a process by which cells degrade macromolecular intracellular material via sequestration in a double-membrane structure, known as an autophagosome, which delivers the enclosed material to a lysosome for degradation.36 Many autophagy-related genes (ATGs) have been identified in mammals, most of which form multi-molecule complexes to regulate autophagosome formation.37,38 Mitochondrial-specific autophagy is known as mitophagy and requires two steps: induction of general autophagy and mitochondrial priming. Mitochondrial priming is facilitated either by the PINK1-Parkin signaling pathway or by the mitophagic receptors NIX and BNIP3.39 BNIP3 regulates mitophagy during hypoxia and is transcriptionally upregulated by HIF-1α.40 Upon expression, BNIP3 localizes to the mitochondria, induces mitochondrial swelling, promotes mitochondrial fission, and stimulates mitochondrial turnover via mitophagy.41,42 It further functions as a tether to stimulate activation of mitophagy by linking damaged mitochondria to LC3 present on nascent autophagosomes.43
A number of studies have demonstrated that CBS depletion induces endothelial dysfunction,1,44 however, the underlying molecular mechanisms are largely unknown. Mitochondria are now appreciated to be targets of H2S signaling15 and, indeed, our group recently reported that CBS activity can mediate mitochondria fusion in ovarian cancer cells.13 Therefore, it would be of great interest to know if the observed endothelial dysfunction in CBS-silenced ECs is associated with mitochondrial dynamic deregulation. So far, to our knowledge, there is no study regarding CBS and its role mitochondrial dynamics in ECs. The present study aims to examine whether CBS can alter mitochondrial function, and investigate its role in mitochondrial dynamics in ECs. A better understanding of these processes will ultimately lead to improvements in human health and could be of clinical importance for the treatment of vascular pathologies.
2 |. MATERIALS AND METHODS
2.1 |. Reagents, cell lines, and culture
siRNA against human CBS was purchased from sigma, St. Louis, MO, USA (SASI_Hs01_00214623), and scrambled control siRNA (1027280) was from QIAGEN, CA, USA. siRNA against human MFN2 was purchased from Ambion®, Grand Island, NY 14072 USA (AM16708). Mitofusin 2(MFN2) (NM_014874) Human Untagged Clone was purchased from Origene technologies, Rockville, MD, USA (SC114726) and pcDNA3.1 was used as empty vector control. EGM bullet kit, 0.25% trypsin-EDTA, and trypsin neutralizing solution were purchased from Lonza (Basel, Switzerland). Oligofectamine, Lipofectin, and OptiMem-I were purchased from Invitrogen (Carlsbad, CA, USA). HUVECs (CC-2517) purchased from Lonza, Basel, Switzerland, were cultured in complete endothelial basal medium (EBM) medium (EBM + EGM kit) and all experiments were performed at Passage 3 and 4. The antibodies details are listed in Supplementary Table 1.
2.2 |. Transfection
For transient gene overexpression, 80% confluent HUVEC/HAOEC cells in 100 mm dishes were transfected with 5 μg plasmid using Lipofectin Transfection Reagent according to the manufacturer’s protocol. For siRNA transfection of HUVECs in 100 mm dishes with scrambled siRNA or CBS siRNA or MFN2 siRNA was carried out using Oligofectamine. The cells were cultured for 48 hours before any subsequent analysis.
2.3 |. Immunoblotting
Immunoblotting analysis was carried on whole cell lysates/mitochondrial fraction/cytosolic fraction of siCtrl and siCBS treated HUVECs/HAOECs in RIPA buffer supplemented with protease-phosphatase mix (Pierce). After protein quantitation using BCA assay (Pierce, Thermo Scientific, MA, USA), the cell lysate (30ug) were separated on 10% or 12% tris-glycine SDS-PAGE gel (1–2 hours at 100 V, Biorad, Hercules, CA) and transferred to PVDF membrane (1 hours at 100 V, Biorad, Hercules, CA). Membranes were blocked in 5% nonfat dry milk in TBS with 0.1% TWEEN-20 (TBST) for 1hr at RT. Primary antibodies were used in dilutions recommended by the manufacturer and secondary antibodies were used at a concentration of 1:10 000. Equal loading was verified by immunoblotting with actin, LDH or VDAC. Details of antibody dilution are given in Supplementary Table 1.
2.4 |. Cell mito-stress analysis
Different parameters of mitochondrial respiration were measured with the 96-well XF analyzer and the XF cell mito stress kit (Seahorse Bioscience, MI). Assay was done as per the manufacturer’s protocol. 12 hours prior to assay, XF sensor cartridges were hydrated with the supplied calibrant (200 μL each well) and incubated at 37°C without CO2 (pH to 7.4 at 37°C). Unbuffered XF assay medium was prepared and transfected cells (as described above) for 24 hours with control or CBS siRNA and were re-plated to the 96-well XF assay plate at a cell density of 3.5 × 104 cells per well in complete EBM media. 30 minutes before the assay, the plates were washed with the assay medium (200 μL, 3×) and incubated at 37°C without CO2. Oligomycin (1 μM), FCCP (2 μM) and a mix of Antimycin A and Rotenone (0.5 μM each) (diluted in assay media from Seahorse as per manufacturer’s protocol) were added into the appropriate ports of the cartridge and calibrated in the instrument. After the calibration of the cartridge, cell culture plate was loaded in the instrument and the assay was run as per the standard template and (OCR) oxygen consumption rate was measured. After the assay, media were aspirated, wells were washed once with PBS (100 μL), 50 μL of RIPA (Boston Biochem, Minneapolis, MN, USA) was added, and lysates were analyzed for total protein content using BCA assay (Pierce, Thermo Scientific, MA, USA). Data were normalized to total protein content.
2.5 |. Mitochondrial membrane potential assay
Mitochondrial membrane potential was measured using TMRE-Mitochondrial Membrane Potential Assay Kit (#ab113852) from Abcam, (Cambridge, MA, USA) as per the manufacturer’s instruction. Briefly, HUVECs were transfected as described and 48 hours post transfection 10 000 cells were replated in a 96-well plate (sterile, tissue culture treated, 96-well plate with clear flat bottom) suitable for fluorescence measurement. About 250 nM positively charged TMRE (tetramethylrhodamine, ethyl ester) dye (provided in the kit and diluted in complete EBM media) was used to label active mitochondria that are negatively charged. FCCP, Carbonyl cyanide p-trifluoro-methoxyphenyl hydrazine (provided in the kit and diluted in complete EBM media), an ionophore that depolarizes mitochondria and prevents binding of the dye to the mitochondria is used as a negative control for the assay. Plate was read at 549 nm excitation and 575 nm emission wavelengths (CLARIOstar, BMG Labtech, NC USA).
2.6 |. Immunofluorescence microscopy
Co-localization of BNIP3/Mitotracker was determined by immunostaining followed by fluorescence microscopy. To assess mitochondrial morphology, cells were stained with MitoTracker Red CMXRos (M7512, Invitrogen, USA). Briefly, HUVECs (30%–40% confluency) were plated on 12 mm coverslips and 48 hours after transfection cells were treated with Mitotracker Red (100 nM) for 15 minutes at 37°C, after three times washing with PBS. The cells were fixed with 4% PFA, permeabilized with 0.1% TritonX-100 in PBS, blocked with 4% BSA in PBS, stained overnight with primary antibody (1:200) in 1% BSA-PBS, washed and stained with Alexa Fluor 488-labeled goat anti-rabbit secondary antibody for 1 hours. DAPI (blue) was used to stain the nucleus. Co-localization of lysotracker/Mitotracker was determined by immunostaining followed by fluorescence microscopy. Briefly, HUVECs (30%–40% confluency) were plated on 12 mm coverslips and 48hrs after transfection cells were treated with Mitotracker Red (100 nM) and lysotracker green (50 nM) for 15 minutes at 37°C, after three times washing with PBS the cells were fixed with 4% PFA and stained with DAPI. Cell images were acquired with a 63× objective using a Zeiss Axio Observer. Z1 (Göttingen, Germany). Co-localization of ER Tracker/Mitotracker was determined by immunostaining followed by fluorescence microscopy. Cells were plated, treated, and imaged as described above. Cells were stained with Mitotracker Red (100 nM) and ER tracker green (E34251, ThermoFisher Scientific, USA) (1 μM) for 15 minutes at 37°C, after three times washing with PBS the cells were fixed with 4% PFA and stained with DAPI.
2.7 |. Confocal microscopy and image analysis
Mitochondrial morphology was determined by immunostaining followed by confocal microscopy. To assess mitochondrial morphology, cells were stained with MitoTracker Red CMXRos (M7512, Invitrogen, USA). Briefly, HUVECs or HAOECs (30%–40% confluency) were plated on 12 mm coverslips and 48 hours after transfection cells were treated with Mitotracker Red (100 nM) for 15 minutes at 37°C, after three times washing with PBS the cells were fixed with 4% PFA and stained with DAPI. Images were collected by confocal microscopy (Zeiss LSM710) and analyzed with ImageJ 1.42. Mitochondria networks were analyzed utilizing a modified method as described by Valente et al using ImageJ’s FIJI distribution and an open source macro tool, MiNA (Mitochondrial Network Analysis), designed by the authors.45 Briefly, images were spilt into their RBG color channels, whereby only the mitochondria channel was analyzed. Images were processed using the “mean” filter before processing by the macro. The macro was run on all default settings, except processing by the “median” filter was deselected. Individual and network mitochondria were counted and network’s branching and branch length were measured. Individual mitochondria are defined as segments which have no branches, whereas network mitochondria are defined as segments which have at least one branching arm.
2.8 |. Real-time PCR
Total RNA was isolated from transfected cells using RNeasy Plus Mini kit (QIAGEN). RNA was first retrotranscribed using iScript cDNA Synthesis kit (Bio-Rad. QRT-PCR was performed on CFX Connect Real Time System (BioRad) and analyzed by ΔΔCT method with 36β4 used as an internal control. To ensure the homogeneity of the PCR products, melting curves were acquired after each reaction. The primers used for qRT-PCR are listed as below: Gene symbol Forward primer sequence Reverse primer sequence
CBS: 5′-CGGCGCTGCTGACCGACATC-3′ 5′-CCCT GCCCCACTCCCAGCAT-3′
MFN2: 5′- CGCTTATCCACTTCCCTCCTC-3′ 5′- CAG GGACATTGCGCTTCACC-3′
BNIP3: 5′- CCTCAGCATGAGGAACACGA-3′ 5′- AA AAGGTGCTGGTGGAGGTT-3′
36β4: 5′-GAGGGAAGGGAATTAGAAAA-3′ 5′-CTTG AACCTGTCTGAAGAG-3′
The comparative Ct method was used to calculate the relative abundance of the mRNA and compared with that of 36β4.13
2.9 |. Tube formation assay
Reduced growth factor Matrigel (BD Biosciences, San Jose, CA, USA) was thawed at 4°C overnight. Thereafter, Matrigel was diluted in 1:1 ratio with cold complete EBM medium and 50 μL of the diluted Matrigel was added per well of a 96-well plate and incubated at 37°C for 30 minutes. 48 hours after transfection, HUVECs were trypsinized and counted and 2 × 104 cells in 100 μL of medium were plated onto Matrigel. Tube formation was captured after 6 hours from plating of cells. Images were acquired using Zeiss Apotome microscope (Carl Zeiss Gm bH, Jena, Germany) in phase contrast mode.
2.10 |. Mitochondrial ROS
The mitochondrial ROS was carried out in HUVECs 48 hours post transfection with scrambled control or CBS siRNA following a previously published procedure with minor modifications.46 In brief, transfected cells were incubated with MitoSOX (Invitrogen) staining (2.5 μmol/L) for 10 minutes at 37°C. Data were acquired with a FACSCalibur (BD Biosciences) and analyzed with FlowJo analytical software.
2.11 |. Cell fractionation
For subcellular fractionation, previously published protocol was followed.47 Briefly, after transfection as describe above, three 100 mm plate for each siCtrl and siCBS was used for subcellular fractionation. For isolation of mitochondria enriched fractions, cells were disrupted in isolation buffer (250 mM sucrose [Sigma, S1888], 1 mM EGTA, 10 mM HEPES, 10 mM Tris-HCl, pH 7.5) using two sets of 40 strokes in a Dounce glass homogenizer (Wheaton, 357538 with “loose” pestle). Homogenates were centrifuged at 800 g for 7 minutes to remove nuclear fraction and unbroken cells. Supernatant was then subjected to centrifugation at 4000 ×g for 15 min, and the pellet was taken as the enriched mitochondrial fraction. Supernatant from this step, which excluded both the nucleus and mitochondria, was considered as cytosolic fraction. Enriched mitochondrial pellet was washed twice with EGTA-free mitochondrial buffer (250 mM sucrose, 10 mM HEPES, 10 mM Tris-HCl, and pH 7.5) and resuspended in an EGTA-free mitochondrial buffer and used for immunoblotting.
2.12 |. Endogenous H2S measurement
Hydrogen sulfide concentrations in HUVECs and HAOEC were measured using methylene blue assay as previously reported.1 Briefly, 48 hours posttransfection, scrambled or CBS siRNA-treated cells were collected by trypsinization and resuspended in 200 μL of PBS (pH 7.4), and then 100 μL was transferred into a tube containing zinc acetate (1% wt/vol, 187.5 μL) and NaOH (12%, 12.5 μL) to trap the H2S for 20 minutes at RT. The remaining cell suspension was used for protein estimation by bicinchoninic acid assay. Finally, 1 mL of H2O (pH 12.8), 200 μL of N,N-dimethylp-phenylenediamine sulfate (20 mM in 7.2 M HCl), and 200 μL of FeCl3 (30 mM in 1.2 M HCl) was added to terminate the reaction. Then the mixture was incubated in dark for 15 minutes at RT, and 600 μL of the mixture was added to a tube with 150 μL of trichloroacetic acid (10% wt/vol) to precipitate protein. After centrifugation at 10 000 ×g for 5 minutes, supernatant was used to take absorbance at 670 nm. The H2S concentration was calculated against a calibration curve of NaHS.
2.13 |. Transmission electron microscopy (TEM).
Transmission Electron Microscopy was performed according to the protocol, as described previously. Briefly, HUVECs were loaded into a 300 Cu mesh carbon-coated formvar grids and were then visualized under TEM. Cells were treated with siCtrl, siCBS, or siMFN2 for 48 hours in complete media (CM) or CM + GYY4137 (1 mM/24 hours). After the incubation, cells were washed with PBS, and the cell pellets were collected with a brief trypsinization after centrifugation at 14 000 ×g for 10 minutes, followed by the fixation with 4% paraformaldehyde (EM grade) and 2% glutaraldehyde (EM grade), in 0.1 M sodium cacodylate buffer overnight at 4°C. According to the previous described protocol, various treatments were performed before the preparation of the ultrathin sections. The prepared sections which were stained with lead citrate and uranyl acetate were viewed under a Hitachi H7600transmission electron microscope at 80 kV equipped with 2k × 2k AMT camera. Images were analyzed using Image J software (NIH) as previously described.48
2.14 |. Data analysis and statistics
All the experiments were repeated independently at least three times and in triplicate where applicable. Data are expressed as mean ± standard deviation (SD). Comparisons between two groups were evaluated using Student’s t-test with equal/unequal variances. For comparisons among multiple groups, we performed ANOVA. If the overall test was significant, we compared each treatment to control with Dunnett’s method for multiple comparisons. For densitometry analysis of immunoblots, Image J software (NIH) was used. P < .05 was considered statistically significant. All tests were two-sided.
3 |. RESULTS
3.1 |. CBS is essential for maintenance of mitochondrial function and cell longevity in ECs
Recently, we reported that silencing CBS in ovarian cancer cells reduced oxygen consumption rate and ATP production.13,49 Here we wanted to investigate whether the silencing of CBS has a similar phenotype in ECs. Oxygen consumption rate (OCR) of cells was measured as an index of the mitochondrial electron transport chain activity (Figure 1A). Compared with siCtrl, depletion of CBS by siRNA significantly decreased basal respiration from 1135.16 pmol/min/mg protein to 886.58 pmol/min/mg protein, respectively, FCCP-induced stimulation of oxygen consumption from 1824.70 pmol/min/mg protein to 1051.42 pmol/min/mg protein, respectively, and ATP production from 646.04 pmol/min/mg protein to 476.97 pmol/min/mg protein, respectively (Figure 1B–D). Furthermore, OCR/ECAR ratio (measure of OXPHOS activity) is also significantly decreased from 2.65 to 1.51 in CBS-silenced cells as compared to siCtrl (Figure 1E). This change is primarily attributed to a decrease in OCR as ECAR levels are unchanged upon CBS silencing (Supplementary Figure 1A). Moreover, the spare respiratory capacity (SRC)—the extra mitochondrial capacity available in a cell to produce energy under stress or nutrient deprived conditions, which is thought to be important for long-term cell survival and function50,51 is significantly decreased in CBS-silenced ECs from 1439.16 pmol/min/mg protein to 641.75 pmol/min/mg protein when compared to siCtrl (Figure 1F). We next measured the mitochondrial membrane potential a known measure of mitochondrial function. To do this, we used tetramethylrhodamine ethyl ester (TMRE), a positively charged fluorescent dye that readily accumulates in relatively negatively charged “active” mitochondria and does not accumulate in “inactive” mitochondria, which have a depolarized membrane. As shown in Figure 1G, mitochondrial membrane potential was significantly reduced in CBS-silenced cells as compared to siCtrl, although the extent of mitochondrial depolarization was not as high as FCCP. Together, these results indicate that CBS is important for mitochondrial function and EC resilience to stress. Moreover, CBS silencing also leads to increased expression of cleaved caspase-3 and Bax as compared to control, this further supports the notion that CBS is required for endothelial cell longevity (Supplementary Figure 1B). These results support our observation of mitochondrial dysfunction upon CBS silencing and associated loss of cell longevity.
FIGURE 1.
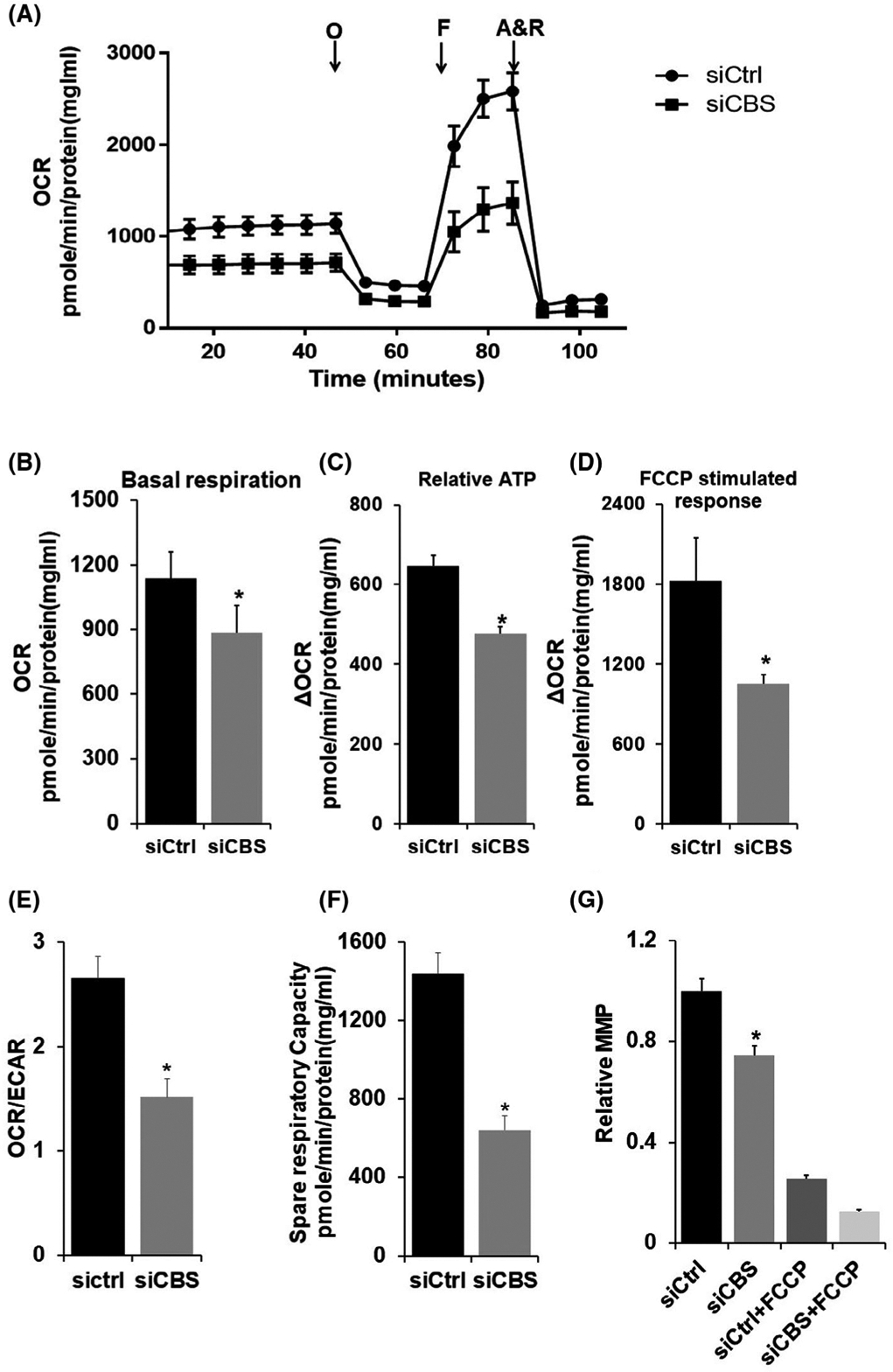
CBS regulates ECs bioenergetics and longevity. A, HUVECs (3.5 × 104) were transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against CBS (siCBS) and the OCR was measured using the Seahorse XF-96 analyzer. B, Basal respiration (before oligomycin), C, relative ATP estimation [change in OCR (Δ OCR) after oligomycin treatment, D, FCCP-stimulated response (maximal respiratory capacity)), E, OCR/ECAR, bar graph represents ratios of O2 consumption rates (OCR, indicator of OXPHOS) to extracellular acidification rates (ECAR, indicator of aerobic glycolysis) at baseline and F, Spare respiratory capacity (SRC) (difference between Basal and Maximum Respiration), derived from the XF trace of three independent experiments are represented as means ± SEM. *P ≥ .05. G, Mitochondrial membrane potential (MMP) was determined using TMRE. Fluorescence was measured in HUVECs transfected with siCtrl or siCBS or treated with 2 μM FCCP, and values are represented in relative MMP (fold change). Experiments were performed three times in triplicate, and values are represented as mean fold change ± SD. *P ≤ .05 is considered statistically significant
3.2 |. Loss of CBS decreases elongated mitochondrial morphology in ECs
Loss of mitochondrial membrane potential (ΔΨm) is reported to trigger dramatic structural changes in mitochondria from a tubular to globular shape, referred to as mitochondrial fragmentation; the resulting globular mitochondria are called swelled or ring/doughnut mitochondria.52 Moreover, we have previously demonstrated a role of CBS in maintaining mitochondria fusion in ovarian cancer cells.13 Therefore, we next examined whether CBS silencing had any effect on mitochondrial morphology alongside mitochondrial function in ECs. To visualize alterations in mitochondrial morphology, we first labeled mitochondria of both siCtrl and siCBS ECs by employing immunofluorescence post-staining of mitochondria with Mitotracker Red CMXRos (Figure 2A). Interestingly, the majority of mitochondria observed in siCtrl were fused, long, filamentous, spaghetti-like structures; whereas CBS-silenced cells showed mainly spherical, donut-shaped mitochondrial morphology. Furthermore, quantification of the changes in mitochondrial morphology rendered upon CBS silencing showed that knockdown cells had significantly decreased unbranched “individual mitochondria” count (n = 26, mean = 50.92 ± 3.45, P = .0417,) compared to siCtrl cells (n = 30, mean = 61.4 ± 3.595) (Figure 2B). Mean network size—defined as number of branches in each mitochondria network in siCBS (n = 29, 6.668 ± 0.6753) was also significantly reduced as compared to siCtrl (n = 33, 14.09 ± 1.671, P = .0002) (Figure 2C). Moreover, mean branch length was also significantly reduced in CBS-silenced ECs (n = 30, 0.5937 ± 0.02975) as compared to siCtrl group (n = 33, 0.7767 ± 0.02225, P < .0001) (Figure 2D). “Individual” mitochondria are defined as mitochondria which display no branching segments, whereas “mitochondria networks” are those mitochondria with at least one branching segment. Although we did notice a difference in individual mitochondria count, one drawback to this analysis method that it counts any mitochondria skeleton with one branching segment (regardless of how small or how few branches) as a “network.” Moreover, donut-shaped, punctated mitochondria can sometimes be skeletonized having a branching segment. This can make it difficult, and therefore inaccurate, to see morphology changes between “individual” mitochondria vs. mitochondrial “networks.” For these reasons, we will only report mitochondria network size and branch length, which have determined to be more accurate readout of morphological changes. Mitochondria network size is determined by the number of branches a network has and mean branch length is the average length of each segment in a network. Taken together, the above results indicate that CBS knockdown induces mitochondria fragmentation, therefore, leading overall smaller networks with shorter branches.
FIGURE 2.
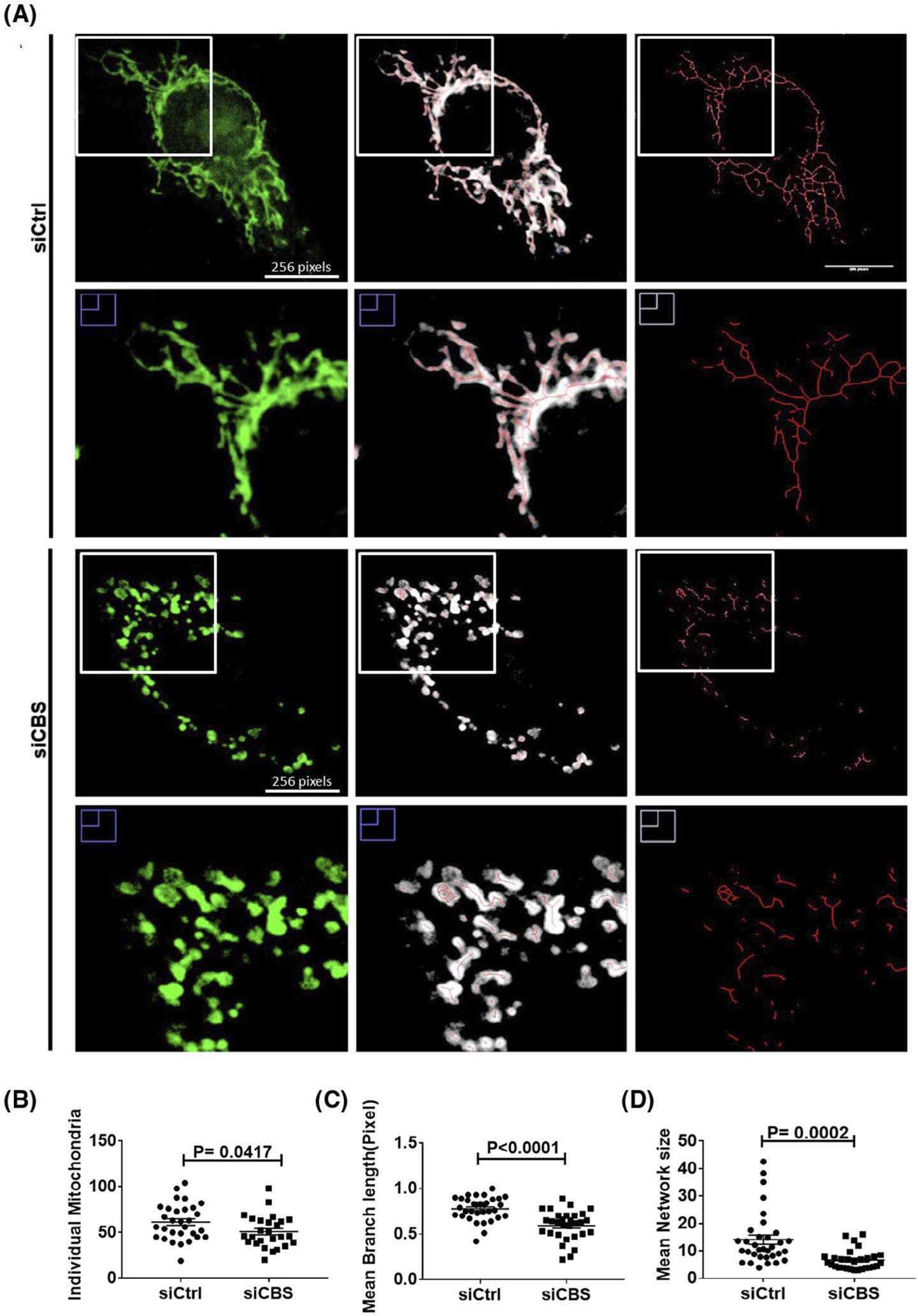
CBS silencing leads to enhanced mitochondrial fission in ECs. A, Mitochondria morphology was determined by Mitotracker Green staining and confocal imaging. The mitochondria channel (green) was isolated and skeletonized for morphology analysis. Images were analyzed using a publicly available ImageJ macro designed by Valente et al.45 B-D, Individual mitochondria are defined as unbranching segments, whereas mitochondria networks are defined as segments with at least one branch, regardless of length. Mitochondria network size is defined as the average number of branches per network. Mean branch length is the average length (in pixels) of all the branches in one network. A Student’s t test (α = 0.05) was used to compare CBS knockdown siCBS (n = 31 cells) with control siCtrl (n = 32 cells) in HUVECs. ~10 cells per treatment group per repeat were consolidated and shown here. All experiments were performed three times and values are represented as means ± SEM. P ≤ .05 is considered statistically significant
3.3 |. CBS silencing downregulates MFN2 expression, thereby resulting in mitochondrial fission
Since CBS silencing causes mitochondrial fission or fragmentation, we next examined whether CBS silencing affected mitochondrial fusion/fission machineries, thereby resulting in altered mitochondrial dynamics. Strikingly, silencing CBS in endothelial cells, HUVECs and HAOECs, significantly reduced mitochondrial outer membrane fusion protein, MFN2, levels as compared to the control group (Figure 3A,B; Supplementary Figure 2A,B). Notably, silencing CBS did not alter MFN2 mRNA levels (Supplementary Figure 2C). Similar to HUVEC, as shown in Figure 2, silencing CBS in HAOEC cells also significantly induced a fragmented mitochondrial morphology, as indicated by a decrease in mitochondrial network size and branch length (Figure 3C,D). Previously, we reported that CBS is the predominant H2S-producing enzyme in ECs and that silencing CBS affects key endothelial phenotypes including cell proliferation.1 Here we further validated our previous findings that upon CBS silencing in both HUVEC and HAOEC, CBS expression was significantly reduced but CSE and 3MST expression remained unaltered (Supplementary Figure 3A,D). Moreover, compared to control, a significant decrease in cellular H2S levels was observed in both cell lines (~50% in HUVEC and ~62% in HAOEC) (Supplementary Figure 3B,E). These results confirm that CBS in the predominant H2S-producing enzyme in ECs. Importantly, in both HUVEC and HAOEC cells there was no apparent change in the expressions of other mitochondrial fusion (MFN1 and OPA1) or fission (DRP1 and FIS1) proteins in CBS-silenced cells as compared to control. (Supplementary Figure 3C,F). Since the effect of CBS silencing was similar in both HUVEC and HAOEC cell lines, we further continued our experiments with only HUVEC cell line. Next, we confirmed the role of MFN2 in mitochondrial dynamics using small interfering RNA (siRNA) approach. We silenced MFN2 in ECs and examined the mitochondrial morphology after post-staining of mitochondria with Mitotracker Red CMXRos. As expected, knockdown of MFN2 in ECs led to enhanced mitochondrial fission compared to control group (Figure 3E–G). To further verify our finding, we re-expressed MFN2 in CBS-silenced ECs (Figure 3H,I) and examined the mitochondrial morphology after post-staining of mitochondria with Mitotracker Red CMXRos. Interestingly, as shown in Figure 3J,K, re-expression of MFN2 in CBS-silenced ECs restored the branching mitochondrial morphology phenotype. Taken together, the above results suggest that CBS maintains mitochondrial health and that loss of CBS induces mitochondria fragmentation through specific downregulation of MFN2.
FIGURE 3.
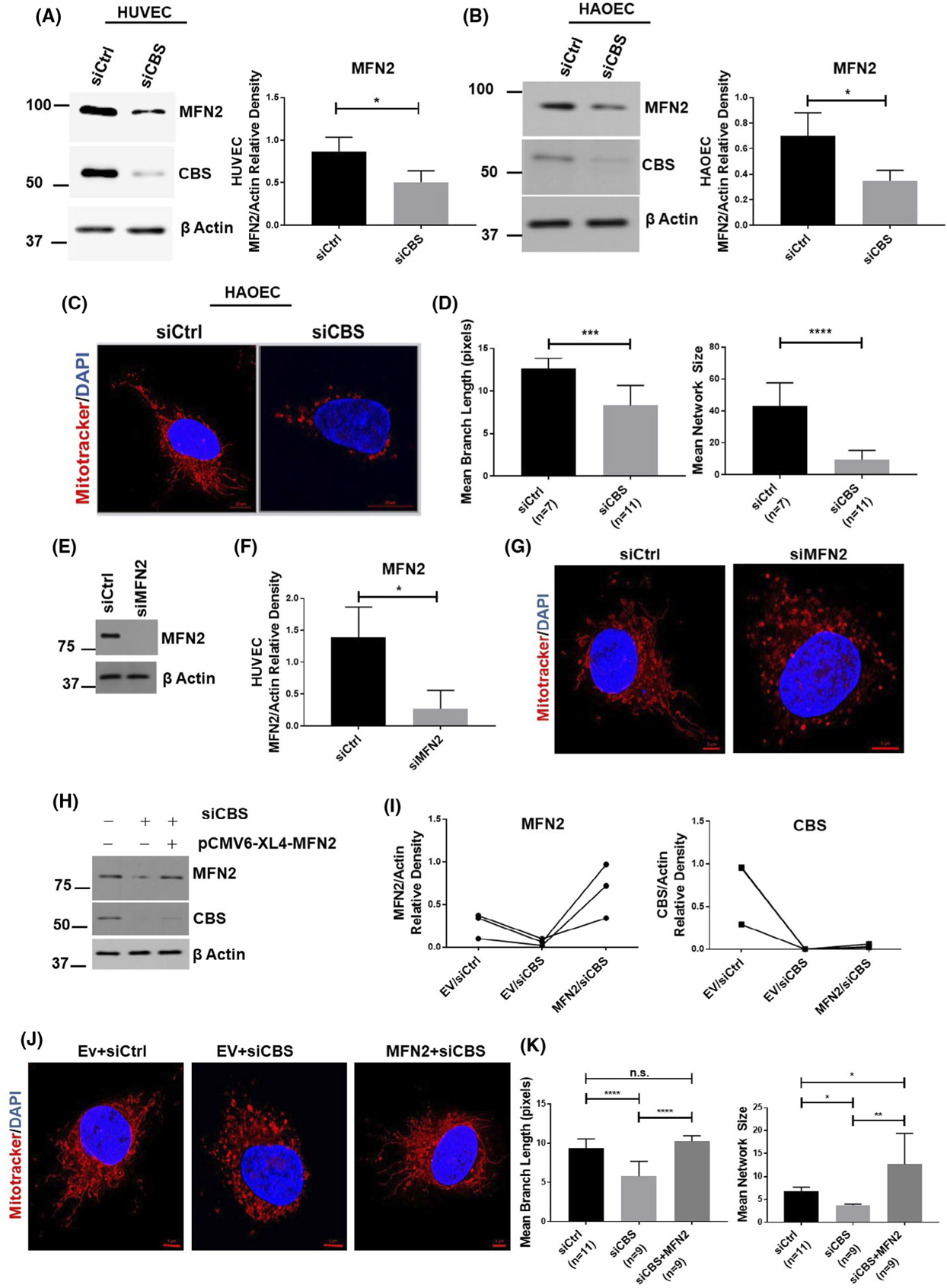
CBS silencing downregulates MFN2 expression. Expression of mitofusin 2 (MFN2) as determined by immunoblotting and densiometric analysis in HUVECs (n = 3) A, and HAOEC cells (n = 3) B,, either transfected with scrambled siRNA (siCtrl) or siRNA against the CBS gene. Efficient gene silencing was determined by immunoblotting with CBS antibody and actin served as a loading control. C, Mitochondrial morphology was depicted by immunofluorescence of siCtrl and siCBS in HAOEC cells, stained with Mitotracker Red CMXRos and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. D, Mitochondria network size is defined as the average number of branches per network. Mean branch length is the average length (in pixels) of all the branches in one network. A Student’s t test (α = 0.05) was used to compare CBS knockdown siCBS (n = 11 cells) with control siCtrl (n = 7 cells) in HUVECs. Representative immunoblot E, and densiometric analysis (n = 3) F, of HUVECs transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against MFN2 (siMFN2) with Actin as the loading control. G, Mitochondrial morphology was depicted by immunofluorescence of siCtrl and siMFN2 cells stained with Mitotracker Red CMXRos and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. H, Expression levels and I, densiometric analysis of MFN2 as determined by immunoblot. Efficient gene silencing was determined by immunoblotting with CBS antibody and actin served as a loading control. J, Mitochondrial morphology was depicted by immunofluorescence of Ev/siCtrl (n = 11 cells), Ev/siCBS (n = 9 cells) and siCBS/MFN2 (n = 9 cells) stained with Mitotracker Red CMXRos and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. K, Mitochondria morphology analysis of J. All immunoblot experiments were performed three times in triplicate and values are represented as means ± SEM. P ≤ .05 is considered statistically significant. Confocal imaging experiments were repeated and data from one set is shown here as means± SEM. P ≤ .05 is considered statistically significant
3.4 |. CBS silencing induces receptor-mediated mitophagy in ECs
We further investigated whether CBS silencing can induce mitophagy or affect mitochondrial biogenesis. Selective mitochondrial autophagy, or mitophagy, is a phenomenon that has recently been characterized and is believed to play a role in the removal of damaged and dysfunctional mitochondria.53,54 We first probed autophagy markers (p62, Beclin, ATG7, ATG5, and LC3 isoforms). Levels of two forms of microtubule-associated protein 1 light chain 3 (LC3) were measured. LC3-I is a cytosolic protein that undergoes multistep processing to become LC3-II, which is incorporated into autophagosome membranes. LC3-I is localized exclusively to the cytosol, whereas LC3-II is tightly bound to the autophagosomal membrane and correlated with autophagic activity.55,56 As shown in Figure 4A–F, CBS silencing significantly increases the LC3B cleavage, however, other autophagy markers remain unaltered. Enhanced LC3-II puncta can also be observed when CBS-silenced ECs were stained with LC3-II antibody as compared to control (Figure 4G,H). Next, we investigated whether this enhanced autophagy is accompanied by mitophagy in CBS-silenced ECs. As shown in Figure 5A,B, there was a significant increase in BNIP3 expression in CBS-silenced ECs, however, there was no significant alteration in BNIP3 mRNA levels, although levels consistently trended upwards (Figure 5C). To further confirm the above finding, we stained both control and CBS-silenced ECs with a BNIP3 antibody. Immunofluorescence images showed a significant increase in BNIP3 staining and localization to the mitochondria in CBS-silenced ECs as compared to the control group (Figure 5D,F).
FIGURE 4.
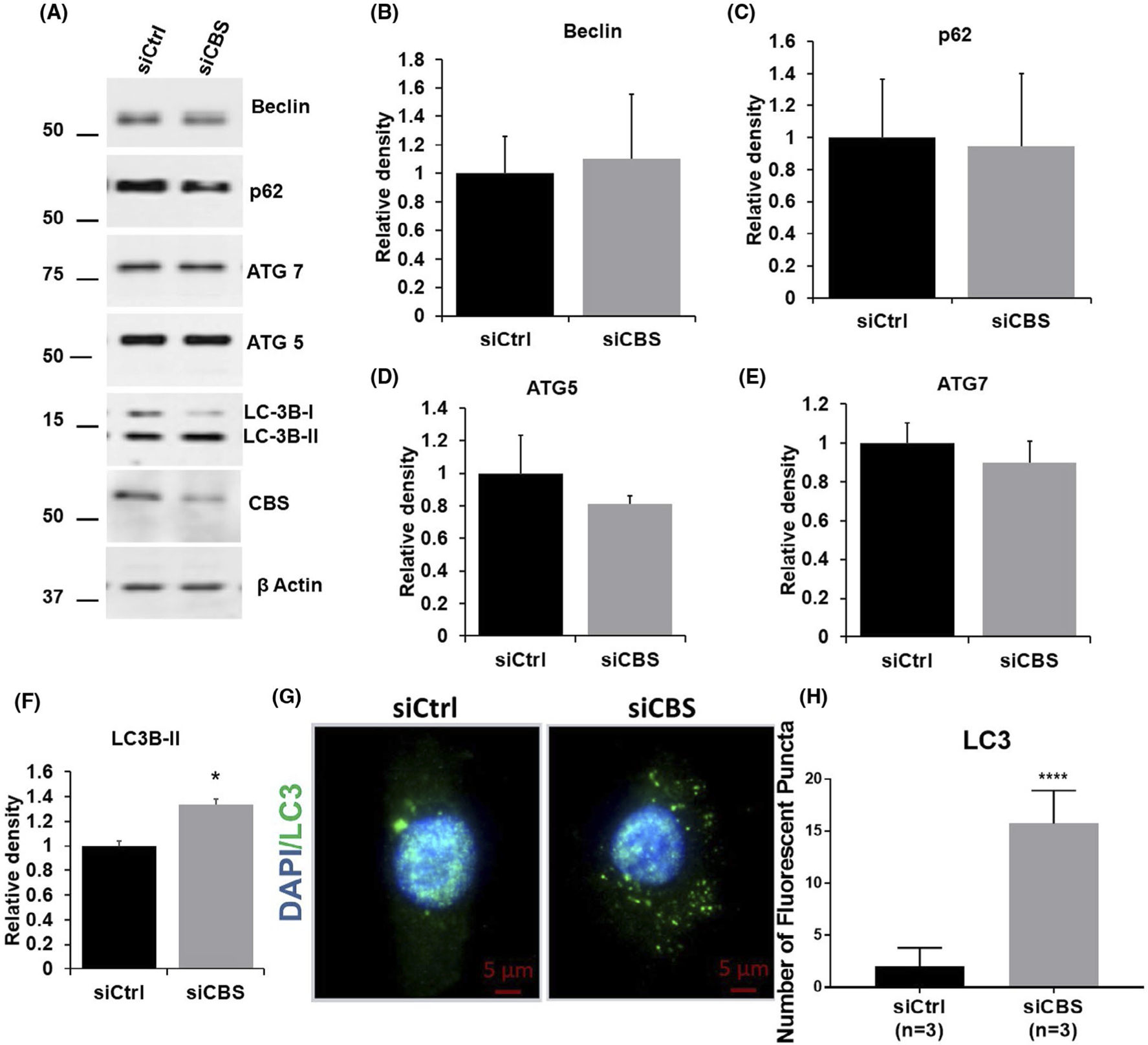
CBS silencing induces autophagy in ECs A, Expression of autophagy markers, beclin, p62, ATG5, ATG7, and LC3 as determined by immunoblotting in HUVECs either transfected with scrambled siRNA (siCtrl) or siRNA against the CBS gene. Efficient gene silencing was determined by immunoblotting with CBS antibody and actin served as a loading control. B-F, are relative density (n = 3) of beclin, p62, ATG5, ATG7, and LC3, respectively. HUVECs were transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against CBS (siCBS), G, Immunofluorescence and H, analysis (n = 3) of HUVECs immunostained with anti-LC3-II antibody and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. All experiments were performed three times in triplicate and values are represented as means ± SEM. For immunofluorescent imaging, n is defined as the average of one experimental repeat. P ≤ .05 is considered statistically significant
FIGURE 5.
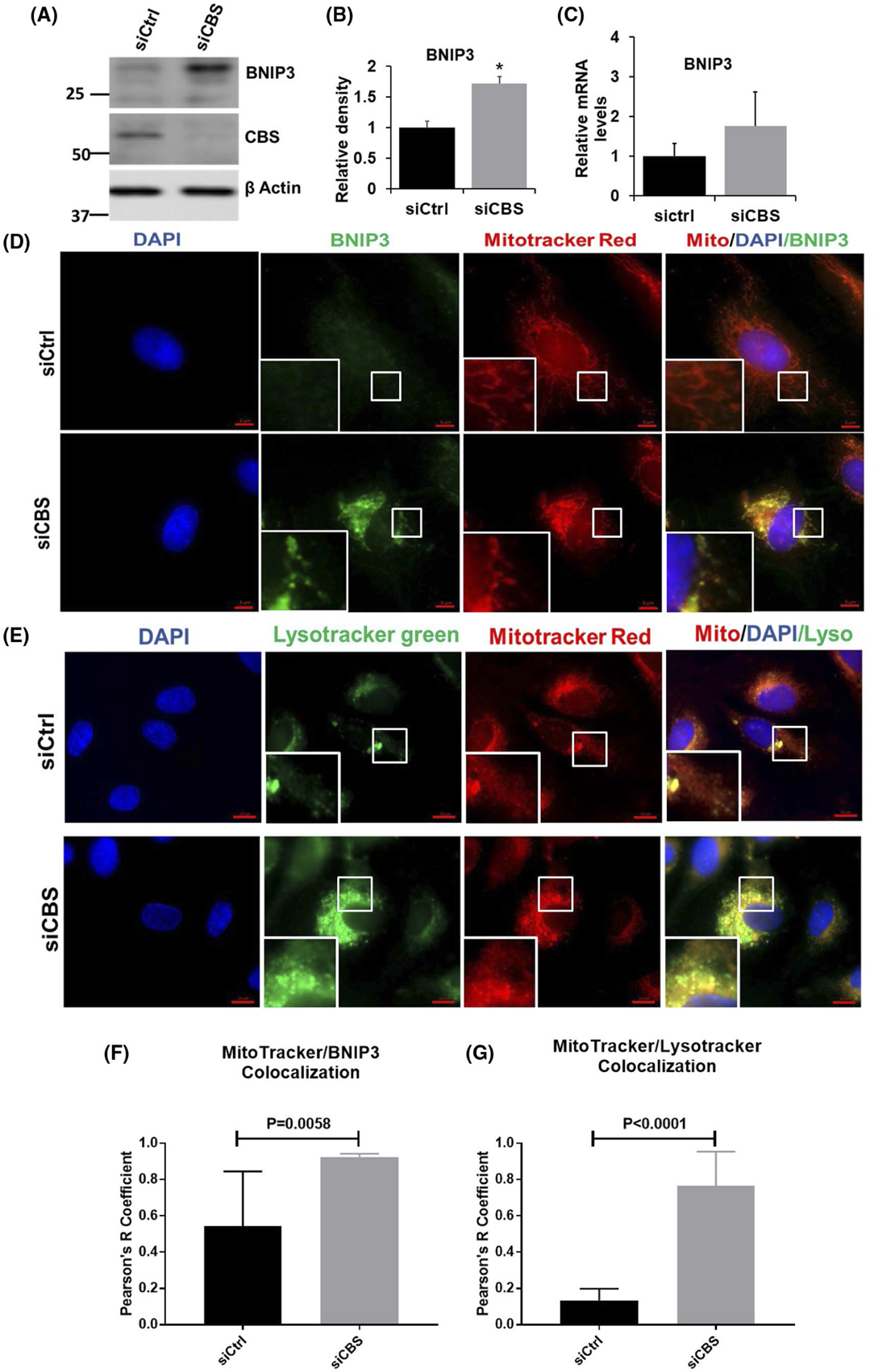
Autophagy in CBS-silenced endothelial cells is accompanied by mitophagy. A, Expression of BNIP3 as determined by immunoblotting in HUVECs either transfected with scrambled siRNA (siCtrl) or siRNA against the CBS gene. Efficient gene silencing was determined by immunoblotting with CBS antibody and actin served as a loading control. B, relative density of BNIP3 (n = 3). C, Relative mRNA level of BNIP3 (n = 3). HUVECs were transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against CBS (siCBS), D, Immunofluorescence and F, colocalization analysis (n = 3) of HUVECs labeled with Mitotracker Red CMXRos followed by immunostaining with anti-BNIP3 antibody and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. E, Immunofluorescence and G, colocalization analysis (n = 3) of HUVECs labeled with Mitotracker Red CMXRos and lysotracker green and counterstained with DAPI (nucleus; blue). Original scale bars, 5 μm. All experiments were performed three times in triplicate and values are represented as means ± SEM. For immunofluorescent imaging, n is defined as the average of one experimental repeat P ≤ .05 is considered statistically significant
To further confirm our observation, we next, resorted to subcellular fractionation to isolate mitochondria from control and CBS-silenced ECs. Purity of mitochondrial and cytosolic fraction was confirmed by immunoblotting against VDAC and LDH, respectively. Immunoblotting against BNIP3 confirmed enhanced expression of BNIP3 localized to the mitochondria in CBS-silenced ECs as compared to control (Supplementary Figure 4A). Next, to rule out the role of Parkin-dependent mitophagy, we probed mitochondrial and cytosolic fractions from CBS-silenced and control ECs to Parkin antibody. There was no apparent difference in the translocation of parkin to the mitochondrial fraction, which further confirmed the Parkin-independent and receptor-mediated mitophagy induced by CBS silencing in ECs (Supplementary Figure 4A). Next, we wanted to investigate whether CBS silencing can affect mitochondrial biogenesis. For this, we first performed immunoblot analysis of NADH-ubiquinone oxidoreductase chain 1(ND1) in cytosolic and mitochondrial fractions from control and CBS-silenced ECs. As shown in Supplementary Figure 3A, there was no significant difference in the expression level of ND1 between control and CBS-silenced ECs. These results confirm that CBS does not affect mitochondrial biogenesis in ECs.
Since the induction of the canonical autophagy pathway requires ATG proteins and mTOR suppression, which are mediated by enhanced ROS production and AMPK activation (that are induced by mitochondria damage and ATP depletion, respectively39), we next investigated the expression of these autophagy regulators by immunoblot. As shown in Supplementary Figure 4B, there is no significant difference in the expression level of phospho-mTOR, mTOR, phospho-p70S6kinase, p70S6kinase, phospho-AMPKα, and AMPKα. These results further confirm BNIP3-mediated mitophagy in CBS-silenced ECs.
Next, to determine whether the observed CBS silencing-induced increases in autophagy were mitochondria-specific autophagy (mitophagy), we used Mitotracker Red and Lysotracker Green to label mitochondria and autophagosomes/lysosomes, respectively, and observe their colocalization by fluorescence microscopy. As the mitochondria are engulfed by autophagosomes/lysosomes during mitophagy, the red fluorescence from Mitotracker and the green fluorescence from lysotracker overlap, producing a yellow color. As shown in Figure 5E,G, colocalization of mitochondria and autophagosomes/lysosomes was significantly increased in CBS-silenced ECs as compared to control ECs. These results further confirm that CBS depletion-induced autophagy is mitochondrial specific, that is, mitophagy.
3.5 |. CBS metabolites can rescue MFN2 levels and enhanced mitophagy
Cystathionine beta synthase enzymatic function leads to synthesis of cystathionine and concomitant release of the gasotransmitter, H2S.49 To determine whether it is the enzymatic function of CBS or the CBS protein itself that is essential in regulating MFN2 expression, we performed rescue experiments by supplementing CBS-silenced cells with well-established H2S donors, GYY4137 (Supplementary Figure 5A) or NaHS. As shown in Figure 6A, treatment with both H2S donors, GYY4137 or NaHS, completely restores MFN2 expression in CBS-silenced cells. Therefore, this suggests that metabolites from the CBS enzymatic pathway are involved in regulating MFN2 expression.
FIGURE 6.
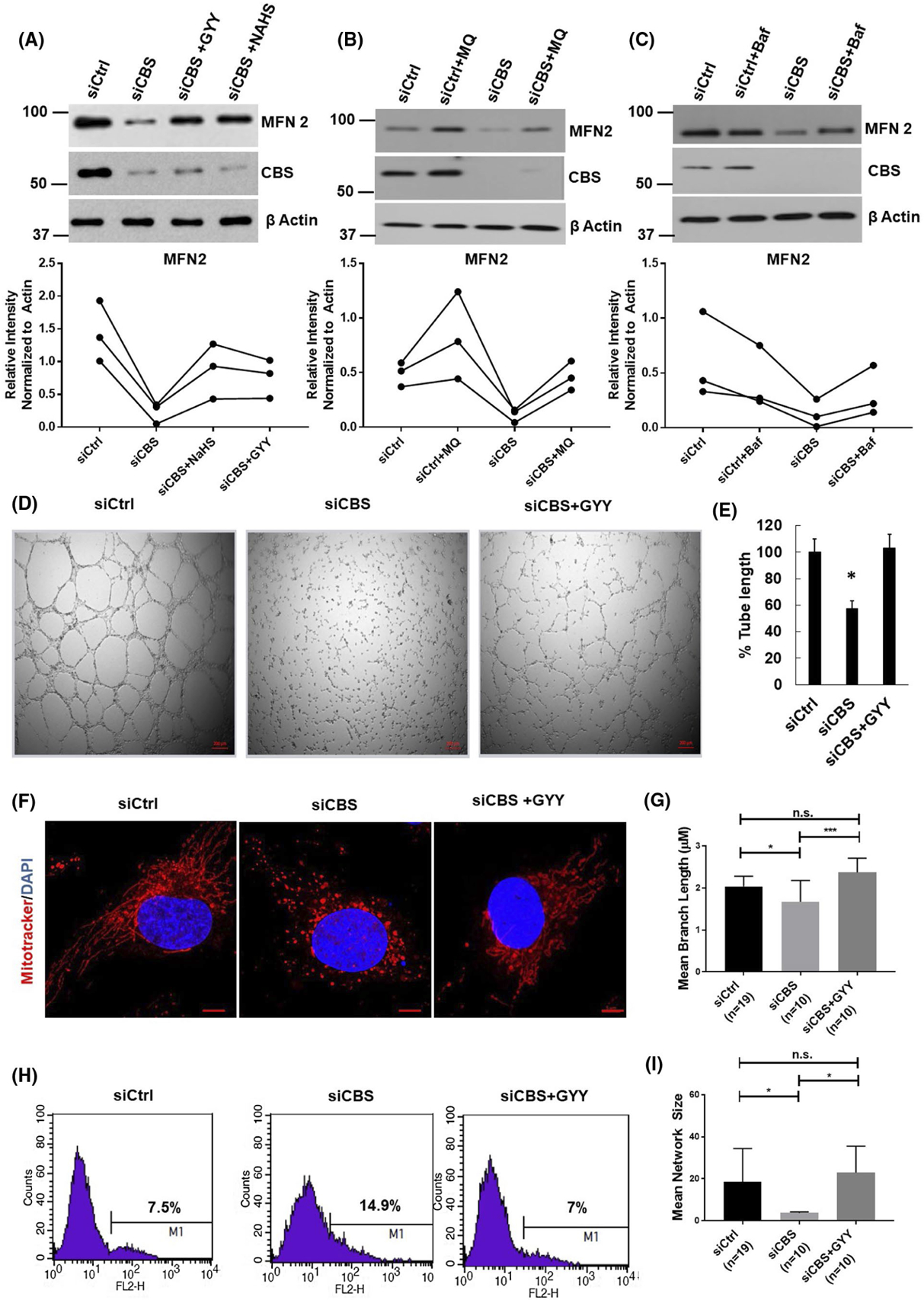
CBS metabolites can rescue MFN2 levels and enhanced mitophagy. A, HUVECs transfected for 48hrs with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and either treated with the H2S donor GYY4137 (GYY) (1 mM/24 hours) or NAHS(2 mM/24 hours) and immunoprobed for MFN2 and CBS protein expression. Representative immunoblot and densiometric analysis provided. All experiments were performed in triplicate and data points from each replicate are connected linearly. B, HUVECs transfected for 48hrs with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and treated with Mitoquinone (MQ) 100 nmol/L/24 hours and immunoprobed for MFN2 and CBS protein expression. Representative immunoblot and densiometric analysis provided. All experiments were performed in triplicate and data points from each replicate are connected linearly. C, HUVEC s transfected for 48hrs with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and treated with Baflomycin (Baf) 50 nM/48 hours and immunoprobed for MFN2 and CBS protein expression. Representative immunoblot and densiometric analysis provided. All experiments were performed in triplicate and data points from each replicate are connected linearly. D, Representative images and E, analysis of tube formation assay of HUVECs transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and treated with the H2S donor GYY4137 (GYY) (1 mM/24 hours) on 2 mg/mL of growth factor-reduced Matrigel. The images were acquired 6 hours after plating HUVECs on Matrigel in complete EBM medium. F, Representative images and G & I, and morphology analysis of HUVECs treated with siCtrl (n = 19 cells) or siCBS (n = 10 cells) siRNA and H2S donor GYY4137 for 24 hours (n = 10 cells). Mitochondrial morphology was depicted by immunofluorescence stained with Mitotracker Red CMXRos and counterstained with DAPI (nucleus; blue). Morphology was analyzed as previously described. Mitochondria network size is defined as the average number of branches per network. Mean branch length is the average length (in pixels) of all the branches in one network Original scale bars, 5 μm. Confocal imaging experiments were repeated and data from one set is shown here as mean± SEM. P ≤ .05 is considered statistically significant H, Analysis of mitochondria oxidative stress. HUVECs transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and treated with the H2S donor GYY4137 (GYY) (1 mM/24 hours) followed by MitoSOX staining and analyzed by a FACS Calibur flow cytometer. A representative histogram depicting mean fluorescence intensity from three independent replicates is shown
We next investigated whether the functional phenotypes in ECs can be rescued by supplementation with GYY4137. As shown in Figure 6D,E, GYY4137 treatment significantly rescues endothelial tube formation on Matrigel in CBS-silenced ECs. We further looked into the mitochondrial morphology after GYY4137 supplementation, as shown in Figure 6F, GYY4137 supplementation was sufficient to restore mitochondrial morphology in CBS-silenced ECs. This is indicated by a rescue of mitochondria network phenotypes upon GYY4137 treatment (Figure 6G,I). These data indicate that significant loss of key endothelial phenotypes by CBS silencing can also be rescued by CBS metabolites.
Since CBS silencing has been known to generate ROS in ECs,1,13 we next investigated the effect of GYY4137 supplementation on ROS generation in CBS-silenced and control cells. As shown in Figure 6H, treatment of slow-releasing H2S donor GYY4137 attenuated mitochondrial oxidative stress caused by knockdown of CBS in ECs. To assess the importance of mitochondrial ROS (mtROS) in CBS silencing-mediated MFN2 downregulation, we treated cells with mtROS scavenger mitoquinone (MQ). MQ treatment successfully quenched ROS in siCBS cells (Supplementary Figure 5B), which lead to increased MFN2 expression in siCtrl cells and fully rescued back MFN2 expression in CBS-silenced ECs (Figure 6B). These results suggest that silencing CBS expression generates mtROS in ECs, likely due to mitochondrial membrane depolarization.
We next clarified whether inhibition of autophagy could rescue MFN2 levels. As shown in Figure 6C, treatment with bafilomycin A1 (Baf) significantly restored MFN2 levels which is comparable to the control group. Together, these results confirm that CBS is important for regulation of MFN2 expression and mitochondrial functions in ECs.
Recently, we have demonstrated that CBS silencing in ovarian cancer leads to MFN2 downregulation by selectively regulating its stability. Silencing CBS creates oxidative stress that activates JNK which in turn phosphorylates MFN2 to recruit the E3 ligase, HUWE1, for its degradation via the ubiquitin-proteasome system. To investigate whether the MFN2 downregulation pathways coincides in ECs and ovarian cancer cells, we first examined the BNIP3 expression levels in control and CBS-silenced CP20 cells. As shown in Supplementary Figure 6A, there is no significant difference in BNIP3 protein levels in control and CBS-silenced ovarian cancer cell CP20, however, there is significant decrease in MFN2 protein levels between the two groups. To further confirm our finding, we next treated the control and CBS-silenced ECs with proteasome inhibitor, MG132. As shown in Supplementary Figure 6B, MG132 treatment was not able to rescue the MFN2 levels in CBS-silenced ECs. These results confirm that MFN2 downregulation in ovarian cancer cells and ECs follow a distinct pathway.
3.6 |. CBS silencing disrupts mitochondria-ER contact
Recently there has been increasing interest in organelle crosstalk—specifically ER-mitochondria coupling, which plays critical roles in Ca2+ homeostasis, metabolism, autophagy, and cell death.57,58 Interestingly, MFN2, which is also present on the ER membrane, has been shown to form a homodimer-linked tether between the ER and mitochondria.48,59 Furthermore, ER-mitochondria tethering has been shown to suppress mitophagy.60 Therefore, we next examined ER-mitochondria coupling following CBS silencing by utilizing a transmission electron microscopy (TEM) approach. We then measured the ER-mitochondria interface length, organelle distance, and mitochondria perimeter to determine the ER-mitochondria contact coefficient (Figure 7A). Analysis of contact coefficients are valuable readouts as they correlate well with various kinds of ER-mitochondria tethering, which we have defined in Supplementary Figure 7A. Interestingly, ER-mitochondria contacts are completely ablated in siCBS-treated cells when compared to siCtrl (Figure 7B,C, and Supplementary Figure 8), which is additionally phenocopied upon MFN2 silencing (Supplementary Figure 7B). Surprisingly, treatment with H2S donor, GYY4137, does not reestablish these contacts (Figure 7B,C), although mitochondria fusion is rescued (Figure 7B,D). These results indicate that CBS-dependent loss of MFN2 initiates mitophagy through disruption of ER-mitochondria contacts.
FIGURE 7.
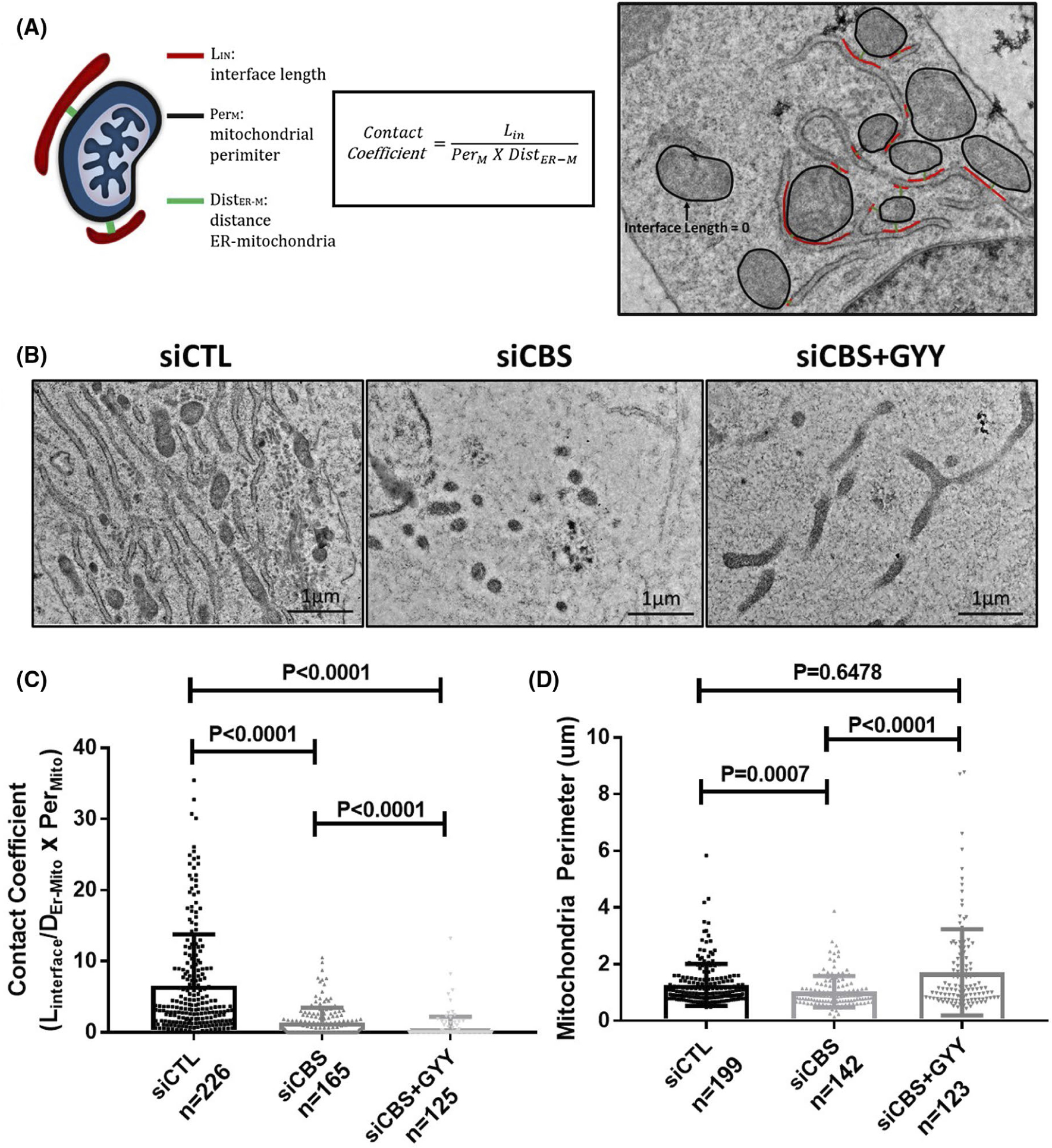
CBS silencing disrupts mitochondria-ER contacts. A, Schematic of ER-mitochondria contact coefficient analysis method and example image of ER-mitochondria contact coefficient analysis. Representative images B, and analysis of ER-mitochondria contact coefficients C, and mitochondria perimeter D, in HUVECs transfected for 48 hours with scrambled siRNA (siCtrl) or siRNA against the CBS gene (siCBS) and treated with the H2S donor GYY4137 (GYY) (1 mM/24 hours). N is defined as each dot and represents one ER-mitochondria contact. Original scale bars, 1 μm. Data are presented as means ± SD P ≤ .05 is considered statistically significant
4 |. DISCUSSION
In the present study, we have demonstrated for the first time that H2S-producing enzyme, CBS, is required for maintenance of mitochondrial function and dynamics in ECs. Our data suggest that alterations in mitochondrial structure can have profound consequences that impact EC function. We also demonstrate that CBS—through mediating MFN2 levels—regulates elongated mitochondrial morphology, which facilitates efficient OXPHOS, favorable redox balance, and ER-mitochondria coupling. Silencing CBS in ECs leads to decreased mitochondrial function, increased mitochondrial fission, and enhanced receptor-mediated mitophagy. Moreover, CBS silencing in ECs reduced the cell longevity and resilience, as elongated mitochondria help better the survival response to nutrient starvation and stress.20–22 Our findings go well with a recent report from Eva Albertini et al. whereby they linked CBS to vascular aging and disease, and demonstrated that CBS deficiency induces endothelial cell senescence, mitochondrial dysfunction, and increased susceptibility of the cells to homocysteine.44
We originally hypothesized that ROS generation and decreased mitochondrial membrane potential were the main culprits for CBS depletion-induced alterations in the EC phenotype. This is because ROS is known to oxidize thiols, lipids, proteins, and nucleic acids, consequently causing oxidative injury to various cellular structures, including the mitochondria.61 There are previous studies that point to the existence of mitochondrial dysfunction in CBS depleted endothelial cells.44 Moreover, previously we have reported that enhanced ROS mediates endothelial cell cytotoxicity and alterations in endothelial cell phenotype in CBS-silenced ECs.1 Interestingly, here we found that silencing of CBS in ECs alters mitochondrial function and dynamics, which is likely a consequence of enhanced ROS, decreased mitochondrial membrane potential, and loss of ER-mitochondria tethering, that altogether culminates in the induction of mitophagy and EC dysfunction (Figure 8).
FIGURE 8.
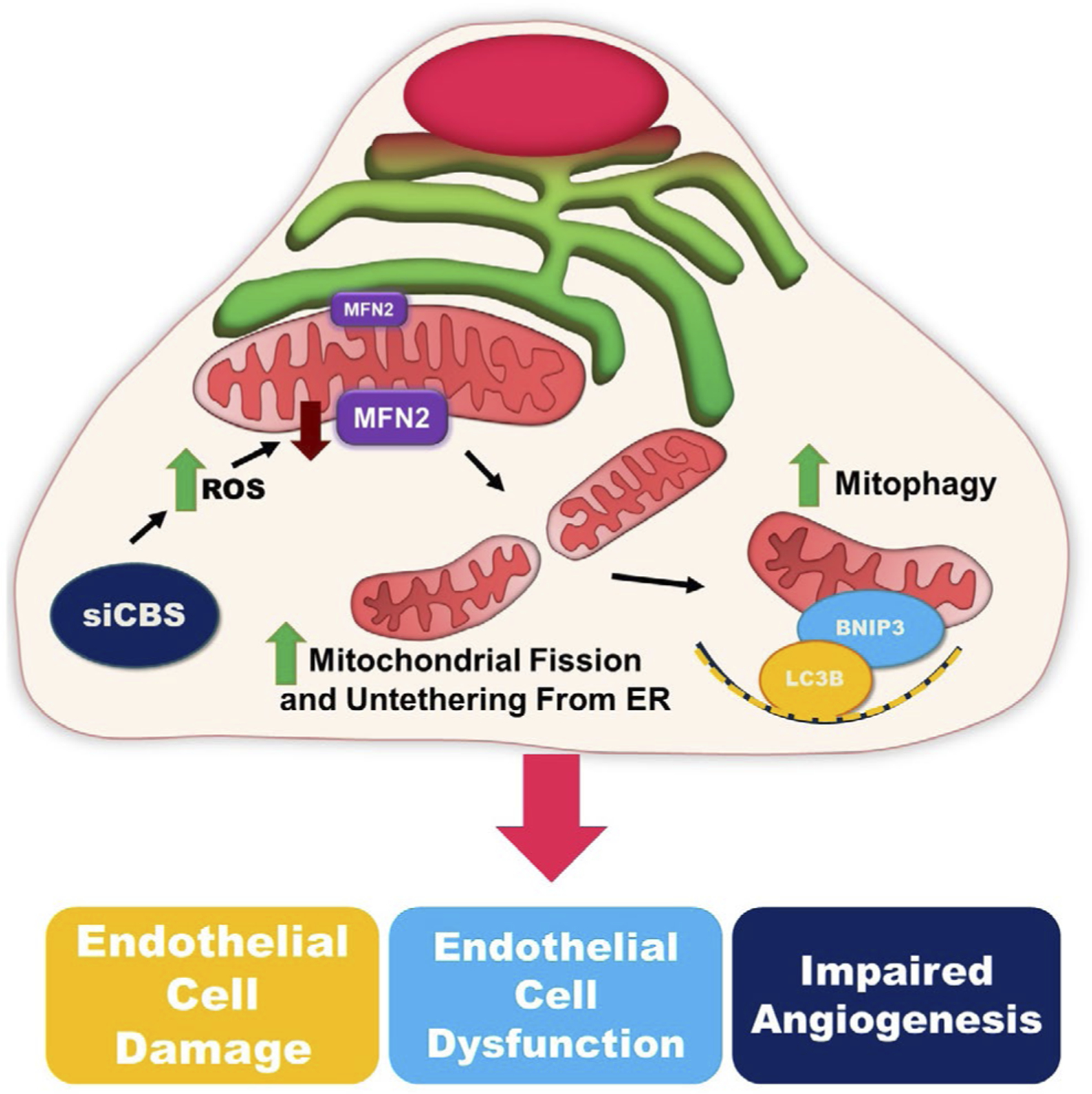
Graphical Abstract—mechanism of CBS-dependent endothelial cell regulation. Silencing CBS in endothelial cells induces ROS accumulation and downregulates MFN2 expression. Loss of MFN2 leads to mitochondria-ER decoupling and an increase in mitochondrial fission. BNIP3 and LC3B are recruited are recruited to the mitochondria where they drive mitophagy and lead to an increase in endothelial cell dysfunction and impaired angiogenesis
Mitochondria are essential hubs of metabolic activity and cell death,62 and participate in macroscopic behaviors or dynamics including fusion, fission, transport, and mitophagy. Although mitochondrial bioenergetics are molecularly distinct from mitochondrial dynamics, recent studies suggest that metabolism and dynamics are highly linked and regulate one another.17 Fusion of mitochondria into linear or tubular networks limits deleterious mutations in mitochondrial DNA (mtDNA),63 induces supercomplexes of the ETC maximizing OXPHOS activity.18,19 In addition, mitochondria elongate as a survival mechanism in response to nutrient starvation and stress, linking fusion to cell longevity.20–22 Mitochondrial fusion is mediated by mitofusin 1 and 2 (MFN1, MFN2), and optic atrophy 1 (OPA1).13 Total deletion in any of these proteins is embryonically lethal and mutations in the genes that encode them lead to several human diseases.16,64 Mitochondrial fission is mediated by dynamin-related protein 1 (DRP1), fission-related proteins like Fission 1 (FIS1), mitochondria fission factor (MFF), and MiD51 and MiD4913 and can generate discrete and fragmented mitochondria that can increase ROS production,23 facilitate mitophagy,24,25 and mediate apoptosis.26 There are several reports that suggest that mitochondrial membrane remodeling is highly responsive to changes in cell metabolism17,65 and deletion of any of the dynamics machinery disturbs OXPHOS and glycolysis.27
In general, ECs are mainly glycolytic given that 85% of ATP generated in ECs is through glycolysis.66 However, there are recent reports that proliferating ECs utilize OXPHOS for fatty acid oxidation (FAO) to sustain DNA synthesis,31 and lymphatic ECs use FAO for epigenetic regulation of lymphatic gene expression.32 Interestingly, it has recently been reported that quiescent ECs can also increase FAO, up to three- to four-fold31–34 and utilize FAO for redox homeostasis.35 Reports suggest that ROS promotes EC activation/dysfunction, thereby leading to increased vascular thrombogenicity, leakiness, and inflammation.67,68 Consistent with a role of CBS in EC redox homeostasis, silencing CBS leads to elevated ROS levels, increased mitochondrial fragmentation, and enhanced mitophagy. Notably, treatment of CBS-silenced ECs with H2S donor GYY4137 and mitochondria-targeted antioxidants, mitoquinone (MQ), could reverse the enhanced ROS production and rescue MFN2 levels and EC tube formation—one of the key EC function phenotypes. Recently, Joanna Kalucka et al35 reported that quiescent ECs require a higher level of redox buffering capacity than proliferating ECs, and that treatment of quiescent ECs with a mitochondrial antioxidant rescues vascular leakage, a key feature of EC dysfunction.
Cystathionine beta synthase is the predominant H2S-producing enzyme in ECs1 and the best-characterized benefit of H2S is the protection of cells against oxidative stress.69,70 H2S is a direct scavenger of ROS and peroxynitrite7 and exert cytoprotection by attenuating mitochondrial ROS production, increasing ATP synthesis, and preventing mitochondrial membrane depolarization in the cardiovascular and nervous system.11 Recent studies have demonstrated that CBS protein is present in mitochondrial fractions of rat livers,12 and ovarian cancer cells,13 and that CSE, another H2S-producing enzyme, can be translocated from the cytosol into the mitochondria in vascular smooth muscle cells.11 There are reports that suggest that H2S produced by mitochondrial CSE or CBS exerts protective effects on mitochondrial function.11,12 In the present study, we observe enhanced mitochondrial oxidative stress, which could be reversed by the H2S donor GYY4137 in CBS-silenced ECs. These results suggest that H2S, generated by CBS, may provide an antioxidant intracellular environment that contributes to the maintenance of normal mitochondrial function and dynamics in ECs. Such a balance is perturbed upon CBS silencing, thereby resulting in an increase in mitochondrial oxidative stress, a loss of mitochondrial membrane potential, altered mitochondrial dynamics, and enhanced mitophagy.
Autophagy is a genetically programmed, evolutionarily conserved catabolic process that degrades cellular proteins, and damaged or excessive organelles.39 General autophagy machinery comprises autophagosome formation which then fuse with lysosomes to form autophagolysosomes where the enveloped contents are degraded.38,71,72 More than 30 autophagy-related genes (ATG) have been identified in yeast and mammals.37,38 Most ATG proteins form multi-molecule complexes to regulate autophagosome formation, which include (1) the ULK1 kinase complex, (2) the Beclin 1-VPS34 class III phosphoinositide 3-kinase (PI3K) complex, (3) the ATG9-ATG2-ATG18 complex, and (4) the ATG5-ATG12-ATG16 and ATG8/LC3 conjugation systems.39 Till now, several organelle-specific autophagy processes have been reported, including specific removal of peroxisomes (pexophagy),73 endoplasmic reticulum (erphagy),74 ribosomes (ribophagy),75 lipid droplets (lipophagy),76 invading microbes (xenophagy),77 and protein aggregates.78 Mitophagy, is the selective autophagic degradation of damaged mitochondria or mitochondria producing excess ROS to prevent the accumulation of mtDNA mutations and additional cellular damage.54,79 It requires two steps: induction of general autophagy and priming of damaged mitochondria for selective auto-phagic recognition. Moreover, mitochondria must undergo fission to generate sufficiently small mitochondria as well as decouple from the ER to be acquired by the autophagosome22,60 ER-mitochondria contacts perform several roles functionally, including marking sites of mitochondria fission, modulating Ca2+ homeostasis and autophagosome generation, and balancing redox status to support EC function.29,58,80 As discussed previously, we have thoroughly demonstrated the role of CBS on mitochondria fission/fusion. Here we also demonstrate for the first time the role of CBS in maintaining ER-mitochondria contacts in ECs by maintaining MFN2 integrity and potentially stabilizing the MFN2 homodimer tether between the ER and mitochondria. Interestingly, treatment with GYY4137 does not rescue back these contacts despite its partial rescue of mitochondria fusion and MFN2 protein levels. This may be due to temporal constraints as all images were acquired 24hrs following GYY4137 treatment. Full contact rescue may take longer to be fully realized. Another possible explanation is that the CBS enzyme itself may be implicated in the tethering process between ER and mitochondria. Therefore, H2S supplementation would not be adequate to induce a full rescue. Such speculations, although thought-provoking, extend beyond the scope of this study and may be the focus of future work.
Mitochondrial priming is mediated either by the Pink1-Parkin signaling pathway or the mitophagic receptors, Nix and BNIP3.39 BNIP3 (Bcl-2/E1B-19 kDa interacting protein 3) contains a Bcl-2 homology 3 (BH3) domain and a carboxyl-terminal transmembrane domain, and acts as a pro-apoptotic mitochondrial protein.81,82 Nix/BNIP3L is a homolog of BNIP3, and they share 53–56% amino acid sequence identity.83 BNIP3 regulates mitophagy during hypoxia, whereas Nix is required for mitophagy during development of the erythroid lineage.84 BNIP3 exists in cells as both a monomeric (~28 kD) and a dimeric form (~60 kD). Notably, both forms of BNIP3 are sufficient to localize to mitochondria and induce cell death.83 BNIP3 expression is transcriptionally up-regulated by HIF-1α in hypoxic conditions.40 Upon expression, BNIP3 localizes to mitochondria, where it collapses mitochondrial membrane potential (ΔΨm), increases generation of ROS, induces mitochondrial swelling, promotes mitochondrial fission, and stimulates mitochondrial turnover via mitophagy.41,42 Reports in cardiac myocytes suggest that genetic knockdown of BNIP3 or loss of function mutations were shown sufficient for suppressing mitochondrial injury and hypoxia-induced cell death.85,86
BCL2 interacting protein 3 functions as a tether to stimulate activation of mitophagy, linking BNIP3 localized on damaged mitochondria to LC3 present on nascent autophagosomes.43 There are reports that phosphorylation of BNIP3 at S17 and S24, which flank the LC3-II interacting region (LIR, WVEL sequence at residues 18–21), promotes mitophagy through enhanced BNIP3-LC3-II interaction.87 BNIP3 is also known to increase the localization of DRP1 to the mitochondria, where it stimulates fragmentation of the mitochondrial network to promote the engulfment of damaged mitochondria.88 Here we present evidence that CBS silencing leads to enhanced expression of LC3 and BNIP3, and colocalization of BNIP3 and lysotracker green with mitochondria (labelled with Mitotracker Red) confirms mitochondria-specific autophagy, that is, mitophagy. Hence, this study identifies BNIP3 as a critical effector of mitophagy in CBS-silenced ECs. Notably, increased mitophagy in CSB silenced ECs seems not to be linked to mitochondrial biogenesis, however, it resulted in increased susceptibility to cell death and failure to survive in stress condition.
Taken together, our data highlight a novel signaling axis that mechanistically links CBS with mitochondrial function and dynamics. This property of CBS has not been previously reported. The present study provides the first evidence that CBS depletion induces mitochondrial fission by a mechanism that impinges upon MFN2 downregulation, ER-mitochondria decoupling, and BNIP3-induced mitophagy. Recently, mitochondrial OXPHOS has gained significant importance in endothelial cell function as reports suggest enhanced FAO in both proliferating as well as quiescent ECs for biomass generation and Redox homeostasis, respectively. In this respect, our finding linking CBS with mitochondrial function and dynamics in ECs is quite significant as loss of CBS function is associated with a wide variety of pathologic conditions in humans, including neurovascular and cardiovascular disorders and could be considered a new therapeutic approach for the intervention of EC dysfunction-related severity.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by National Institutes of Health Grant 1R01 CA220237-01A1, 2CA136494, CA213278 (to P.M.) and CA157481 (to R.B.) and 1R01HL120585 (to both PM and RB). We thank the Peggy and Charles Stephenson Cancer Center at the University of Oklahoma Health Sciences Center for a seed grant and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103639 for the use of Histology and Immunohistochemistry Core, which provided immunohistochemistry and image analysis service.
Funding information
National Institutes of Health, Grant/Award Number: 1R01 CA220237-01A1, 2CA136494, CA213278, CA157481 and 1R01HL120585
Abbreviations:
- AMPK
5′ AMP-activated protein kinase
- AOAA
aminooxyacetic acid
- ATG
autophagy genes
- ATP
adenosine triphosphate
- Baf
bafilomycin A1
- BNIP3
BCL2 interacting protein 3
- BSA
bovine serum albumin
- CBS
cystathionine beta synthase
- CSE
cystathionine gamma lyase
- DRP1
dynamin-related protein 1
- EC
endothelial cell
- ECAR
extracellular acidification rate
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FCCP
carbonyl cyanide p-trifluoro-methoxyphenyl hydrazine -mitochondrial uncoupler
- FIS1
fission 1
- HIF-1α
hypoxia inducible factor 1 alpha
- H2S
hydrogen sulfide
- JNK
c-Jun N-terminal kinase
- KATP
ATP-sensitive K+ channel
- LC3
MAP1
- LC3B
microtubule-associated proteins 1A/1B light chain 3B
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- MFF
mitochondria fission factor
- MFN2
mitofusin 2
- MQ
mitoquinone
- ND1
NADH-ubiquinone oxidoreductase chain 1
- OCR
oxygen consumption rate
- OPA1
optic atrophy 1
- OXPHOS
oxidative phosphorylation
- PINK1
PTEN-induced kinase 1
- ROS
reactive oxygen species
- SRC
spare respiratory capacity
- TMRE
tetramethylrhodamine ethyl ester
- VDAC
voltage-dependent anion channel (VDAC)/mitochondrial porin
- ΔΨm
mitochondrial membrane potential
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Saha S, Chakraborty PK, Xiong X, et al. Cystathionine beta-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016;30:441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- 3.Beard RS Jr, Bearden SE. Vascular complications of cystathionine beta-synthase deficiency: future directions for homocysteine-to-hydrogen sulfide research. Am J Physiol Heart Circ Physiol. 2011;300:H13–H26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yap S, Naughten ER, Wilcken B, Wilcken DE, Boers GH. Vascular complications of severe hyperhomocysteinemia in patients with homocystinuria due to cystathionine beta-synthase deficiency: effects of homocysteine-lowering therapy. Semin Thromb Hemost. 2000;26:335–340. [DOI] [PubMed] [Google Scholar]
- 5.Brattstrom L, Wilcken DE. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr. 2000;72:315–323. [DOI] [PubMed] [Google Scholar]
- 6.Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J. Cellular redox state and endothelial dysfunction in mildly hyper-homocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:34–41. [DOI] [PubMed] [Google Scholar]
- 7.Whiteman M, Armstrong JS, Chu SH, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. [DOI] [PubMed] [Google Scholar]
- 8.Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12:1065–1077. [DOI] [PubMed] [Google Scholar]
- 10.Papapetropoulos A, Pyriochou A, Altaany Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106:21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA. 2012;109:2943–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc Natl Acad Sci USA. 2013;110:12679–12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakraborty PK, Murphy B, Mustafi SB, et al. Cystathionine beta-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018;32:4145–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szabo C, Coletta C, Chao C, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy B, Bhattacharya R, Mukherjee P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J 2019;33(12):13098–13125. 10.1096/fj.201901304r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. [DOI] [PubMed] [Google Scholar]
- 17.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank M, Duvezin-Caubet S, Koob S, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. [DOI] [PubMed] [Google Scholar]
- 25.Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. [DOI] [PubMed] [Google Scholar]
- 27.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Q, Chan SY. Mitochondrial and metabolic drivers of pulmonary vascular endothelial dysfunction in pulmonary hypertension. Adv Exp Med Biol. 2017;967:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safiedeen Z, Rodriguez-Gomez I, Vergori L, et al. Temporal cross talk between endoplasmic reticulum and mitochondria regulates oxidative stress and mediates microparticle-induced endothelial dysfunction. Antioxid Redox Signal. 2017;26:15–27. [DOI] [PubMed] [Google Scholar]
- 30.Marcu R, Zheng Y, Hawkins BJ. Mitochondria and angiogenesis. Adv Exp Med Biol. 2017;982:371–406. [DOI] [PubMed] [Google Scholar]
- 31.Schoors S, Bruning U, Missiaen R, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong BW, Wang X, Zecchin A, et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature. 2017;542:49–54. [DOI] [PubMed] [Google Scholar]
- 33.De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Vandekeere S, Kalucka J, et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017;36:2334–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalucka J, Bierhansl L, Conchinha NV, et al. Quiescent endothelial cells upregulate fatty acid beta-oxidation for vasculoprotection via redox homeostasis. Cell Metab. 2018;28(881–894):e13. [DOI] [PubMed] [Google Scholar]
- 36.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. [DOI] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Cregg JM, Dunn WA Jr, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. [DOI] [PubMed] [Google Scholar]
- 38.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. [DOI] [PubMed] [Google Scholar]
- 39.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo K, Searfoss G, Krolikowski D, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. [DOI] [PubMed] [Google Scholar]
- 41.Hamacher-Brady A, Brady NR, Logue SE, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. [DOI] [PubMed] [Google Scholar]
- 42.Ghavami S, Eshragi M, Ande SR, et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010;20:314–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albertini E, Koziel R, Durr A, Neuhaus M, Jansen-Durr P. Cystathionine beta synthase modulates senescence of human endothelial cells. Aging (Albany NY). 2012;4:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119:315–326. [DOI] [PubMed] [Google Scholar]
- 46.Dey A, Xiong X, Crim A, et al. Evaluating the mechanism and therapeutic potential of PTC-028, a novel inhibitor of BMI-1 function in ovarian cancer. Mol Cancer Ther. 2018;17:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banerjee Mustafi S, Aznar N, Dwivedi SK, et al. Mitochondrial BMI1 maintains bioenergetic homeostasis in cells. FASEB J. 2016;30:4042–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naon D, Zaninello M, Giacomello M, et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA. 2016;113:11249–11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhattacharyya S, Saha S, Giri K, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE. 2013;8:e79167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans. 2009;37:1385–1388. [DOI] [PubMed] [Google Scholar]
- 51.van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyazono Y, Hirashima S, Ishihara N, Kusukawa J, Nakamura KI, Ohta K. Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission-independent manner. Sci Rep. 2018;8:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homo-logue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanida I Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–2214. [DOI] [PubMed] [Google Scholar]
- 57.Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. [DOI] [PubMed] [Google Scholar]
- 58.Gomez-Suaga P, Paillusson S, Miller CCJ. ER-mitochondria signaling regulates autophagy. Autophagy. 2017;13:1250–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Filadi R, Greotti E, Pizzo P. Highlighting the endoplasmic reticulum-mitochondria connection: focus on Mitofusin 2. Pharmacol Res. 2018;128:42–51. [DOI] [PubMed] [Google Scholar]
- 60.McLelland G, Goiran T, Yi W, et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018;7:e32866 10.7554/elife.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang CN, Liu YJ, Duan GL, et al. CBS and CSE are critical for maintenance of mitochondrial function and glucocorticoid production in adrenal cortex. Antioxid Redox Signal. 2014;21:2192–2207. [DOI] [PubMed] [Google Scholar]
- 62.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116:2763–2774. [DOI] [PubMed] [Google Scholar]
- 64.Archer SL. Mitochondrial fission and fusion in human diseases. N Engl J Med. 2014;370:1074. [DOI] [PubMed] [Google Scholar]
- 65.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. [DOI] [PubMed] [Google Scholar]
- 66.Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol. 2017;79:43–66. [DOI] [PubMed] [Google Scholar]
- 67.Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascu Pharmacol. 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 68.Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H(2) O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279:C21–C30. [DOI] [PubMed] [Google Scholar]
- 69.Moody BF, Calvert JW. Emergent role of gasotransmitters in ischemia-reperfusion injury. Med Gas Res. 2011;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakao A, Sugimoto R, Billiar TR, McCurry KR. Therapeutic anti-oxidant medical gas. J Clin Biochem Nutr. 2009;44:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mizushima N Autophagy: process and function. Genes Dev. 2007;21:2861–2873. [DOI] [PubMed] [Google Scholar]
- 72.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- 74.Reef S, Zalckvar E, Shifman O, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–475. [DOI] [PubMed] [Google Scholar]
- 75.MacIntosh GC, Bassham DC. The connection between ribophagy, autophagy and ribosomal RNA decay. Autophagy. 2011;7:662–663. [DOI] [PubMed] [Google Scholar]
- 76.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol. 2009;19:359–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. [DOI] [PubMed] [Google Scholar]
- 79.Goldman SJ, Taylor R, Zhang Y, Jin S. Autophagy and the degradation of mitochondria. Mitochondrion. 2010;10:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen G, Ray R, Dubik D, et al. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273:12415–12421. [DOI] [PubMed] [Google Scholar]
- 83.Chen G, Cizeau J, Vande Velde C, et al. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10. [DOI] [PubMed] [Google Scholar]
- 84.Ney PA. Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta. 2015;1853: 2775–2783. [DOI] [PubMed] [Google Scholar]
- 85.Dhingra R, Margulets V, Chowdhury SR, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci USA. 2014;111:E5537–E5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dhingra A, Jayas R, Afshar P, et al. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med. 2017;112:411–422. [DOI] [PubMed] [Google Scholar]
- 87.Zhu Y, Massen S, Terenzio M, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.