
FIGURE 1.
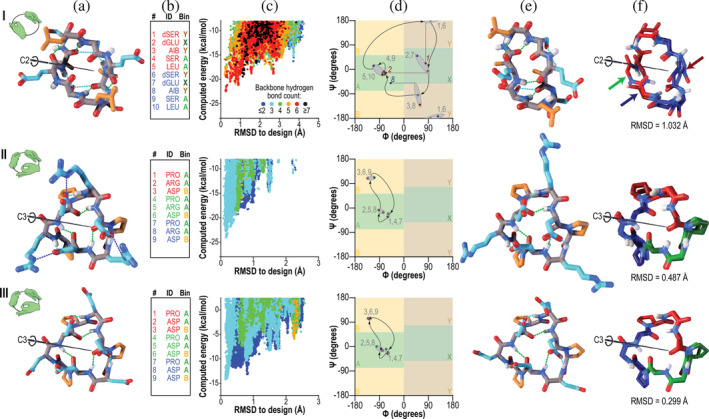
Designed peptides with cyclic (C2 or C3) symmetries. Columns: (a) Computer models produced by Rosetta design. Backbone‐backbone, backbone‐sidechain, and sidechain‐sidechain hydrogen bonds are shown as green, cyan, and blue dashed lines, respectively. Backbone atoms are shown in grey, polar side‐chains in cyan, and apolar side‐chains in orange. The C2 or C3 symmetry axis is shown as a black rod. Hand icons depict the symmetry. (b) Designed amino acid sequences and Ramachandran bin strings. Amino acids are coloured red and blue (C2 symmetry) or red, green, and blue (C3 symmetry) to indicate different repeating units. (c) Computed energy landscape of design, in which each point represents a structure prediction trajectory, with computed energy plotted against RMSD to the designed structure. Colors represent the number of backbone hydrogen bonds. (d) Ramachandran map representation of the designed structures (grey points) compared to the experimentally‐determined structures (points colored by symmetry lobe, as in column b). Grey numbers indicate sequence positions, and curved arrows show the progression through the sequence. Grey ovals group the designed and observed backbone angles, as a guide to the eye. In the case of peptide C2‐1, three residues, indicated with red and blue numbers, showed considerable deviation in φ (horizontal dashed lines) or ψ (vertical dashed line). (e) Structures determined by X‐ray crystallography. Colors are as in column a. (f) Overlay of the designed model (lighter colors) with the X‐ray crystal structure (darker colors). Symmetric lobes are shown in colors matching sequences in column b. Side‐chains other than those of proline are omitted for clarity. Rows: (I) Peptide C2‐1. The backbone heavy‐atom RMSD between crystal structure and design was 1 Å, mainly due to shifts in ψ of residue 1 and φ of residue 2 (red dashed lines in column d and blue and green arrows in column f) which together rotated the amide bond between residues 1 and 2 by 180°, and in φ of residue 8, which reoriented a backbone carbonyl (red arrow) in the absence of the hydrogen bond that would have been donated to it by the rotated amide proton. Despite these changes, much of the structure overlaid well on the design. (II and III) Peptides C3‐2 and C3‐3, which shared a common backbone configuration. Crystal contacts resulted in somewhat different conformations of polar side‐chains, but the backbone heavy‐atom RMSDs to the designs were 0.5 and 0.3 Å, respectively, yielding near‐perfect alignment in both cases (column e)