
Figure 2 –
TERT promoter regulation: a new therapeutic avenue in cancer?
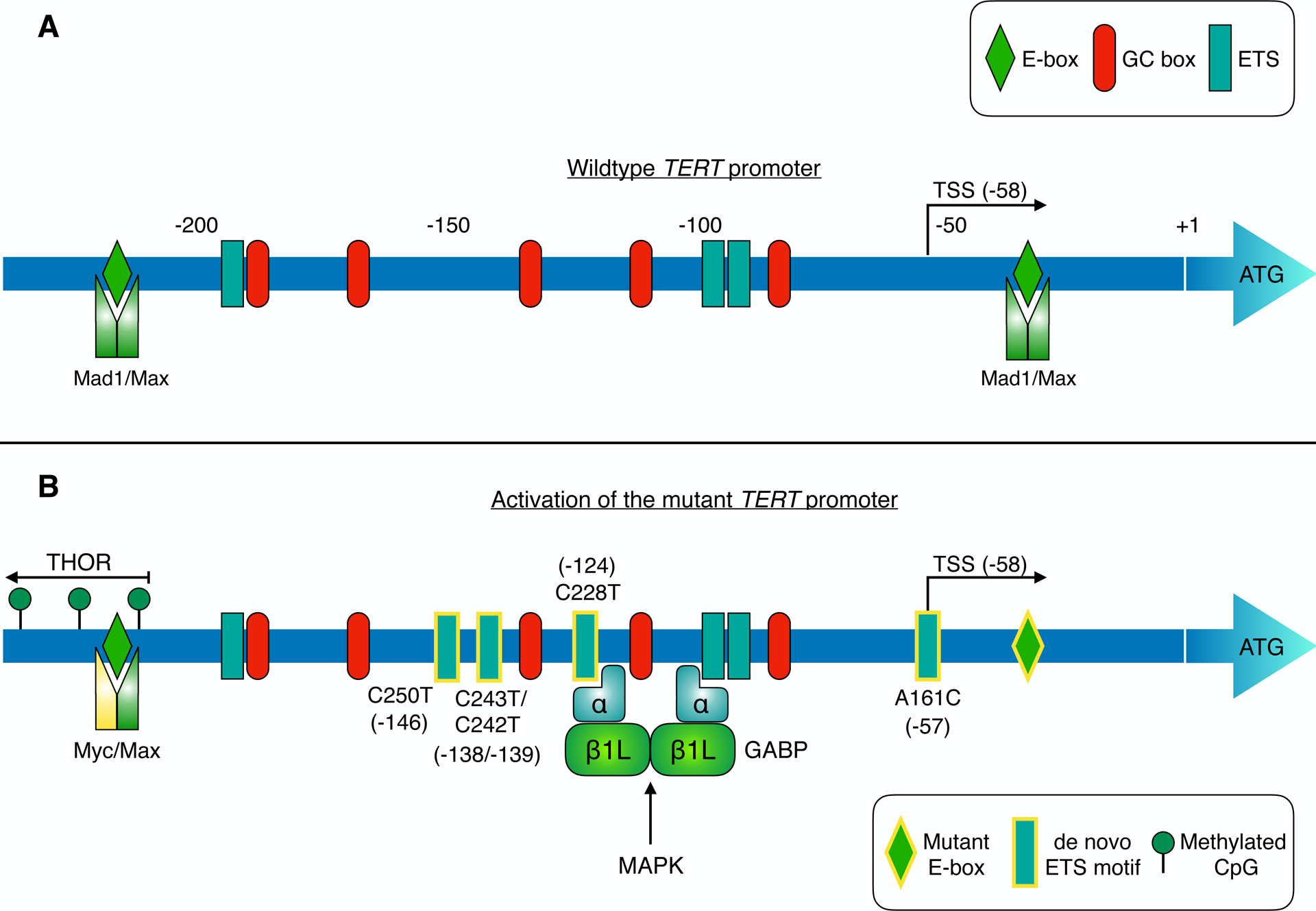
The TERT core promoter region harbours multiple binding sites for SP1, Myc-Max-Mad1 and ETS family transcription factors (corresponding to GC boxes, E-boxes and ETS motifs respectively; panel A). TERT transcription is suppressed in telomerase-negative cells by different mechanisms including repressive chromatin modifications and binding of Mad1/Max to E-boxes in the core promoter [95] [120]. Telomerase expression is reactivated in cancers (panel B) via diverse mechanisms, such as TERT promoter mutation, hypermethylation, and TERT gene amplification [14] [27]. Mutually exclusive point mutations in the TERT promoter generate de novo binding sites (depicted with yellow-bordered rectangles) for ETS transcription factors in ~15–25% of tumours. These de novo ETS motifs can be bound by distinct ETS family members in different tumour types, thereby enhancing TERT transcription (see Table 1). The activity of these TFs can be regulated by oncogenic pathways such as MAPK. For instance, in glioblastoma, heterotetrameric GABP is recruited to de novo ETS sites in the mutant promoter, enhancing TERT expression [18]. GABP activation of the mutant TERT promoter is responsive to MAPK stimulation [121]. Similarly, ETS1 activation of TERT is suppressed by MEK inhibition in BRAF-mutant melanoma models [102]. Methylation of CpG sites upstream of the core promoter upregulates TERT expression in cancer. This TERT hypermethylated oncological region (THOR) spans from approximately −217 to −649, relative to the ATG start codon [27]. Point mutation of a Myc-Max-Mad1 binding site (yellow-bordered diamond) in clear cell renal cell carcinoma may impair repression of TERT transcription by Mad1 [122].