

SUMMARY
Endometriosis affects 1 in 10 women and is characterized by the presence of abnormal endometrium at ectopic sites. ARID1A mutations are observed in deeply invasive forms of the disease, often correlating with malignancy. To identify epigenetic dependencies driving invasion, we use an unbiased approach to map chromatin state transitions accompanying ARID1A loss in the endometrium. We show that super-enhancers marked by high H3K27 acetylation are strongly associated with ARID1A binding. ARID1A loss leads to H3K27 hyperacetylation and increased chromatin accessibility and enhancer RNA transcription at super-enhancers, but not typical enhancers, indicating that ARID1A normally prevents super-enhancer hyperactivation. ARID1A co-localizes with P300 at super-enhancers, and genetic or pharmacological inhibition of P300 in ARID1A mutant endometrial epithelia suppresses invasion and induces anoikis through the rescue of super-enhancer hyperacetylation. Among hyperactivated super-enhancers, SERPINE1 (PAI-1) is identified as an essential target gene driving ARID1A mutant endometrial invasion. Broadly, our findings provide rationale for therapeutic strategies targeting super-enhancers in ARID1A mutant endometrium.
Graphical Abstract
In Brief
ARID1A mutations are observed in deeply invasive endometriosis. Here, Wilson et al. show that ARID1A prevents H3K27 hyperacetylation of super-enhancers. Inhibition of P300 in ARID1A-mutant endometrium rescues super-enhancer hyperacetylation and inhibits invasion. SERPINE1 (PAI-1) super-enhancer hyperacetylation drives the invasion of ARID1A-mutant endometrium. Invasive endometriosis may be sensitive to super-enhancer-targeted therapies.
INTRODUCTION
The endometrium (the inner lining of the uterus) is composed of epithelia and stroma that continually proliferate, differentiate, and shed throughout the menstrual cycle in anticipation of pregnancy (Gellersen and Brosens, 2014; Mihm et al., 2011). The multiple rounds of tissue regression and regeneration that occur throughout a woman’s reproductive years make the endometrium particularly prone to disease (Gargett et al., 2012; Syed et al., 2020; Teixeira et al., 2008). As cyclical tissue breakdown, re-epithelialization, and stromal restoration occurs, the maintenance of proper cell identity is thought to be an important feature of a healthy endometrium (Gellersen and Brosens, 2014). Alterations in normal endometrial function result in numerous conditions, including benign diseases, such as endometrial hyperplasia (Montgomery et al., 2004), adenomyosis (Maheshwari et al., 2012), and endometriosis (Zondervan et al., 2018, 2020), as well as endometrial cancer (Morice et al., 2016) and ovarian cancer (Kurman and Shih, 2016).
Mutations in the SWI/SNF subunit ARID1A (BAF250A) were first identified in ovarian clear-cell carcinoma and ovarian endometrioid carcinoma, two epithelial ovarian cancer subtypes associated with endometriosis (Jones et al., 2010; Wiegand et al., 2010). Inactivating ARID1A mutations have been identified in numerous other endometrial pathologies (Mao and Shih, 2013; Wu et al., 2014). ARID1A levels are lower in eutopic endometrium, and it is required for embryo implantation in the uterus (Kim et al., 2015). ARID1A mutations are observed in deep ovarian and deep infiltrating endometriosis (Anglesio et al., 2017; Borrelli et al., 2016; Lac et al., 2019a, 2019b; Samartzis et al., 2012; Suda et al., 2018). ARID1A mutations are also observed in atypical endometrial hyperplasia (Mao et al., 2013; Werner et al., 2013) and endometrial cancer (Guan et al., 2011; Wiegand et al., 2011).
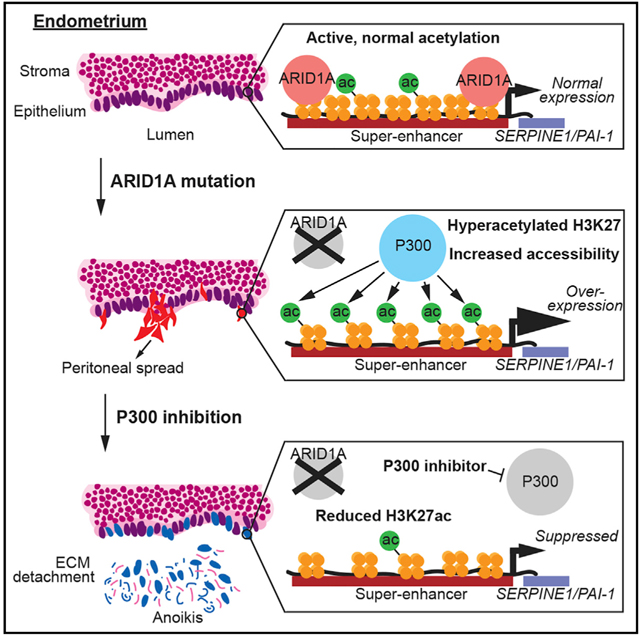
Endometriosis is characterized by the growth and spread of abnormal endometrial tissue at sites outside of the uterus (Bulun, 2009; Giudice and Kao, 2004). The identification of high-frequency ARID1A somatic mutations in deep ovarian endometriosis supports epidemiological and experimental evidence linking endometriosis to endometriosis-associated ovarian cancer (Pearce et al., 2012). ARID1A mutations may increase the risk of endometriosis and malignant transformation by providing a selective advantage to displaced endometrial cells undergoing retrograde menstruation (Suda et al., 2018). Alterations in endometrial cell identity, such as the transdifferentiation of endometrial epithelium, promote the acquisition of invasive cell properties, a feature often observed in mesenchymal cells (Bartley et al., 2014; Bilyk et al., 2017; Yang and Yang, 2017). Cellular invasion requires cells to migrate, degrade the extracellular matrix, and survive under anchorage-independent conditions (Kalluri and Weinberg, 2009; Mareel and Leroy, 2003). These properties allow abnormal endometrial cells to spread locally or colonize distal sites. In this study, we identify a mechanism by which ARID1A represses invasive phenotypes by antagonizing P300 activity at super-enhancers (SEs).
RESULTS
ARID1A Co-localizes with H3K27ac and Is Associated with SEs
Although ARID1A is mutated in several disorders of the endometrial epithelium, little is known about how ARID1A loss alters the epigenomic landscape in these cells. Here, in an unbiased approach, we examined chromatin features from both control and ARID1A-depleted cells and built a genome-wide segmentation model of unique chromatin states (ChromHMM). We profiled several post-translational histone modifications by chromatin immunoprecipitation sequencing (ChIP-seq), including H3K4me1, H3K4me3, H3K27me3, H3K27ac, and H3K18ac, following ARID1A-depletion in 12Z human endometrial epithelial cells (Zeitvogel et al., 2001). These data were used in conjunction with assay for transposase-accessible chromatin sequencing (ATAC-seq) and total RNA-seq datasets from ARID1A-depleted 12Z cells (Wilson et al., 2019), which allowed us to build a comprehensive model of chromatin state transitions accompanying ARID1A loss (Figures 1A and 1B). A series of genomic feature enrichment tests allowed us to annotate the predicted biological function of each of the 18 chromatin states, including 8 distinct classes of enhancer elements segregated by combinatorial chromatin features (Figures 1C–1F). Using ARID1A ChIP-seq data (Wilson et al., 2019), we observed that ARID1A binding is most strongly associated with highly active regulatory elements marked by H3K27ac, including SE chromatin states (S11–S13) and other highly active enhancer states (S14) (Figure 1G–H).
Figure 1. ARID1A Is Associated with Highly Active Regulatory Elements Marked by H3K27ac.
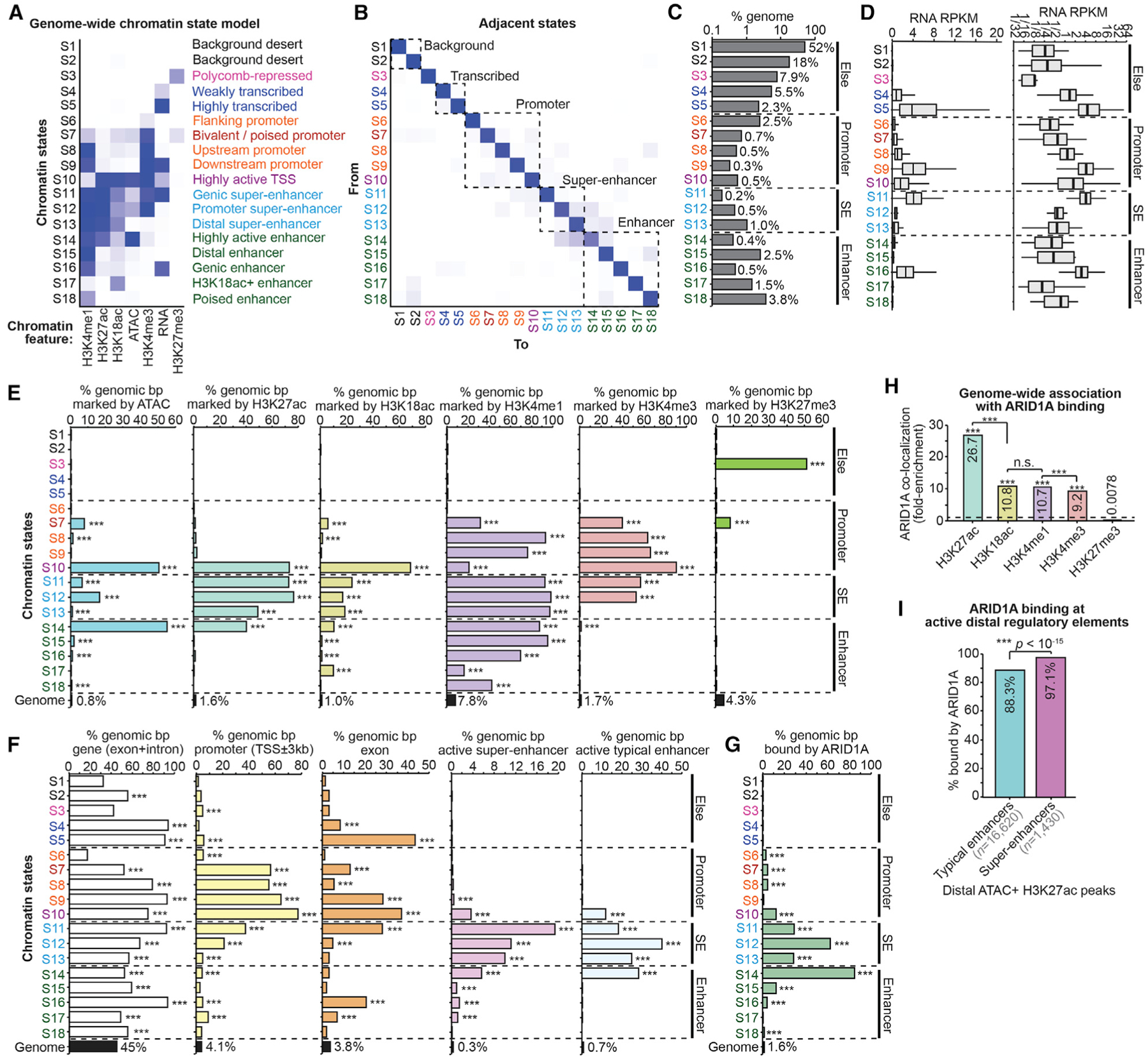
(A) Chromatin state model generated by ChromHMM. A total of 18 states were identified through genomic profiling of 7 chromatin features in ARID1A wild-type and knockdown 12Z cells: total RNA-seq, ATAC-seq, and H3K27me3, H3K4me3, H3K4me1, H3K18ac, and H3K27ac ChIP-seq. Genome was segmented into 200-bp intervals based on state classifications. Darker heatmap colors indicate higher relative enrichment for each chromatin feature in that state. Right-side labels are inferred biological functions of each state based on combinatorial chromatin features and genome ontology annotation.
(B) Heatmap displaying chromatin state adjacency frequencies (how often 2 chromatin states neighbor each other). The darker color indicates more frequent state neighboring.
(C) Percentage of genome coverage for each chromatin state.
(D) Total RNA quantification of each chromatin state as reads per kilobase per million mapped reads (RPKM) per 200-bp genomic interval. Left, linear scale; right, log2 scale.
(E) Percentage of genome coverage per chromatin state for all other measured chromatin features. The statistic is hypergeometric enrichment compared to whole genome.
(F) Percentage of genome coverage per chromatin state for other genomic features. Active SEs and TEs are distal H3K27ac peaks marked by ATAC, as defined in Figure S1A. The statistic is hypergeometric enrichment.
(G) Percentage of genome coverage per chromatin state for ARID1A binding. The statistic is hypergeometric enrichment.
(H) Genome-wide association between ARID1A binding and profiled histone modifications. Enrichments are displayed as fold-enrichment, per genomic base pair. The statistic is hypergeometric enrichment. Pairwise enrichment statistics computed by the chi-square test.
(I) Association between ARID1A binding and TEs versus SEs, per H3K27ac peak, as defined in Figure S1A. The statistic is 2-tailed Fisher’s exact test. ***p < 0.001.
SEs are enhancer clusters that control the transcription of genes involved in cellular identity and thus play key roles in developmental and disease processes (Lovén et al., 2013; Whyte et al., 2013). Enhancers are characterized by abundant H3K27ac and accessible chromatin (Calo and Wysocka, 2013). We used both H3K27ac ChIP-seq and ATAC-seq data to identify 18,050 putative active enhancers (Figure S1A). From this set of active enhancers, we used the Rank Ordering of Super-Enhancers (ROSE) algorithm to identify active SE and observed 413 unique SEs that contained 1,430 sites marked by H3K27ac and ATAC (Figure S1A). Active distal enhancer regions (located further than 3 kb from a transcription start site [TSS]) not categorized as SEs were designated as typical enhancers (TEs) (n = 16,620) (Figure S1A). We observed greater H3K27ac signal at SE peaks relative to TE peaks (Figure S1B). SEs comprised three H3K27ac peaks on average (Figure S1C). ARID1A associated with the majority of both SEs and TEs, but was bound to a higher proportion of SEs than TEs (Figure 1I), suggesting a role for ARID1A in the regulation of active SEs.
ARID1A Prevents Super-Enhancer Hyperacetylation
To further understand the role of ARID1A in chromatin regulation, we analyzed the effects of ARID1A depletion on chromatin state classification and the abundance of histone modifications. ChromHMM modeling revealed that most chromatin states do not display substantial reprogramming following ARID1A loss (Figure 2A). SEs and other enhancer states bound by ARID1A typically did not change state, although some highly active enhancers (S14) gained further activation characteristics (S14 > S13, S14 > S12), while others lost active marks (S14 > S15) (Figure 2B). Among the histone modifications tested, H3K27ac displayed the greatest proportion of differentially regulated sites following ARID1A loss (Figures 2C and S1D–S1H). Interestingly, ARID1A loss did not affect H3K27me3 occupancy genome-wide (Figure S1F), even though SWI/SNF is known to antagonize polycomb chromatin silencing in other cellular contexts (Bracken et al., 2019).
Figure 2. ARID1A Prevents H3K27 Hyperacetylation at SEs.
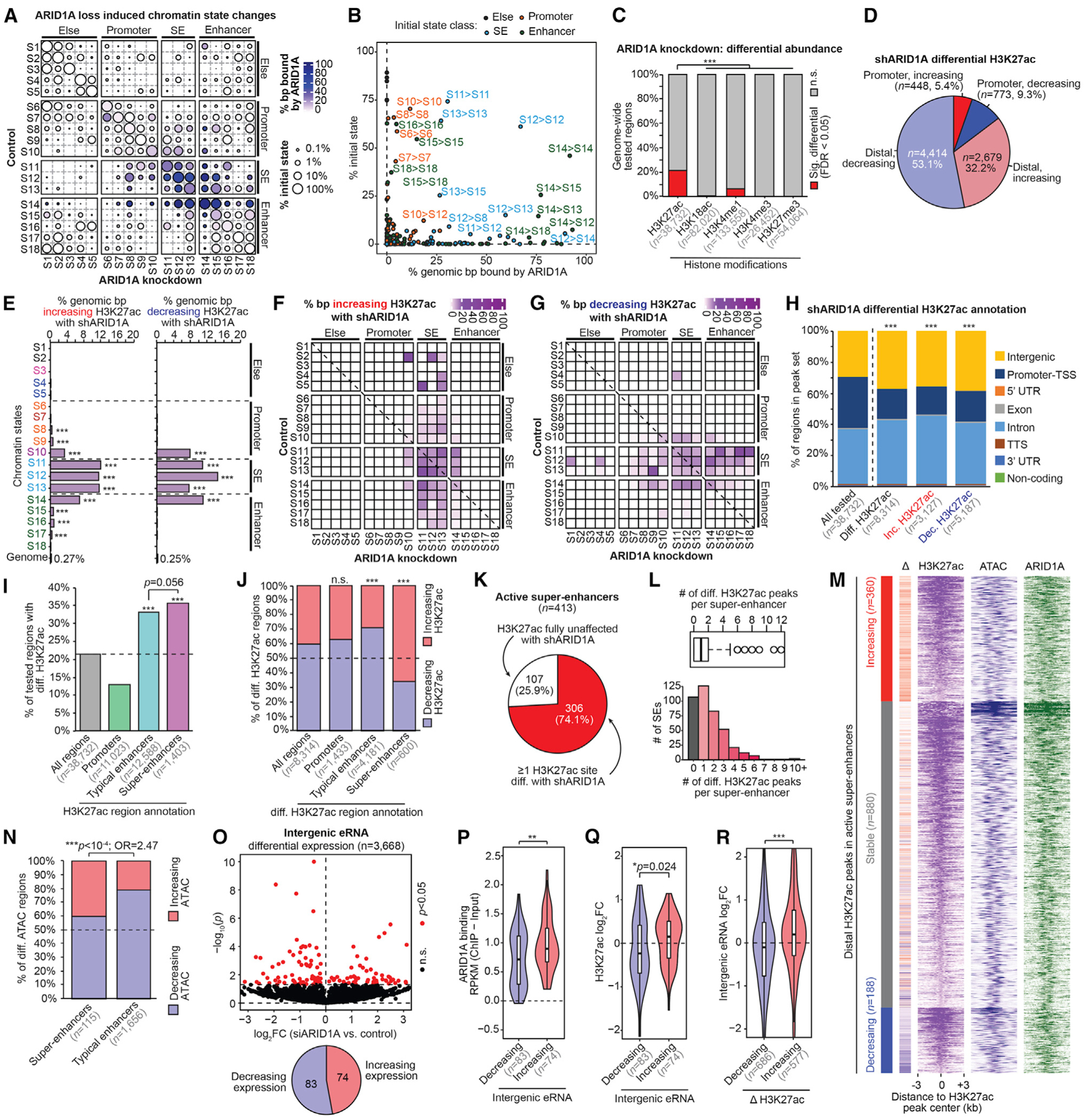
(A) Map of chromatin state changes following ARID1A loss. For each state-state change, circle size depicts the relative amount of that state change compared to the initial genome-wide state representation ([genomic bp initial → final]/[genomic bp initial]), and color indicates the proportion bound by ARID1A.
(B) Scatterplot of the 2 features quantified in (A) for each state-state change. Each dot representing a state-state change is further colored by its initial state class: S1–S5, “else”; S6–S10, “promoter”; S11–S13, “SE”; S14–S18, “enhancer.” The most prominent state-state changes are labeled as [initial] > [final].
(C) Proportion of genome-wide regions displaying significant (FDR < 0.05) differential abundance following ARID1A loss for each histone modification. Tested regions are the union of replicate-overlapping peak sets per assay. The pairwise statistic is the 2-tailed Fisher’s exact test.
(D) Gene proximity and directionality of significant differential H3K27ac sites (FDR < 0.05, n = 8,314).
(E) Genomic enrichment for (left) increasing H3K27ac or (right) decreasing H3K27ac following ARID1A loss at each chromatin state compared to the whole genome. The statistic is hypergeometric enrichment.
(F) Map of chromatin state changes as in (A), but overlaid color feature is the proportion of state-state base pairs displaying increasing H3K27ac.
(G) Map of chromatin state changes as in (F), but for decreasing H3K27ac.
(H) Distribution of genomic features of all tested H3K27ac regions compared to differential (total, increasing, or decreasing). The statistic is the chi-square test.
(I) Enrichment of differential H3K27ac among promoters, TE or SE, compared to all tested H3K27ac regions. The statistic is hypergeometric enrichment and pairwise 2-tailed Fisher’s exact test.
(J) Proportion of increasing versus decreasing H3K27ac at significant differential regions binned by promoter, SE, and TE, compared to all differential regions. The statistic is hypergeometric enrichment.
(K) Percentage of active SE (n = 413) with at least 1 H3K27ac peak displaying differential H3K27ac upon ARID1A loss.
(L) Number of differential H3K27ac regions per SE depicted as a boxplot in the style of Tukey (top) or a histogram (bottom). The median number of differentially acetylated regions per SE is 1.
(M) Signal heatmap at distal H3K27ac peaks located within SEs, segregated by differential H3K27ac status: increasing (n = 360), decreasing (n = 188), or stable (n = 880). Each peak subset is ranked by H3K27ac signal in the control cells. Delta corresponds to H3K27ac log2 fold change (log2FC) from small hairpin ARID1A (shARID1A) versus control: red values, increased H3K27ac; blue, decreased H3K27ac.
(N) Proportion of increasing versus decreasing differential ATAC regions located within SEs and TEs following ARID1A loss. The statistic is the 2-tailed Fisher’s exact test.
(O) Volcano plot displaying DE intergenic eRNA (n = 3,668) following ARID1A loss. Intergenic eRNA regions were selected from the 18,050 distal ATAC + H3K27ac peaks (Figure S1A), which did not overlap gene bodies and had detectable RNA. The x axis is log2FC upon ARID1A loss; the y axis is DE significance. Significant (p < 0.05) DE eRNA marked in red. The pie chart displays the ratio of intergenic eRNA significantly increasing or decreasing expression upon ARID1A loss.
(P) ARID1A binding at intergenic enhancer sites with decreasing (n = 83) or increasing (n = 74) eRNA expression. The statistic is the 2-tailed, unpaired Wilcoxon test.
(Q) Change (log2FC) in H3K27ac abundance at intergenic sites of increasing (n = 74) or decreasing (n = 83) eRNA expression following ARID1A loss. The statistic is the 2-tailed, unpaired Wilcoxon test.
(R) Change (log2FC) in eRNA expression at intergenic enhancer sites, with increasing (n = 577) or decreasing (n = 686) H3K27ac upon ARID1A loss. The statistic is the 2-tailed, unpaired Wilcoxon test.
*p < 0.05, **p < 0.01, and ***p < 0.001.
Next, we examined the H3K27ac changes occurring in ARID1A-deficient cells. The majority of differential H3K27ac sites were found among distal elements and, among those sites, we observed decreased acetylation following ARID1A loss (Figure 2D). Furthermore, most H3K27ac changes occurred at SE and highly active enhancer chromatin states where ARID1A is bound (Figures 2E–2G). Intriguingly, sites that gained H3K27ac following ARID1A loss tended to become SE states (S11–S13) (Figure 2F), while sites that lost H3K27ac tended to transition from SE to other enhancer states (S14–S18) (Figure 2G). Consistently, H3K27ac sites at promoters (within 3 kb of a TSS) were less likely to be affected by ARID1A loss than distal intergenic and intronic elements (Figures 2H and S1I–S1L), and SEs were marginally more likely to show changes in H3K27ac than TEs (Figure 2I). However, while most active TEs displayed decreased H3K27ac, most active SEs displayed increased H3K27ac following ARID1A loss (Figure 2J), suggesting a specific role for ARID1A in preventing H3K27ac hyperacetylation at SEs. Among the 413 active SEs, 74.1% displayed differential H3K27ac at one or more sites following ARID1A loss (Figures 2K and 2L). H3K27ac was increased at 360 peaks within active SEs following ARID1A loss (Figure 2M). Compared to TEs, SEs also displayed a greater proportion of sites with increased chromatin accessibility upon ARID1A loss (Figure 2N).
Enhancer RNA (eRNA) transcription promotes enhancer activity through enhancer-promoter communication and chromatin looping, and eRNA is associated with SEs (Ko et al., 2017). We explored the role of eRNA at ARID1A-regulated SEs as a marker of enhancer activity, and, among 3,668 intergenic enhancers with detectable eRNA expression, we observed 157 differentially expressed (DE) eRNAs upon ARID1A loss (Figure 2O). Among these, ARID1A binding was stronger at sites with upregulated eRNA following ARID1A loss (Figure 2P). Furthermore, upregulated eRNAs were associated with increased H3K27ac (Figures 2Q and 2R). Collectively, these data support a role for ARID1A in restricting SE activity, such that ARID1A loss results in H3K27 hyperacetylation, increased chromatin accessibility, and eRNA expression.
ARID1A and P300 Co-occupy Highly Active SEs
Having observed a role for ARID1A in preventing H3K27ac at SE, we next asked whether ARID1A is associated with P300, a histone acetyltransferase (HAT) that acetylates H3K27 and H3K18 residues (Jin et al., 2011; Schiltz et al., 1999) and has known roles at SEs (Pott and Lieb, 2015). We used the Enrichr tool to screen ENCODE (Encyclopedia of DNA Elements) ChIP-seq datasets for factors with overlapping sets of target genes (Chen et al., 2013; Kuleshov et al., 2016) and identified P300 as the top factor likely to co-regulate DE genes following endometrial ARID1A loss (Figures S2A and S2B). We performed P300 ChIP-seq in wild-type 12Z cells and identified 25,096 P300 binding sites throughout the genome, enriched within several chromatin states (Figures 3A and 3B). Intriguingly, P300 is more associated with active TSS (S10) than ARID1A (Figure 3B, compared to Figure 1G), and P300 binding was enriched and co-bound with ARID1A at promoters (Figures S2C–S2M). Known roles for P300 in enhancer regulation (Long et al., 2016) led us to study ARID1A and P300 co-regulation at distal sites. We observed 2,609 distal sites with both ARID1A and P300 binding (Figure 3C). Chromatin accessibility marks the sites of regulatory activity (Kornberg and Lorch, 1992), and ARID1A is associated with open chromatin states (Kelso et al., 2017). Among P300-bound, accessible sites, ARID1A is associated more with the co-regulation of distal sites than promoters (Figure 3D).
Figure 3. P300 and ARID1A Co-regulate H3K27ac at Highly Active SEs.
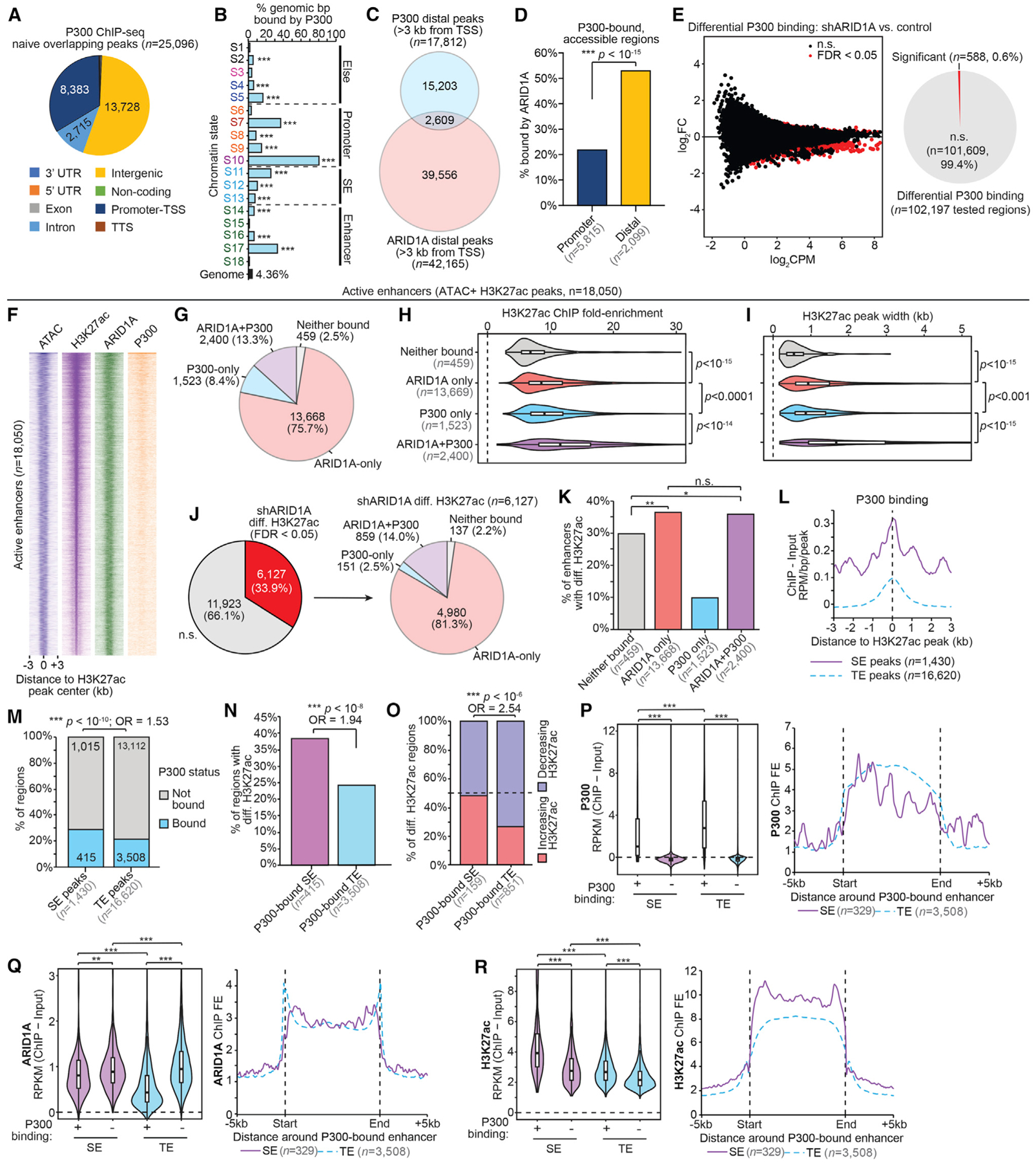
(A) Genomic annotation of replicate-overlapping P300 ChIP broad peaks in wild-type 12Z (FDR < 0.05, n = 25,096 peaks).
(B) Enrichment for P300 binding (control cells) among chromatin states compared to whole genome. The statistic is hypergeometric enrichment.
(C) Proportional Euler diagram displaying overlap between distal regions bound by ARID1A (n = 42,165) and P300 (wild-type cells, n = 17,812).
(D) ARID1A binding among accessible P300-bound sites. P300 bound sites (wild-type cells) were first segregated by promoter versus distal status, then filtered for accessibility (ATAC). The statistic is the 2-tailed Fisher’s exact test.
(E) Differential P300 ChIP-seq following ARID1A loss. At left is an MA plot revealing differential binding, with significant sites (FDR < 0.05) highlighted in red. The x axis is signal abundance quantified as log2 counts per million (log2CPM), and the y axis is the log2FC difference of P300 binding in shARID1A versus control conditions (n = 2 ChIP replicates). At right is the ratio of tested sites binned by differential binding significance. Further analyses of P300 binding use the control condition data (F–R).
(F) Signal heatmap displaying chromatin accessibility (ATAC), H3K27ac, and binding of ARID1A and P300 at enhancers (n = 18,050), centered on H3K27ac peak ± 3 kb. Enhancers were ranked by total H3K27ac signal.
(G) Proportion of active enhancers (n = 18,050) bound by ARID1A, P300, both, or neither.
(H) H3K27ac ChIP peak signal (fold enrichment, FE) relative to input at active enhancers segregated by ARID1A and P300 binding. The statistic is the 2-tailed, unpaired Wilcoxon test.
(I) H3K27ac ChIP peak width at active enhancers segregated by ARID1A and P300 binding. The statistic is the 2-tailed, unpaired Wilcoxon test.
(J) Ratio of enhancers (n = 18,050) displaying differential H3K27ac following ARID1A loss (left), and further segregation by ARID1A and P300 binding status (n = 4,681) (right).
(K) Proportion of differential H3K27ac regions among enhancers bound by ARID1A, P300, both, or neither. The statistic is the 2-tailed Fisher’s exact test.
(L) P300 ChIP signal at distal SE and TE H3K27ac peaks. The x axis is the distance to the H3K27ac peak center. The y axis is signal as ChIP – Input RPM per base pair per peak.
(M) Proportion of distal SE and TE H3K27ac peaks bound by P300. The statistic is the 2-tailed Fisher’s exact test.
(N) Proportion of P300-bound SE and TE regions displaying differential H3K27ac upon ARID1A loss. The statistic is the 2-tailed Fisher’s exact test.
(O) Proportion of increasing versus decreasing H3K27ac at differential SE and TE regions bound by P300. The statistic is the 2-tailed Fisher’s exact test.
(P–R) Violin plots (left) of ChIP signal for P300 (P), ARID1A (Q), and H3K27ac (R) at distal H3K27ac peaks in SE and TE regions further binned by P300 binding. Peak n’s from left to right: 415, 1,015, 3,508, and 13,112. The statistic is the 2-tailed, unpaired Wilcoxon test. Meta peak profiles (right) for P300 (P), ARID1A (Q), and H3K27ac (R) at P300-bound SE (entire SE region, n = 329) and P300-bound TE (n = 3,508).
*p < 0.05, **p < 0.01, and ***p < 0.001.
Chromatin remodeling enzymes regulate both the recruitment and catalytic activity of the histone modifying enzymes (Clapier and Cairns, 2009; Swygert and Peterson, 2014). Given the changes in H3K27ac in ARID1A-deficient cells, we tested whether P300 localization was affected by ARID1A loss using ChIP-seq. We observed no change in P300 binding following ARID1A loss at >99% of sites (Figure 3E), suggesting that ARID1A loss does not greatly affect P300 recruitment.
We then explored the role of ARID1A and P300 co-localization at enhancers. Among the 18,050 putative active enhancers, the majority were bound predominantly by ARID1A without P300 (Figures 3F and 3G). However, ARID1A-P300 co-bound enhancers displayed greater H3K27ac peak signal and broader H3K27ac peak distribution (Figures 3H and 3I). Among enhancers that display differential H3K27ac, ARID1A was again bound without P300 at the majority of sites (Figure 3J), although the enrichment of ARID1A with or without P300 at enhancers with differential H3K27ac was not significantly different (Figure 3K). We next considered the role of P300 binding and ARID1A co-regulation at SEs, and found that P300 binding was observed at a greater proportion of distal SE peaks than TE peaks (Figures 3L and 3M). Among P300-bound sites, differential H3K27ac following ARID1A loss was more frequently observed at SE than TE (Figure 3N). Furthermore, among P300 bound sites with differential H3K27ac, a greater number of SEs than TEs displayed increased H3K27ac (Figure 3O). Lastly, we compared ARID1A, P300, and H3K27ac levels at SEs versus TEs that are either P300 bound or not bound. At enhancers where P300 is bound, P300 binding is strongest at TEs compared to SEs (Figure 3P). However, ARID1A binding signal is stronger at P300-bound SEs (Figure 3Q), where the H3K27ac signal is highest, compared to TEs (Figure 3R). These results collectively suggest that ARID1A differentially regulates SE through P300-dependent H3K27ac deposition in normal endometrium.
P300 HAT Activity Is Required for ARID1A Mutant Cell Invasion
ARID1A loss in the endometrial epithelium leads to collective invasion when combined with an activating PIK3CA mutation (Wilson et al., 2019). To explore the functional relationship between ARID1A and P300, we used small interfering RNAs (siRNAs) targeting P300 (siP300), ARID1A (siARID1A), or non-targeting siRNAs (control). Knockdown of ARID1A and/or P300 in 12Z cells (Figure 4A) had no effect on cell growth or proliferation (Figures S3A and S3B). ARID1A loss increased cell invasion, and P300 loss alone had no effect, but co-knockdown of ARID1A and P300 completely rescued ARID1A mutant cell invasion (Figure 4B). Invasion was not observed in 12Z treated with broad-spectrum histone deacetylase inhibitors, suggesting that invasion does not depend solely on a global increase in histone acetylation (Figures S3C and S3D). These results demonstrate an essential role for P300 in driving invasive phenotypes in ARID1A mutant endometriotic cells.
Figure 4. P300 Promotes Invasion and Survival of ARID1A Mutant Endometriotic Epithelia.
(A) Western blot analysis as indicated in 12Z cells, representative of 2 independent experiments.
(B) Invasion of 12Z following indicated treatments. Representative images and total invaded cell numbers are shown (scale bar, 500 μm). Means ± SDs, n = 3. Unpaired, 2-tailed t test.
(C) Survival of mice based on time until vaginal bleeding. LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl (n = 16) median (μ1/2) 107 days. LtfCre0/+; (Gt) R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl (n = 12) median 143 days (p = 0.0018, Mantel-Cox test). LtfCre0/+; Ep300fl/fl mice were aged to 187 days, and no phenotypes were observed (n = 6).
(D) Histology and IHC using indicated antibodies (n ≥ 2 mice) in endometrium (scale bar, 200 μm). KRT8 was a positive control for endometrial epithelium. The arrowheads indicate epithelia.
(E) Quantification of H3K27ac and H3K18ac IHC, ratio of H-scores of epithelia to stroma. Means ± SDs, n = 4–8 mice, unpaired, 2-tailed t test.
(F) Western blot of H3K27ac following A-485 treatment of 12Z for 24 h and densitometry of H3K27ac relative to H3, normalized to control (vehicle). Means ± SDs, n = 3–5 independent replicates per condition. Unpaired, 2-tailed t tests were performed in comparison to the vehicle treatment condition. Irrelevant lanes were removed from the image; see Figure S7.
(G) Viability assay for cells treated with A-485, normalized cell counts relative to vehicle control. Raw data are presented in Figure S4C. Half-maximal inhibitory concentration (IC50) values were not significantly different between 12Z untreated and control shRNA, or between control shRNA and shARID1A (unpaired, 2-tailed t test). Means ± SDs, n = 4.
(H) Invasion of 12Z following indicated cell treatments. Representative images and total invaded cell numbers are shown (scale bar, 500 μm). Means ± SDs, n = 4. Unpaired, 2-tailed t tests performed in comparison to siARID1A + vehicle.
(I) Caspase 3/7 activity of indicated cell treatments. Means ± SDs, n = 3. Unpaired, 2-tailed t test.
(J) Ratio of dead to live cells after 16 h in Matrigel. Means ± SDs, n = 6. Unpaired, 2-tailed t test.
*p < 0.05, **p < 0.01, and ***p < 0.001.
To determine whether P300 loss rescues the invasive phenotype in vivo, we crossed Ep300 conditional knockout mice (Kasper et al., 2006) with our LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl model, resulting in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl (Figures S3E and S3F). LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice displayed an increased survival compared to LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice (Figure 4C). LtfCre0/+; Ep300fl/fl mice displayed no phenotype (Figures 4C and S3G). P300 expression was lost in the endometrial epithelium of LtfCre0/+; Ep300fl/fl and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice by immunohistochemistry (IHC) (Figure 4D). Increased expression of the apoptotic marker cleaved caspase 3 was observed in the endometrial epithelium of LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice, but not LtfCre0/+; Ep300fl/fl mice, indicating a specific effect of P300 loss on ARID1A and PIK3CA mutant endometrium (Figure 4D). The epithelial layer in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice appeared to desquamate from the endometrial stroma, and cleaved caspase 3+, desquamated epithelial cells were observed throughout the lumen of the uterus (Figure S3H). P300 loss suppressed the proliferation occurring in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl epithelia (Figure S3I). While LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl endometrial epithelium invade the myometrium, the presence of endometrial glands in the myometrium was not observed in LtfCre0/+; (Gt) R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice. Similar to ARID1A-deficient 12Z cells, we observed the loss of H3K27ac, but not H3K18ac, in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice, suggesting that P300 loss leads to a specific reduction of H3K27ac in endometrial epithelial cells (Figures 4D and 4E). These results implicate P300 HAT activity in LtfCre0/+; (Gt) R26Pik3ca*H1047R; Arid1afl/fl lesion development.
We next explored the role of P300 HAT activity in promoting ARID1A mutant phenotypes. A-485 is a small-molecule P300/CREB-binding protein (CBP) HAT inhibitor (Lasko et al., 2017; Weinert et al., 2018). We tested the efficacy of A-485 in 12Z cells and observed a dose-dependent reduction in H3K27ac with significant inhibition at 316 nM (Figure 4F). In both ARID1A-deficient and wild-type states, we observed a limited effect of A-485 on cell growth and viability (Figures 4G and S4A–S4D). These results suggest that A-485 treatment results in the inhibition of P300 HAT activity at low concentrations without an effect on cell health.
Next, we tested the efficacy of A-485 in inhibiting P300-dependent, ARID1A mutant invasive phenotypes. We observed a significant reduction in ARID1A mutant invasion at concentrations that did not inhibit cell growth, with significant decreases in invasion at 10 nM A-485 and a complete rescue of the phenotype at 100 nM A-485 (Figures 4H and S4E), while the migration phenotype was inhibited at 31 nM and completely rescued at 316 nM (Figure S4F). Since apoptosis is induced in LtfCre0/+; (Gt) R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl mice, we considered that P300 HAT inhibition may lead to anchorage-dependent cell death or anoikis (Paoli et al., 2013). We tested whether A-485 induces anoikis under non-adherent conditions, and we observed increased caspase 3/7 activity in ARID1A-deficient cells following A-485 treatment, suggesting that A-485 induces anoikis (Figure 4I). Furthermore, we observed an increase in cell death in ARID1A-deficient cells embedded in Matrigel following A-485 treatment (Figure 4J). In mice, coexisting mutations in ARID1A and PIK3CA are required for lesion formation, so we wanted to determine whether the effect of A-485 in ARID1A-deficient cells was modulated by phosphatidylinositol 3-kinase (PI3K) activation. In cells with an overexpression of PIK3CAH1047R and ARID1A loss, we observed a similar inhibition of invasion and migration and an induction of anoikis (Figure S5). These results suggest that the inhibition of P300 HAT activity via low-dose A-485 treatment blocks invasion and promotes anoikis of ARID1A-deficient endometriotic cells.
P300 HAT Inhibition Reverses H3K27 Hyperacetylation at a Subset or SEs in ARID1A-Deficient Endometrial Cells
To explore how ARID1A and P300 co-regulate H3K27ac, we used the targeted genome profiling approach cleavage under targets and release using nuclease (CUT&RUN) (Skene et al., 2018). H3K27ac CUT&RUN showed significant overlap with H3K27ac ChIP-seq (Figure 5A). To determine the effects of P300 loss or HAT inhibition on H3K27ac in ARID1A-deficient cells, we next compared differential H3K27ac among 12Z cells treated with siARID1A versus control, and also compared cells co-treated with siARID1A + siP300 or 1 μM A-485 versus siARID1A alone (Figure 5B). Notably, the genome-wide effects of 1 μM A-485 on H3K27ac in siARID1A cells highly overlapped with siP300, validating that A-485 affects P300 targets. We identified 6,521 regions of H3K27ac that were affected by ARID1A loss and further affected by P300 loss or A-485 treatment (Figure 5C). Among these 6,521 intersecting regions, the majority of H3K27ac sites showed an additive increase or decrease in H3K27ac with combination treatments: decreased acetylation following ARID1A loss and further decreases with P300 loss or inhibition (n = 3,005) or increased acetylation following ARID1A loss and further increases with P300 loss or inhibition (n = 1,455) (Figures 5D and 5E). However, a subset of sites displayed increased H3K27ac following ARID1A loss, which was rescued by further P300 loss or A-485 treatment (“gain reversal,” n = 1,132) (Figures 5D and 5E). Interestingly, the gain reversal sites had the lowest levels of H3K27ac in control cells compared to other groups (Figure 5F), suggesting ARID1A normally limits acetylation at these sites. Furthermore, a large proportion of gain reversal regions are bound by ARID1A, while “acetylation gain” sites were infrequently bound by ARID1A (Figure 5G). This was further supported by genomic annotation showing that gain reversal sites were found at intergenic regions and introns, were enriched for SEs and other highly active enhancer chromatin states, and contained the highest proportion of active SE regions (Figures 5H–5K), suggesting that gain reversal sites contain SE elements at which ARID1A antagonizes P300 HAT activity toward H3K27ac.
Figure 5. ARID1A Antagonizes P300 HAT Activity at a Subset of Active SEs.
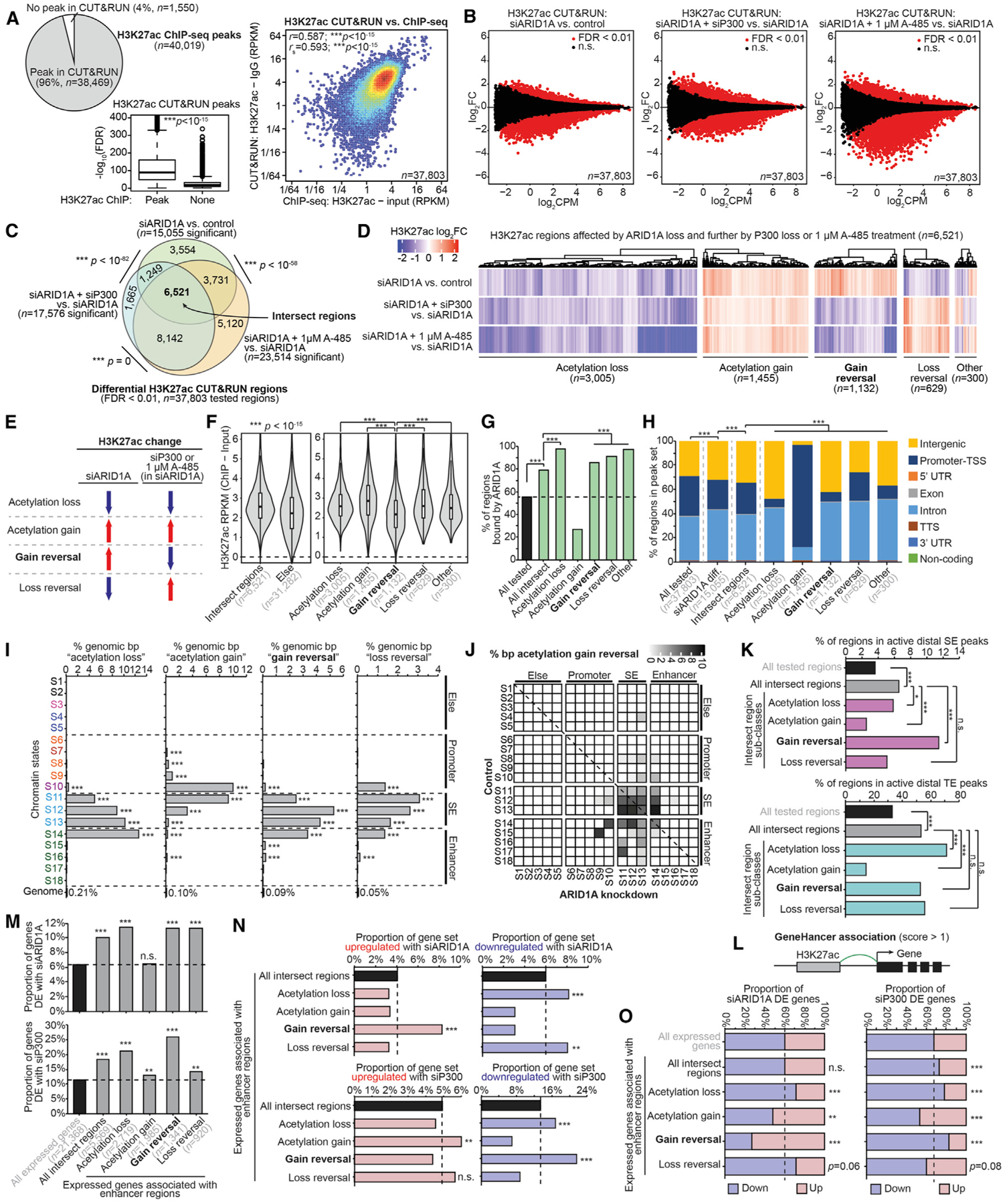
(A) Comparison of H3K27ac CUT&RUN and ChIP-seq. Left, pie chart displaying theproportion of H3K27ac ChIP-seq replicate-overlapping peaks (n = 40,019) identified by CUT&RUN versus not identified. Center, CUT&RUN signal at replicate-overlapping peaks quantified by −log10(FDR), displayed as a boxplot in the style of Tukey with outliers. CUT&RUN peaks are further segregated by whether they were also identified by ChIP-seq. The statistic is an unpaired, 2-tailed Wilcoxon test. Right, correlation of CUT&RUN versus ChIP signal at 37,803 consensus peaks identified by ChIP used for differential analysis. RPKM signal values are further log2 transformed for plotting. The statistics are Pearson and Spearman correlations.
(B) MA plots for H3K27ac CUT&RUN comparisons: left, siARID1A versus control; center, siARID1A + siP300 versus siARID1A; right, siARID1A + 1 μM A-485 versus siARID1A. A total of 37,803 consensus peaks previously identified by H3K27ac ChIP were used for differential testing, and significant (FDR < 0.01) regions are marked in red.
(C) Proportional Euler diagrams displaying overlapping differential H3K27ac regions between the comparisons in (B). The statistic is hypergeometric enrichment.
(D) Clustering of H3K27ac log2FC values among 6,521 intersect regions (C). H3K27ac classes are defined by directionality patterns.
(E) Diagrammatic explanation of H3K27ac classes identified in (D). “Acetylation loss” sites (n = 3,005) display decreasing H3K27ac with siARID1A and further decrease with siP300 or 1 μM A-485 treatment. “Acetylation gain” sites (n = 1,455): increasing H3K27ac with siARID1A and further increase with siP300 or 1 μM A-485 treatment. “Gain reversal” sites (n = 1,132): increasing H3K27ac with siARID1A and decrease with further siP300 or 1 μM A-485 treatment. “Loss reversal” sites (n = 629): decreasing H3K27ac with siARID1A that increase with siP300 or 1 μM A-485 treatment.
(F) H3K27ac ChIP-seq signal quantification at intersect regions versus else, and the 5 intersect region classes defined in (D) and (E). Statistic is unpaired, 2-tailed Wilcoxon test.
(G) Genomic enrichment for ARID1A binding at H3K27ac intersect regions and subclasses. The statistic is hypergeometric enrichment.
(H) Genomic annotation of various H3K27ac regions and intersect subclasses. The statistic is chi-square.
(I) Genomic enrichment for H3K27ac intersect region classes at each chromatin state, compared to the whole genome. The statistic is hypergeometric enrichment.
(J) Map of chromatin state changes following ARID1A loss overlaid by the proportion of state-state base pairs displaying acetylation gain reversal as the color feature.
(K) Enrichment for H3K27ac intersect regions and subclasses at (top) active distal SE peaks and (bottom) active TE peaks. The statistic is hypergeometric enrichment.
(L) Diagram of GeneHancer database usage to associate H3K27ac enhancer regions with genes.
(M) Enrichment for differential gene expression following (top) siARID1A or (bottom) siP300 (in siARID1A cells) treatment among expressed genes associated with H3K27ac enhancer regions by GeneHancer. The statistic is hypergeometric enrichment.
(N) Enrichment for (left) upregulated versus (right) downregulated genes following (top) siARID1A versus (bottom) siP300 (in siARID1A cells) treatment among enhancer-associated genes as in (M). The statistic is hypergeometric enrichment.
(O) Distribution of upregulated versus downregulated genes in enhancer-associated gene classes as in (M) and (N) for (left) siARID1A or (right) siP300 (in siARID1A cells) DE genes. The statistic is hypergeometric enrichment.
To understand how increased P300 HAT activity affects transcriptional processes in ARID1A-deficient cells, we performed RNA-seq following knockdown of P300, ARID1A, or both in 12Z cells. We used the GeneHancer database (Fishilevich et al., 2017) to associate regions of differential H3K27ac targets (Figure 5L). Genes linked to the gain reversal cluster were enriched for genes with differential expression (DE) following ARID1A knockdown versus control and for differential expression following ARID1A and P300 co-knockdown versus ARID1A knockdown alone (Figure 5M). Specifically, genes linked to gain reversal regions were more likely to be upregulated following ARID1A knockdown relative to control and downregulated following ARID1A and P300 co-knockdown relative to ARID1A knockdown alone (Figures 5N and 5O).
We reasoned that upregulated genes driving ARID1A-deficient invasion would be rescued upon P300 loss or A-485 treatment. To narrow down a smaller subset of genes responsible for P300-dependent invasion in ARID1A-deficient cells, we performed additional RNA-seq using 100 nM A-485, a lower dose that has no effects on cell health or global H3K27ac reduction, but significantly inhibits invasion and migration (Figures 4F–4H, S3B, and S4F). While P300 loss resulted in the differential expression of 2,657 genes (false discovery rate [FDR] < 0.0001), 100 nM A-485 treatment resulted in the differential expression of only 566 genes, suggesting a more specific effect (Figure 6A). Concordantly dysregulated genes between siP300 and A-485 overlapped, providing additional validation of this approach (Figure 6A). To determine gene regulation by SEs, we identified 3 groups of SE-regulated genes: active genes with a promoter directly within an SE (Figure 6B) (Whyte et al., 2013), active genes with a promoter within 50 kb of an SE (Figure 6C) (Sanyal et al., 2012), and active genes linked to SEs through the GeneHancer database (Figure 6D). In all cases, SE-regulated genes were enriched among DE genes with ARID1A loss and further P300 loss or HAT inhibition (Figures 6B–6D). To identify genes implicated in ARID1A mutant invasion, we compared overlapping genes sets from siARID1A versus control, siARID1A + siP300 versus siARID1A, and siARID1A + 100 nM A-485 versus siARID1A comparisons and identified a set of 138 “triple intersect” genes (Figure 6E). These correspond to genes affected by ARID1A loss and further affected by P300 loss or inhibition of P300 HAT activity. This gene set was enriched for the hallmark epithelial-to-mesenchymal transition pathway and Gene Ontology (GO) gene sets related to invasive phenotypes (Figures 6F and 6G). Among the 138 triple intersect genes, we identified 50 genes that were upregulated by ARID1A loss and further suppressed by P300 loss or low-dose A-485-mediated HAT inhibition (Figure 6H). Of these, 16 genes were associated with H3K27ac gain reversal enhancers, and 3 gene loci have associated SE elements. Only SERPINE1 was identified as displaying gene expression reversal following ARID1A loss and further 100-nM A-485 treatment, association with H3K27ac gain reversal enhancer elements, and regulation by a SE.
Figure 6. Inhibition of P300 Histone Acetyltransferase (HAT) Activity Reverses the Expression of a Subset of ARID1A-Regulated Genes.
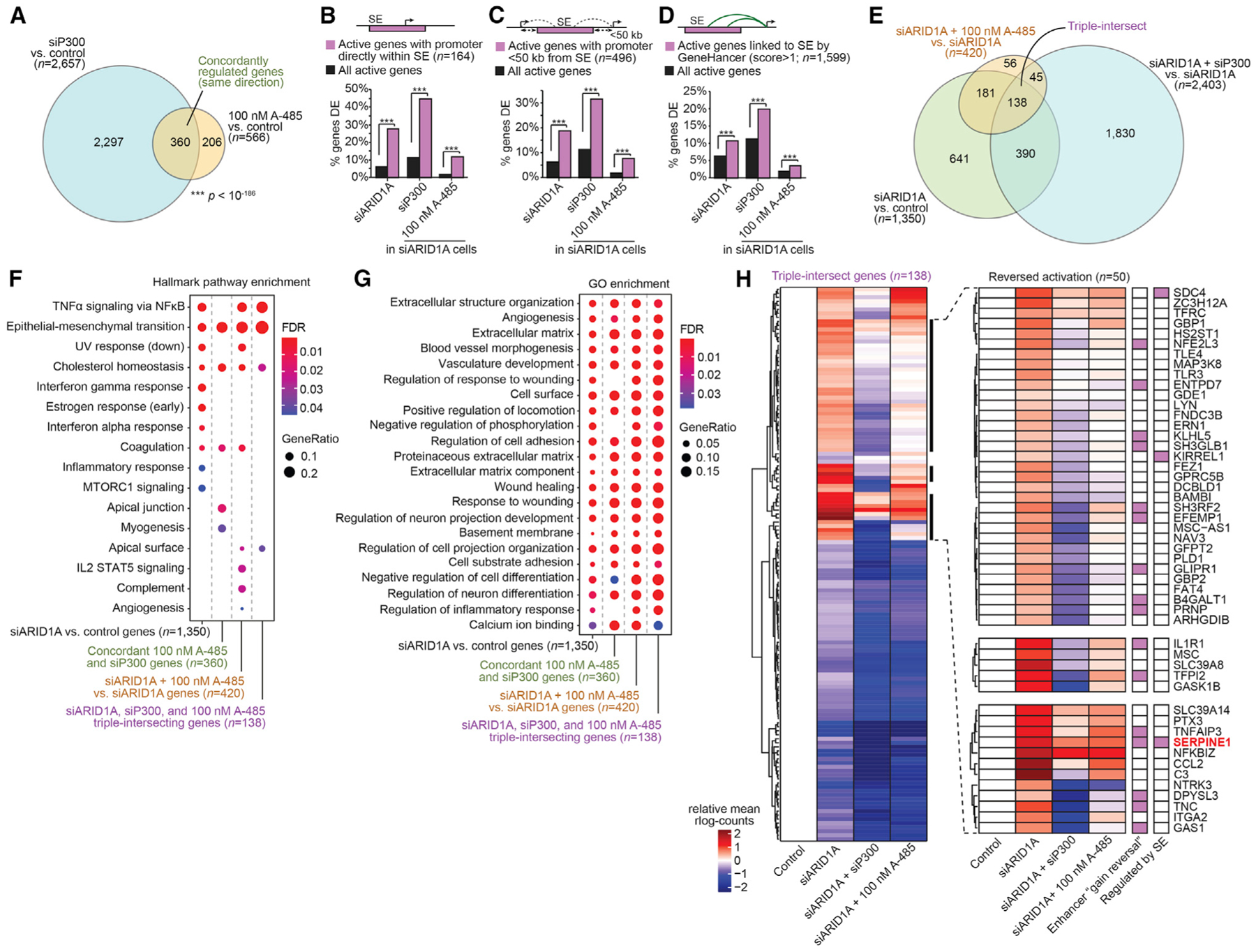
(A) Proportional Euler diagram displaying concordant, overlapping DE genes (FDR < 0.0001) by siP300 or 100 nM A-485 treatment (p < 10−186). The statistic is hypergeometric enrichment.
(B–D) Enrichment of DE genes affected by ARID1A loss, P300 loss, or A-485 treatment for (B) genes with active promoters directly inside of SE (n = 164), (C) promoters within 50 kb of a SE (n = 496), or (D) genes linked to SE by the GeneHancer database (n = 1,599). The statistic is hypergeometric enrichment.
(E) Proportional Euler diagram displaying overlap of DE genes (FDR < 0.0001) in indicated comparisons. “Triple intersect” genes refer to the full intersection of all noted gene expression comparisons.
(F and G) Gene set enrichment analysis for (F) MSigDB Hallmark pathways and (G) Gene Ontology (GO) biological process terms on various DE genes clades identified in (A) and (E).
(H) Heatmap for relative expression of triple intersect genes (n = 138, as in E), highlighting genes in which 100 nM A-485 reverses ARID1A loss-driven upregulation (right, n = 50). Red values: increased expression relative to control; blue: decreased expression relative to control. The rightmost columns indicate association with acetylation gain reversal enhancers (Figures 5D and 5E) or regulation by SE, in purple.
***p < 0.001.
SERPINE1 Promotes ARID1A Mutant Cell Invasion
The serine protease inhibitor, SERPINE1 (also known as plasminogen activator inhibitor type 1 [PAI-1]), is a member of the urokinase plasminogen activator (uPA) system (Smith and Marshall, 2010). This system regulates extracellular fibrin proteolysis and influences cell invasion, migration, and ECM remodeling (Duffy, 2004). SERPINE1 is a biomarker for endometriosis, with high levels of expression observed in ovarian and deep infiltrating endometriosis (Alotaibi et al., 2019; Gilabert-Estellés et al., 2003; Ramón et al., 2005; Ye et al., 2017). We examined a published RNA-seq dataset of human endometrial organoids and observed that SERPINE1 was upregulated in organoids derived from ectopic endometrial tissue compared to healthy endometrial tissue (log2 fold change [FC] = 3.86, FDR = 0.051) (Boretto et al., 2019). In 12Z cells, the SERPINE1 SE was ranked in the top 5% of active SEs (Figure 7A), and it displayed H3K27 hyperacetylation upon ARID1A loss, which was reversed by further P300 loss or A-485 treatment (Figure 7B). Notably, SERPINE1 was the most significant upregulated gene upon ARID1A loss (Figure 7C), and P300 co-knockdown or HAT inhibition rescued SERPINE1 expression (Figures 7D and 7E). SERPINE1 was also upregulated in the endometrial epithelium of LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice (Figure 7F). LtfCre0/+; (Gt) R26Pik3ca*H1047R; Arid1afl/fl eutopic endometrial epithelia and ectopic lesions showed increased SERPINE1 by IHC, which was not observed in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl (Figures 7G and 7H). Among a cohort of deep infiltrating and ovarian endometriosis tissue samples (Alotaibi et al., 2019), samples with a loss of ARID1A expression displayed the highest expression of SERPINE1 by IHC (Figures S6A and S6B).
Figure 7. Hyperactivation of SERPINE1 SE Promotes ARID1A Mutant Cell Invasion.
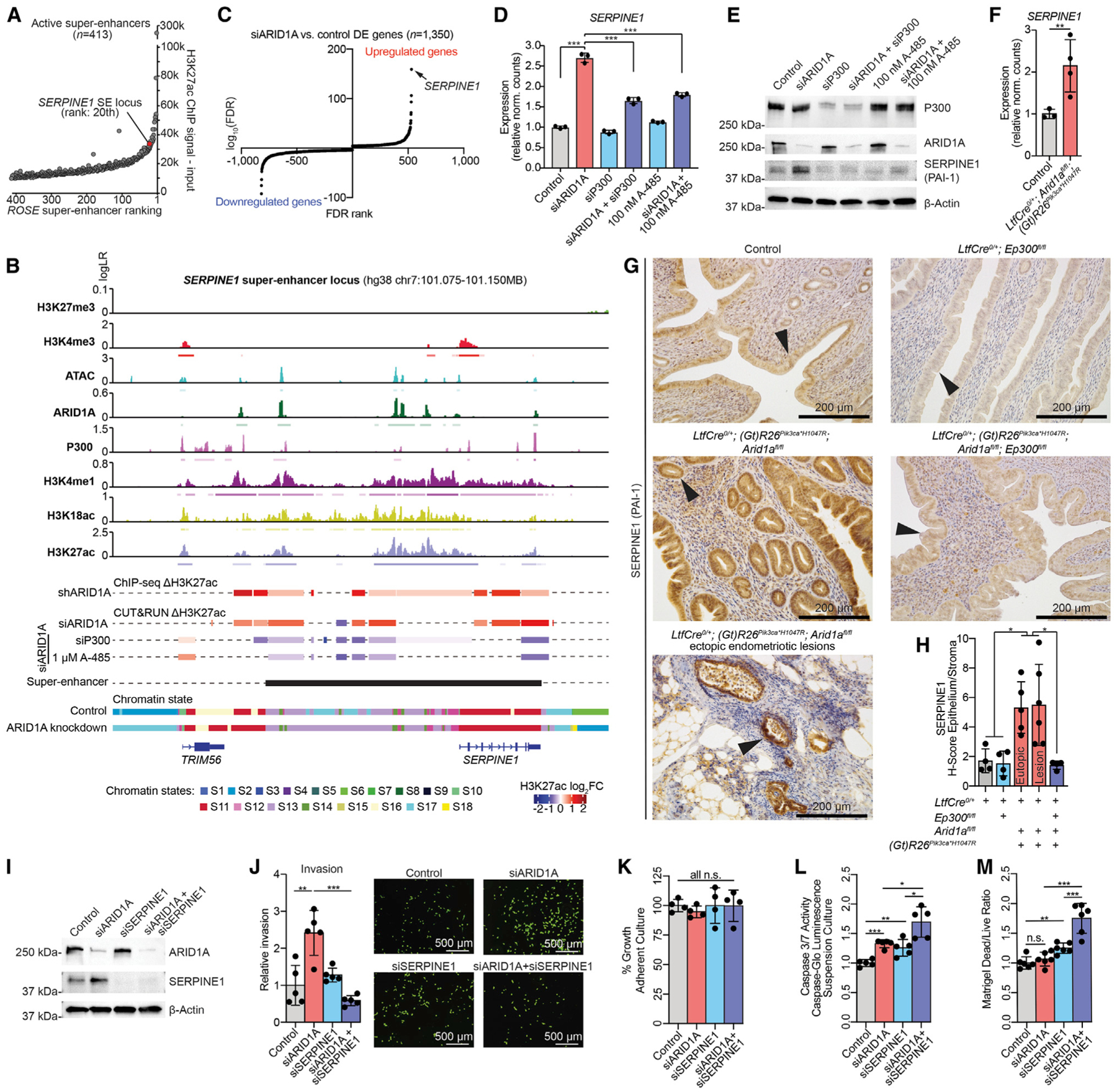
(A) ROSE ranking of active SEs (n = 413). The SERPINE1 SE locus is ranked 20 out of 413 based on H3K27ac levels.
(B) Genomic snapshot of ChIP and ATAC signals alongside differential H3K27ac and chromatin state annotations at the SERPINE1 SE locus. For signal tracks, the y axis represents assay signal-to-noise presented as log-likelihood ratio (logLR) as reported by MACS2, and small bars below the tracks represent replicate-overlapping peaks. H3K27ac log2FC colored bars denote significant differential H3K27ac regions (FDR < 0.05 for ChIP, FDR < 0.01 for CUT&RUN). ROSE active SE locus is represented by the black bar.
(C) Significance (log10FDR, y axis) of DE genes following ARID1A loss, ranked by FDR value (x axis). SERPINE1 is the most significantly upregulated gene (arrow).
(D) Expression of SERPINE1 (RNA-seq) following indicated 12Z cell treatments. Means ± SDs, n = 3. The statistic is DESeq2 FDR.
(E) Western blot analysis as indicated in 12Z cells, representative of 2 independent experiments.
(F) Relative expression of SERPINE1 by RNA-seq. Means ± SDs, n = 3 control mice and n = 4 mutant mice. The statistic is DESeq2 FDR.
(G) IHC of SERPINE1 in endometrium of indicated genotypes; n = 4–5 mice per condition.
(H) Quantification of IHC staining, ratio of H-scores of epithelia to stroma. Means ± SDs, n = 4–5 mice, unpaired, 2-tailed t test.
(I) Western blot analysis as indicated in 12Z cells, representative of 2 independent experiments.
(J) Invasion of 12Z following indicated treatment. Representative images and total invaded cell numbers are shown (scale bar, 500 μm). Means ± SDs, n = 5, unpaired, 2-tailed t test.
(K) Measurement of cell growth following indicated treatments. Means ± SDs, n = 4. No significant differences, unpaired, 2-tailed t test.
(L) Caspase-Glo assay of 12Z in suspension following indicated treatments. Means ± SDs, n = 5, unpaired, 2-tailed t test.
(M) Ratio of dead to live cells after 24 h in Matrigel. Means ± SDs, n = 6, unpaired, 2-tailed t test.
*p < 0.05, **p < 0.01, and ***p < 0.001.
To determine whether SERPINE1 promotes the invasion of ARID1A-deficient cells, we inactivated SERPINE1 via siRNA transfection in 12Z (Figure 7I). While there was no change in invasion with SERPINE1 loss alone, SERPINE1 loss suppressed the invasive phenotype of ARID1A-deficient cells (Figure 7J). SERPINE1 loss had no effect on adherent cell growth (Figures 7K and S3B). In non-adherent conditions, ARID1A and SERPINE1 co-knockdown resulted in increased caspase 3/7 activity (Figure 7L) and increased death in cells suspended in Matrigel (Figure 7M), indicating that SERPINE1 is required for anoikis resistance in ARID1A mutant cells. These results suggest that ARID1A prevents hyperacetylation of the SERPINE1 SE in the wild-type state, while ARID1A loss results in P300-dependent hyperacetylation and increased activity of the SERPINE1 SE, increased SERPINE1 transcription, and the acquisition of invasive phenotypes. P300 inhibition in ARID1A-deficient cells suppresses H3K27 hyperacetylation of the SERPINE1 SE, resulting in decreased SERPINE1 expression and anoikis.
DISCUSSION
In this study, we demonstrate that ARID1A prevention of SE hyperactivation plays an essential physiological role in maintaining endometrial tissue homeostasis and preventing cell invasion. ARID1A mutant cell invasion has been described in other diseases and malignancies, but the functional link between ARID1A loss, SE hyperactivation, and the subsequent acquisition of P300-dependent invasiveness is unique to the endometrium (Lakshminarasimhan et al., 2017; Li et al., 2017; Sun et al., 2017; Yan et al., 2014). Retrograde menstruation is thought to play a role in the spread of abnormal endometrial tissue to ectopic sites. ARID1A mutations may predispose displaced endometrial cells to forming endometriotic lesions by promoting the acquisition of invasive phenotypes in a cell-autonomous manner (Wilson et al., 2019, 2020). Our findings suggest that epigenetic dysregulation of SEs promotes endometrial invasion and survival at ectopic sites. Alterations in SE activity may be an important feature of endometriotic epithelium.
The SWI/SNF-mediated regulation of SEs may be cell type specific. SWI/SNF subunit SMARCB1 can antagonize chromatin accessibility at SEs in mouse embryonic stem cells (Langer et al., 2019), while SMARCB1 loss in rhabdoid tumors impairs SWI/SNF binding to TEs, not SEs (Wang et al., 2017). SWI/SNF can regulate Myc expression in acute myeloid leukemia through interactions with a lineage-specific SE (Shi et al., 2013), as has been described among other enhancers (Alver et al., 2017). In mouse embryonic fibroblasts, the deletion of SWI/SNF family members has been shown to reduce H3K27ac at enhancers (Alver et al., 2017). SWI/SNF can promote chromatin accessibility at enhancers (Kelso et al., 2017; Vierbuchen et al., 2017). In embryonic stem cells, mutations in SWI/SNF catalytic subunit Brahma result in enhancer reprogramming (Gao et al., 2019). In breast cancer, ARID1A binds and represses enhancers containing estrogen receptor-binding elements through co-recruitment of HDAC1, and ARID1A loss results in H4 acetylation, BRD4 recruitment, and subsequent transcription (Nagarajan et al., 2020). However, in that study, ARID1A knockout did not result in differential H3K27ac (Nagarajan et al., 2020). Our data suggest a distinct role for ARID1A-P300 antagonism in the regulation of SE chromatin accessibly and H3K27ac deposition in the endometrial epithelium.
We previously showed that ARID1A genome-wide binding is enriched at promoters, and promoter chromatin accessibility increases following ARID1A loss (Wilson et al., 2019). Although ARID1A is enriched at promoters and we observed ARID1A-P300 co-binding at these sites, we demonstrate here that ARID1A-P300 antagonism uniquely occurs at SEs, which show hyperacetylated H3K27, increased chromatin accessibility, and eRNA transcription following ARID1A loss. Both SERPINE1 and SDC4 have large SEs spanning the promoter region, although we also show that SE regulation by ARID1A, P300 and A-485 affects the transcription of genes located up to 50 kb away. Interestingly, although ARID1A is bound at the SERPINE1 promoter and multiple sites throughout the 41-kb SERPINE1 SE, increased H3K27ac is observed throughout the majority of the SE interval in ARID1A-deficient cells, suggesting that ARID1A regulates chromatin over large chromatin domains.
Recently, there has been interest in the therapeutic inhibition of SE activity in several diseases. Small-molecule inhibitors of SE factors, particularly the BET bromodomain inhibitor JQ1, have undergone clinical trials for multiple cancer types (Shin, 2018). BRD4 interacts with H3K27ac-rich SE regions, and the disruption of BRD4 bromodomain-SE interactions using small molecules can decrease oncogene expression (Sengupta et al., 2015). The inhibition of histone acetylation represents a growing area of interest in small-molecule therapeutics (Simon et al., 2016). Targeted disruption of P300 HAT activity at SE may have therapeutic utility in endometrial diseases.
Several studies have demonstrated a relationship between SERPINE1/PAI-1 expression and endometriosis (Bruse et al., 1998, 2004). SERPINE1 promoter polymorphisms linked to high levels of PAI-1 expression have been reported in endometriosis (Bedaiwy et al., 2006; Ramón et al., 2005). Relative to other types of endometriosis, PAI-1 expression is increased in deep infiltrating (Alotaibi et al., 2019) and ovarian endometriosis (Gilabert-Estellés et al., 2003; Ramón et al., 2005). ARID1A mutations exist in both deep infiltrating and ovarian endometriosis (Anglesio et al., 2017; Suda et al., 2018), and our clinical cohort suggests that ARID1A loss leads to PAI-1 overexpression in endometriosis. As a secreted factor, elevated plasma PAI-1 levels have been observed in women with recurrent pregnancy loss or preeclampsia, and the secretion of PAI-1 from endometriotic lesions may contribute to endometriosis-associated infertility and pain (Ye et al., 2017). PAI-1 negatively regulates fibrinolysis and plays a role in endometrial hemostasis during menstruation (Davies and Kadir, 2012; Mehta and Shapiro, 2008). In addition to its roles in cell adhesion and migration, it remains possible that increased PAI-1 affects menstrual clotting and promotes fibrosis or scar tissue formation in endometriosis. Lastly, PAI-1 may serve as a biomarker for invasive ARID1A mutant endometriosis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronald Chandler (rlc@msu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the sequencing data generated in this manuscript is GEO: GSE148474. Previously published datasets analyzed herein are also available at GEO: GSE121198.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse care, use, and genotyping
All mice were maintained on an outbred genetic background using CD-1 mice (Charles River). (Gt)R26Pik3ca*H1047R, LtfCre (Tg(Ltf-iCre)14Mmul) and Ep300fl alleles were purchased from The Jackson Laboratory and identified by PCR using published methods (Adams et al., 2011; Daikoku et al., 2014; Kasper et al., 2006). Arid1afl allele was distinguished by PCR as previously described (Chandler et al., 2015). Genotyping primers are listed in Key Resources Table. Endpoints were vaginal bleeding, severe abdominal distension, and signs of severe illness including dehydration, hunching, jaundice, ruffled fur, signs of infection, or non-responsiveness. Sample sizes for each genotype were chosen based on the proportions of animals with vaginal bleeding between each experimental group and Kaplan-Meyer log rank test for survival differences. All mice analyzed in the study were between 6 and 32 weeks old. In cases where a mobility endpoint occurred, tissues were collected at the time of vaginal bleeding, including LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl (μ1/2 = 107 days) and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl; Ep300fl/fl (μ1/2 = 143 days) mice. In cases where the animal did not reach a morbidity endpoint or show reduced survival, tissues were collected at comparable time points (between 90 and 150 days) from age-matched, littermate control mice from the mutant crosses. Uteri were collected at time of sacrifice and placed immediately into neutral-buffered formalin at 4°C. After 24 hr, tissues were washed with PBS and 50% EtOH, placed in 70% EtOH, and weight measurements were recorded. Mice were housed at the Michigan State University Grand Rapids Research Center in accordance with protocols approved by Michigan State University. Michigan State University is registered with the U.S. Department of Agriculture (USDA) and has an approved Animal Welfare Assurance from the NIH Office of Laboratory Animal Welfare (OLAW). MSU is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-ARID1A/BAF250A (D2A8U) | Cell Signaling Technology | Cat# 12354; RRID: AB_2637010 |
| Rabbit monoclonal anti-b-Actin (D6A8) | Cell Signaling Technology | Cat# 8457; RRID: AB_10950489 |
| Rabbit monoclonal anti-Akt (pan) (C67E7) | Cell Signaling Technology | Cat# 4691; RRID: AB_915783 |
| Rabbit monoclonal anti-Phospho-Akt (Ser473) (D9E) | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Rabbit monoclonal anti-Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) |
Cell Signaling Technology | Cat# 4858; RRID: AB_2721245 |
| Rabbit monoclonal anti-Cleaved Caspase-3 (Asp175) (D3E9) | Cell Signaling Technology | Cat# 9579; RRID: AB_10897512 |
| Mouse monoclonal anti-p300 (NM11) | Santa Cruz Biotechnology | Cat# sc-32244; RRID: AB_628076 |
| Rabbit monoclonal anti-p300 (D8Z4E) | Cell Signaling Technology | Cat# 86377; RRID: AB_2800077 |
| Rat monoclonal anti-TROMA-I | Developmental Studies Hybridoma Bank |
Cat# TROMA-I; RRID: AB_531826 |
| Mouse monoclonal anti-PAI-1 (SERPINE1) (C-9) | Santa Cruz Biotechnology | Cat# sc-5297; RRID: AB_628154 |
| Rabbit polyclonal anti-PAI-1 (SERPINE1) | Abcam | Cat# ab66705; RRID: AB_1310540 |
| Rabbit monoclonal anti-Histone H3 (D1H2) XP | Cell Signaling Technology | Cat# 4499; RRID: AB_10544537 |
| Rabbit polyclonal anti-H3K27ac | Active Motif | Cat# 39133; RRID: AB_2561016 |
| Rabbit polyclonal anti-H3K18ac | Abcam | Cat# ab1191; RRID: AB_298692 |
| Rabbit polyclonal anti-H3K4me1 | Abcam | Cat# ab8895; RRID: AB_306847 |
| Rabbit monoclonal anti-H3K4me3 (C42D8) | Cell Signaling Technology | Cat# 9751; RRID: AB_2616028 |
| Rabbit monoclonal anti-H3K27me3 (C36B11) | Cell Signaling Technology | Cat# 9733; RRID: AB_2616029 |
| Rabbit monoclonal anti-Ki67 (D3B5) | Cell Signaling Technology | Cat# 12202; RRID: AB_2620142 |
| Donkey anti-Rabbit IgG, Biotin-SP-conjugated | Jackson ImmunoResearch Labs | Cat# 711-065-152; RRID: AB_2340593 |
| Donkey anti-Rat IgG, Biotin-SP-conjugated | Jackson ImmunoResearch Labs | Cat# 712-065-153; RRID: AB_2315779 |
| Goat anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Horse anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID: AB_330924 |
| Donkey anti-rabbit IgG, IRDye 800CW conjugated antibody | LI-COR Biosciences | Cat# 926–32213; RRID: AB_621848 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Normal Donkey Serum | Jackson ImmunoResearch Labs | Cat# 017-000-121 |
| Bovine Serum Albumin (IgG-Free, Protease-Free) | Jackson ImmunoResearch Labs | Cat# 001-000-161 |
| Intercept Blocking Buffer (TBS) | LI-COR Biosciences | Cat# 927–60001 |
| Sodium Citrate | Sigma-Aldrich | Cat# C8532 |
| Tris | VWR Life Science | Cat# 0497 |
| Signal Stain Ab Diluent | Cell Signaling Technology | Cat# 8112L |
| Animal-Free Blocking Solution (5X) | Cell Signaling Technology | Cat# 15019L |
| VECTASTAIN Elite ABC-HRP Kit (Peroxidase, Standard) | Vector Laboratories | Cat# PK-6100 |
| ImmPACT DAB substrate kit | Vector Laboratories | Cat# SK-4105 |
| Hematoxylin QS | Vector Laboratories | Cat# H-3404 |
| Proteinase K | ThermoFisher | Cat# EO0491 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8340 |
| A-485 | Tocris | Cat# 6387 |
| Trichostatin A | Tocris | Cat# 1406 |
| SAHA | Tocris | Cat# 4652 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833 |
| Vybrant Dye Cycle Ruby Stain | ThermoFisher | Cat# V10309 |
| CUTANA pAG-MNase | EpiCypher | Cat# 15–1016 |
| BioMag Plus Concanavalin A | Bangs Laboratories | Cat# BP531 |
| Critical Commercial Assays | ||
| Lipofectamine RNAiMAX Transfection Reagent | Life Technologies | Cat# 13778150 |
| FuGENE HD Transfection Reagent | Promega | Cat# E2311 |
| PureLink HiPure Plasmid Maxiprep Kit | Invitrogen | Cat# K210006 |
| qPCR Lentivirus Titration Kit | abm | Cat# LV900 |
| SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) | Cell Signaling | Cat# 9003 |
| ChIP DNA Clean & Concentrator Kit | Zymo Research | Cat# D5201 |
| Microplate BCA Protein Assay Kit – Reducing Agent Compatible | Thermo Scientific | Cat# 23252 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat# 8090 |
| Annexin V-FITC Kit | Miltenyi Biotec | Cat# 130-092-052 |
| Viability/Cytotoxicity Assay Kit for Animal Live & Dead Cells | Biotium | Cat# 30002-T |
| KAPA Hyper Prep Kit (v5.16) | Kapa Biosystems | Cat# KR0961 |
| Bioo Scientific NEXTflex Adapters | Bioo Scientific | Cat# NOVA-401001 |
| Kapa Illumina Library Quantification qPCR assays | Kapa Biosystems | Cat# KK4824 |
| TG NextSeq® 500/550 High Output Kit v2 (150 cycles) | Illumina | Cat# TG-160–2002 |
| NovaSeq 6000 SP Reagent Kit (100 cycles) | Illumina | Cat# 20027464 |
| Mycoplasma PCR Detection Kit | Applied Biological Materials | Cat# G238 |
| Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit | ThermoFisher | Cat# C10420 |
| NucleoSpin Gel and PCR Clean-up Kit | Takara | Cat# 740609.50 |
| Deposited Data | ||
| 12Z cell line wild-type P300 ChIP-seq dataset (P300_ChIP: GSE148470) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line shRNA-treated P300 ChIP-seq dataset (diff_P300_ChIP: GSE148471) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line shRNA-treated H3Kac ChIP-seq dataset (diff_H3Kac_ChIP: GSE148472) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line siRNA-transfection and A-485 treatment RNA-seq dataset (RNA: GSE148473) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line siRNA-transfection and A-485 treatment CUT&RUN dataset (diff_H3K27ac_CUTNRUN: RNA: GSE157731) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line shRNA-treated H3K4me1 ChIP-seq dataset (diff_H3me1_ChIP: GSE157732) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| 12Z cell line shRNA-treated H3K27me3 and H3K4me3 ChIP-seq dataset (diff_H3K4me3_H3K27me3_ChIP: GSE157735) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE148474. |
| In vivo mouse EPCAM-sorted endometrial epithelium RNA-seq dataset (Mouse_RNA-seq: GSE129784) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| In vivo mouse EPCAM-sorted endometrial epithelium ATAC-seq dataset (Mouse_ATAC-seq: GSE129783) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| 12Z cell line ATAC-seq dataset (12Z_ATAC-seq: GSE129780) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| 12Z cell line siRNA-transfection RNA-seq dataset (12Z_RNA-seq: GSE129782) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| 12Z cell line siRNA and plasmid-transfection RNA-seq dataset (12Z_1A_PI3K_RNA-seq: GSE129779) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| 12Z cell line ARID1A ChIP-seq dataset (12Z_ChIP-seq: GSE129781) | Deposited to Gene Expression Omnibus | GEO accession: SuperSeries GSE121198. |
| Experimental Models: Cell Lines | ||
| Human: 12Z human endometriosis | Laboratory of Asgi Fazleabas | RRID: CVCL_0Q73 |
| Human: Lenti-X™ 293T embryonic kidney cells | Clontech | Cat# 632180; RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tg(Ltf-iCre)14Mmul | Jackson Laboratory | Cat# 026030 |
| Mouse: (Gt)Rosa26Pik3ca*H1047R | Jackson Laboratory | Cat# 016977 |
| Mouse: Arid1afl | Chandler et al., 2015 | N/A |
| Mouse: Ep300fl | Jackson Laboratory | Cat# 025526 |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | Cat# D-001810 |
| SMARTpool: ON-TARGETplus ARID1A siRNA | Dharmacon | Cat# L-017263–00 |
| SMARTpool: ON-TARGETplus EP300 siRNA | Dharmacon | Cat# L-003486–00 |
| SMARTpool: ON-TARGETplus SERPINE1 siRNA | Dharmacon | Cat# L-019376–01 |
| LtfCre common primer: AACTAGCACACCTGGTTGAGG | Jackson Laboratory | Primer 21216 |
| LtfCre wild type reverse genotyping primer: CTTCTTGGGAGGCAGTGAAC | Jackson Laboratory | Primer 21217 |
| LtfCre mutant reverse genotyping primer: CAGGTTTTGGTGCACAGTCA | Jackson Laboratory | Primer 21218 |
| Rosa26 common genotyping primer: CTGGCTTCTGAGGACCG | Jackson Laboratory | Primer 21306 |
| Rosa26 mutant reverse genotyping primer: CGAAGAGTTTGTCCTCAACCG | Jackson Laboratory | Primer 21307 |
| Rosa26 wild type reverse genotyping primer: AATCTGTGGGAAGTCTTGTCC | Jackson Laboratory | Primer 21310 |
| Ep300 forward genotyping primer: GTGAGTTGATGTCCCTGTCG | Jackson Laboratory | Primer 20733 |
| Ep300 reverse genotyping primer: CAGACACCCTCTTGCACTCA | Jackson Laboratory | Primer 20734 |
| Arid1a common genotyping primer: CTAGGTGGAAGGTAGCTGACTGA | Chandler et al., 2015 | N/A |
| Arid1a wild type reverse genotyping primer: TACACGGAGTCAGGCTGAGC | Chandler et al., 2015 | N/A |
| Arid1a mutant reverse genotyping primer: AGAGTAACTAATAACTGCTGGAGGATG | Chandler et al., 2015 | N/A |
| Recombinant DNA | ||
| pBabe puro | AddGene | Cat# 1764 |
| pBabe puro HA PIK3CA H1047R | AddGene | Cat# 12524 |
| MISSION® pLKO.1-puro Non-Target shRNA Control Plasmid DNA | Sigma | Cat# SHC016 |
| ARID1A MISSION® shRNA Plasmid DNA | Sigma | Cat# TRCN0000059091; NM_006015.3–7163s1c1 |
| ARID1A MISSION® shRNA Plasmid DNA | Sigma | Cat# TRCN0000059090; NM_006015.3–1702s1c1 |
| ARID1A MISSION® shRNA Plasmid DNA | Sigma | Cat# TRCN0000059089; NM_006015.3–2287s1c1 |
| pNHP | Manfredson Lab | N/A |
| pHEF-VSVG | Manfredson Lab | N/A |
| Software and Algorithms | ||
| Prism 8 | Graphpad | https://www.graphpad.com |
| ImageJ 1.52k | National Institutes of Health | https://imagej.nih.gov/ij |
| Adobe Illustrator CC 24.1 | Adobe | https://www.adobe.com |
| Excel 16.16.20 | Microsoft | https://products.office.com/excel?legRedir=true&CorrelationId=ab2af84b-7abf-4e89-94c5-ce16df5245a1 |
| NIS Elements Advanced Research 4.30.02 | Nikon | https://www.microscope.healthcare.nikon.com/ |
| Excel 16.16.2 | Microsoft | https://products.office.com/excel?legRedir=true&CorrelationId=1025d416-560d-4d7a-aee3-00a26279ffe2 |
| R 3.5.0 | R Core Team, 2018 | https://www.r-project.org/ |
| Trim Galore! 0.4.1 | Krueger, F. | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| FastQC 0.11.3 | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| MultiQC 1.6 | Ewels et al., 2016 | https://multiqc.info/ |
| cutadapt 1.15 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| STAR 020201 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESeq2 1.22.2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| IHW: Independent Hypothesis Weighting 1.10.1 | Ignatiadis et al., 2016 | http://bioconductor.org/packages/release/bioc/html/IHW.html |
| Bowtie2 2.2.6 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SAMtools 1.7 | Li et al., 2009 | http://www.htslib.org/doc/samtools.html |
| BEDtools 2.24.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS 2.1.0 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| csaw 1.16.1 | Lun and Smyth, 2016 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| ggplot2 3.3.0 | Wickham, 2016 | https://ggplot2.tidyverse.org/ |
| preseqR 4.0.0 | Daley and Smith, 2013 | https://cran.r-project.org/web/packages/preseqR/index.html |
| ATACseqQC 1.6.4 | Ou et al., 2018 | https://bioconductor.org/packages/release/bioc/html/ATACseqQC.html |
| GenomicRanges 1.34.0 | Lawrence et al., 2013 | |
| eulerr 6.1.0 | Larsson, 2020 | https://cran.r-project.org/web/packages/eulerr/index.html |
| ROSE: Rank Ordering of Super-Enhancers | Whyte et al., 2013; Lovén etal., 2013 | https://bitbucket.org/young_computation/rose/src/master/ |
| HOMER 4.10.3 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| biomaRt 2.38.0 | Durinck et al., 2005, 2009 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| GeneHancer database 4.4 | Fishilevich et al., 2017 | https://www.genecards.org/GeneHancer_version_4-4 |
| ComplexHeatmap 1.20.0 | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| edgeR 3.24.3 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| ENCODE hg38 blacklist | Amemiya et al., 2019 | https://github.com/Boyle-Lab/Blacklist |
| TxDb.Hsapiens.UCSC.hg38.knownGene 3.4.0 | Bioconductor Core Team and Bioconductor Package Maintainer, 2016 | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsapiens.UCSC.hg38.knownGene.html |
| Picard Tools 2.0.1 | Broad Institute | http://broadinstitute.github.io/picard/ |
| GENCODE v28 | Frankish et al., 2019 | https://www.gencodegenes.org/human/release_28.html |
| ChromHMM | Ernst and Kellis, 2017 | http://compbio.mit.edu/ChromHMM/ |
| Other | ||
| Corning® Transwell® polycarbonate membrane cell culture inserts | Sigma-Aldrich | Cat# 3422 |
| Cultrex PathClear Basement Membrane Extract | R & D Systems | Cat# 3432-005-01 |
| Culture-Insert 4 Well in m-Dish 35 mm high ibiTreat | ibidi | Cat# 80466 |
| 96-well Cellstar Cell-Repellent plate | Greiner Bio-one | Cat# 655970 |
| white 96-well plate | costar | Cat# 3610 |
Cell lines
12Z immortalized human endometrial epithelial cells (Zeitvogel et al., 2001) were maintained in DMEM/F12 media supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin/streptomycin (P/S). The 12Z cells were provided by the laboratory of Asgi Fazleabas, and cell line validation was performed by IDEXX BioResearch, finding the result that the 12Z cell line has a unique profile not found in the current public databases. A recent study found 12Z cells to be an authentic and pure endometriosis cell line based on marker analysis and short tandem repeat profiling (Romano et al., 2020). Lenti-X™ 293T (Clontech) cells were maintained in DMEM +110 mg/L Sodium Pyruvate (GIBCO) supplemented with 10% FBS, 1% L-glutamine, 1% P/S. 12Z and Lenti-X 293T cells were regularly tested for mycoplasma using the Mycoplasma PCR Detection Kit (Applied Biological Materials). No commonly mis-identified cell lines were used in this study.
METHOD DETAILS
Histology and immunohistochemistry
For indirect immunohistochemistry (IHC), 10% neutral buffered formalin (NBF)-fixed paraffin sections were processed for heat-based antigen unmasking in 10 mM sodium citrate [pH 6.0]. Sections were incubated with antibodies at the following dilutions: 1:200 ARID1A (D2A8U) (12354, Cell Signaling); 1:1000 P300 (86377, Cell Signaling); 1:400 Phospho-S6 (4585, Cell Signaling); 1:100 KRT8 (TROMA1, DHSB); 1:200 Cleaved Caspase-3 (9579, Cell Signaling); 1:400 Ki67 (12202, Cell Signaling); 1:200 H3K27ac (39133, Active Motif); 1:200 H3K18ac (ab1191, Abcam); 1:1000 PAI-1 (SERPINE1) (ab66705, Abcam). TROMA-I antibody was deposited to the DSHB by Brulet, P./Kemler, R. (DSHB Hybridoma Product TROMA-I). Biotin-conjugated secondary antibodies were donkey anti-rabbit IgG (711-065-152, Jackson Immuno-research Lab) and donkey anti-rat IgG (#705-065-153, Jackson Immuno-research Lab). VECTASTAIN Elite ABC HRP Kit (Vector) was used for secondary antibody detection. Sections for IHC were lightly counter-stained with Hematoxylin QS or Methyl Green (Vector Labs). Routine Hematoxylin and Eosin (H&E) staining of sections was performed by the Van Andel Research Institute (VARI) Histology and Pathology Core. A VARI animal pathologist reviewed histological tumor assessments.
To determine H-scores from mouse slides, one field of view (20X) on a Nikon Eclipse Ni-U upright microscope per mouse from a slide stained with antibody (SERPINE1, H3K27ac, H3K18ac) was used. Epithelial and stromal cells were assigned a value from 0 to 3 indicating intensity of staining (no staining = 0, low staining = 1, moderate staining = 2, and strong staining = 3) and the proportion of cells was determined for each staining intensity. For Ki67, a value of 1 (positive staining) or 0 (negative staining) was assigned to determine the number of Ki67+ cells.
For human endometriosis tissue samples, SERPINE1 (PAI-1) IHC was carried out as published (Alotaibi et al., 2019). Briefly, IHC using the EnVision+ Dual Link system (Dako) and 3,3-diaminobenzidine (DAB) was performed, using mouse monoclonal PAI-1 antibody C-9 (sc 5297, Santa Cruz). PAI-1 expression was evaluated in endometriotic epithelium and stroma using the Histoscore calculation. Areas of endometriosis epithelium and stroma were first scanned at low power (×10) and then analyzed at high power (×40) to evaluate the staining intensity and estimate the proportion of positive cells. ARID1A IHC was used as a surrogate of loss-of-function alterations (Khalique et al., 2018; Trizzino et al., 2018) using a Dako Omnis automated immunostainer (Agilent Technologies) and the anti-ARID1A rabbit monoclonal D2A8U (Cell Signaling Technology).
Transfections
12Z cells were seeded at a density of 30,000 cells/mL in DMEM/F12 media supplemented with 10% FBS and 1% L-glutamine. The following day, cells were transfected with 50 nM siRNA (Dharmacon, ON-TARGETplus Non-targeting Pool, human ARID1A #8289 SMARTpool, human P300 #3486 SMARTpool, human SERPINE1 #19376 SMARTpool) using the RNAiMax (ThermoFisher) lipofectamine reagent according to the manufacturer’s instructions at a ratio of 1:1 volume:volume in OptiMEM (GIBCO). After 24 hr, the media was replaced. For plasmid co-transfection experiments, cells were transfected the following day with 500ng pBabe vector containing PIK3CAH1047R (pPIK3CAH1047R) or pBabe empty vector using the FuGene HD transfection reagent (Promega) according to the manufacturers’ instructions at a ratio of 2:1 volume:mass, and media was replaced after 4 hr. The pPIK3CAH1047R was a gift from Jean Zhao (Addgene plasmid 12524) (Zhao et al., 2005). In A-485 co-treatment studies, A-485 was included in the media 24 hr-post transfection in 0.1% DMSO. 48 hr after transfection, media was replaced with DMEM/F-12 media supplemented with 0.5% FBS, 1% P/S and 1% L-glutamine. Cells were collected 72 hr-post siRNA transfection using the Quick-RNA Miniprep Kit (Zymo Research) for RNA, RIPA buffer (Cell Signaling) for whole cell lysate, or histone extraction.
Generation and use of lentiviral shRNA particles
Lentiviral particles expressing shRNAs were produced in 293T cells according to the manufacturers’ instructions. Lenti-X™293T cells were transfected with lentiviral packaging mix composed of pNHP and pVSVG (generous gifts from Dr. Fredric Manfredsson) and MISSION pKLO.1 plasmid containing non-targeting shRNA (control) or pooled ARID1A shRNAs (shARID1A) (Sigma) using poly-ethylenimine (PEI) in DMEM + 4.5g/L D-Glucose, 110mg/L Sodium Pyruvate, 10% FBS, 1% L-glutamine. After 24 hr, media was replaced with DMEM/F12, 10% FBS, 1% L-glutamine, 1% P/S. Viral particles were collected after 48 and 96 hr, and viral titers were calculated using the qPCR Lentiviral Titration Kit (ABM).
For lentiviral transduction of 12Z cells, cells were treated with a multiplicity of infection of 100 units per cell. After 24 hours, media was replaced. For plasmid co-transfection experiments, cells were transfected the following day with 500ng pBabe vector containing PIK3CAH1047R (pPIK3CAH1047R) or pBabe empty vector using the FuGene HD transfection reagent (Promega) according to the manufacturers’ instructions at a ratio of 2:1 volume:mass, and media was replaced after 4 hr. In A-485 co-treatment studies, A-485 was included in the media 24 hr-post transfection in 0.1% DMSO. To generate stable expression cell lines, transduced cells were treated with 600 ng/mL puromycin (Sigma) for three weeks.
Histone extraction
Cells were washed with PBS and scraped in PBS containing 5 mM sodium butyrate. Cells were centrifuged and resuspended in TEB buffer (phosphate buffered saline supplemented with 0.5% Triton X-100, 5 mM sodium butyrate, 2 mM phenylmethylsulfonyl fluoride, 1 × protease inhibitor cocktail) and incubated on a 3D spindle nutator at 4°C for 10 min. Cells were centrifuged at 3,000 RPM for 10 min at 4°C. TEB wash step was repeated once. Following second wash, pellet was resuspended in 0.2 N HCl, and incubated on 3D spindle nutator at 4°C overnight. The following day, samples were neutralized with 1:10 volume 1M Tris-HCl pH 8.3. Sample was centrifuged at 3,000 RPM for 10 min at 4°C, and supernatant containing histone proteins was collected.
Western blotting
Protein whole cell lysates and histone extracts were quantified using the Micro BCA Protein Assay Kit (ThermoFisher) and a FlexSystem3 plate reader. Protein lysates were run on a 4%–15% gradient SDS-PAGE gel (BioRad) and transferred to PVDF membrane using the TransBlot Turbo system (BioRad). Primary antibodies dilutions were 1:1,000 ARID1A (D2A8U) (12354, Cell Signaling); 1:100 P300 (NM11) (sc-32244, Santa Cruz); 1:1,000 β-Actin (8457, Cell Signaling); 1:100 PAI-1 (sc-5297, Santa Cruz); 1:1,000 Akt (4691, Cell Signaling); 1:2,000 Phospho-Akt (Ser473) (4060, Cell Signaling). Horseradish peroxidase (HRP) conjugated secondary antibodies (Cell Signaling) were used at a dilution of 1:2,000. Clarity Western ECL Substrate (BioRad) was used for protein band visualization, and western blot exposures were captured using the ChemiDoc XRS+ imaging system (BioRad).
For histone extracts, samples were run on a 15% SDS-PAGE gel and transferred to nitrocellulose membrane in 20 mM sodium phosphate pH 6.7 at 400 mA for 90 min. Primary antibody dilutions were 1:2,000 Histone H3 (4499, Cell Signaling); 1:1,000 H3K27ac (Active Motif, 39133). Donkey anti-rabbit IgG, IRDye 800CW conjugated secondary antibody (LI-COR Biosciences) was used at a dilution of 1:10,000 and fluorescence imaging was performed using the LI-COR Odyssey CLx imaging system (LI-COR Biosciences). Uncropped western blot images are collected in Figure S7.
Transwell invasion assay
12Z cells were seeded in 6-well dishes at a density of 50,000 cells per well. After 24 hr, cells were transfected with siRNA as described above. For drug treatment experiments, cells were treated drug 24 hr after transfection. At 48 hr post-transfection, cells were trypsinized, and 100 μL of cell mixture containing 30,000 cells and 0.3 mg/mL Matrigel was seeded into transwell plates (8 μm pore polycarbonate membrane, Corning) pre-coated with 100 μL of 0.3 mg/mL Matrigel. After 1 hr, serum-free DMEM/F12 1% P/S, 1% L-glutamine media was added to the top chamber and DMEM/F12, 5% FBS, 1% P/S, 1% L-glutamine was added to the bottom chamber. For drug studies, drug was included in both top and bottom chamber media. After 16 hr, transwell units were transferred to plates containing 2 μg/mL calcein-AM in DMEM/F12. After 1 hr, media was aspirated from the top chamber and unmigrated cells were removed with a cotton swab. Images were collected using a Nikon Eclipse Ti microscope in five non-overlapping fields per well. ImageJ software (National Institutes of Health) was used to quantify cells based on size and intensity.
Matrigel viability assay
12Z cells were seeded in 6-well dishes at a density of 50,000 cells per well. After 24 hr, cells were transfected with siRNA as described above. For drug treatment experiments, cells were treated drug 24 hr after transfection. At 48 hr post-transfection, cells were trypsinized, and 50 μL of cell mixture containing 10,000 cells and 0.3 mg/mL Matrigel was seeded into 96-well plates pre-coated with 100 μL of 0.3 mg/mL Matrigel. After 1 hr, 50 μL of serum-free DMEM/F12 1% P/S, 1% L-glutamine media was added. For drug studies, 100 nM A-485 or vehicle was included in the media. After 16 or 24 hr, 2 μg/mL calcein-AM and 4 μg/mL ethidium homodimer III were added. Wells were imaged using a Nikon Eclipse Ti microscope, and ImageJ software (National Institutes of Health) was used to quantify cells based on size and intensity.
Migration assay
12Z cells were seeded into 35mm dishes containing 4-well culture inserts at a density of 4,000 cells per well. Cells were treated with lentiviral particles expressing non-targeting shRNA (control) or shARID1A at a multiplicity of infection of 100, 24 hr after seeding. Media was replaced with serum-free DMEM/F12 containing 1% L-glutamine and 1% P/S including drug or vehicle after 24 hr. Culture inserts were removed and serum-free media containing vehicle or drug was replenished after 16 hr. At 0 and 24 hr of migration, images were taken using a Nikon Eclipse Ti microscope. Distances between migration fronts were measured using NIS Elements Advanced Research software at 16 different points 100 μm apart. Migration distance was calculated by subtracting the average distance across migration fronts at 24 hr from the average distance at 0 hr. Cells were counted within a window surrounding the 1050 mm2 migration area.
Viability assay
Cells were seeded in 6-well plates at a density of 100 cells/well. After 24h, cells were treated with A-485 at concentrations from 10 nM to 100 μM. After 6 days, cells were stained with crystal violet and counted.
Cell growth assay
Cells were seeded at a density of 4,000 cells per well in a 96-well plate. After 24 hr, cells were transfected as described above. After 24 hr, cells were treated with drugs for 48–72 hr. Cells were incubated with 2 μg/mL calcein-AM for 1 hr and fluorescence was measured using a SpectraMax i3× (Molecular Devices).
Cell Suspension Caspase-Glo Assay
The Caspase-Glo 3/7 Assay (Promega) was used according to the manufacturer’s instructions. Following transfection (48 hr) and drug treatment (24 hr) cells were seeded at 10,000 cells per well in a 96-well Cellstar Cell-Repellent plate (Greiner Bio-one) in serum-free DMEM/F12, 1% L-glutamine, 1% P/S containing A-485 or vehicle. After 24 hr, cells were treated with Caspase-Glo at a ratio of 1:1 and incubated at 37°C for 1 hr. Cells were then transferred to a white 96-well plate (costar) and luminescence was measured using a SpectraMax i3x (Molecular Devices).
Annexin V assay
Expression of Annexin V was measured by flow cytometry using the Annexin V-FITC Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Flow cytometry was performed using a BD Accuri C6 flow cytometer (BD Biosciences) and analyzed using FlowJo v10 software (BD Biosciences).
Cell Cycle assay
The Click-iT Plus EdU Flow cytometry Assay Kit (Invitrogen) was used for cell cycle assays. Cells were treated with 10 μM of EdU for 2 hours in culture media. Cells were harvested by trypsinization and washed in 1% BSA in PBS. Cells were resuspended in 100 μL of ice cold PBS, and 900 μL of ice cold 70% ethanol was added dropwise while vortexing. Cells were incubated on ice for two hours. Cells were washed with 1% BSA in PBS and then treated with the Click-iT Plus reaction cocktail including Alexa Fluor 488 picolyl azide according to the manufacturer’s instructions for 30 min. Cells were washed with 1X Click-iT permeabilization buffer and wash reagent, and then treated with 5 μM of Vybrant Dye Cycle Ruby Stain (ThermoFisher) diluted in 1% BSA in PBS for 30 min at 37°C. Flow cytometry was performed using a BD Accuri C6 flow cytometer (BD Biosciences) and analyzed using FlowJo v10 software (BD Biosciences).
Construction and Sequencing of Directional mRNA-seq Libraries
RNA samples were collected 72 hr following siRNA transfection using the Quick-RNA Miniprep Kit (Zymo Research). Libraries were prepared by the VARI Genomics Core from 500 ng of total RNA using the KAPA mRNA HyperPrep kit (v4.17) (Kapa Biosystems). RNA was sheared to 300–400 bp. Prior to PCR amplification, cDNA fragments were ligated to IDT for Illumina unique dual adapters (IDT DNA Inc). Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies), QuantiFluor® dsDNA System (Promega), and Kapa Illumina Library Quantification qPCR assays (Kapa Biosystems). Individually indexed libraries were pooled and 100 bp, single end sequencing was performed on an Illumina NovaSeq6000 sequencer using an SP, 100 cycle sequencing kit (Illumina) and each library was sequenced to an average raw depth of 35M reads. Base calling was done by Illumina RTA3 and output of NCS was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.9.0.
Chromatin Immunoprecipitation
12Z cells were crosslinked 72 hr post-transduction with lentiviral particles containing control shRNAs or ARID1A-targeting shRNAs (differential P300, H3K18ac, H3K27ac, H3K27me3, H3K4me3 and H3K4me1 ChIP-seq) or untreated cells were used (wild-type P300 ChIP-seq). For crosslinking, cells were treated with 1% formaldehyde in cell culture media for 15 min at room temp. Formaldehyde was quenched by the addition of 0.125 M Glycine, and cells were washed with PBS. 4×106 crosslinked cells were used per IP for H3K4me3 and H3K27me3, and 1×107 crosslinked cells were used per IP for all other antibodies. Chromatin from crosslinked cells was fractionated by digestion with micrococcal nuclease using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) per the manufacturers’ instructions. IPs were performed in duplicate per antibody and condition by adapting established methods (Boyd and Farnham, 1997).
For P300 IPs, nuclei were resuspended in nuclear lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA [pH 8.0], 1% SDS) and sonicated for 30 s. Protein G magnetic beads (Cell Signaling) were preconjugated with antibody overnight at 4°C in wash buffer (1X PBS, 0.5% BSA, 0.02% Tween-20). Antibody used was 5 μg P300 (sc-32244, Santa Cruz). Fractionated chromatin was diluted into IP buffer (0.01%SDS, 1.1% TrionX-100, 1.2 mM EDTA [pH 8.0], 16.7 mM Tris-HCl [pH 8.0], 167 mM NaCl) and incubated with preconjugated antibody/Dynabeads overnight at 4°C. Samples were washed at 4°C with high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA [pH 8.0], 20 mM Tris-HCl [pH 8.0], 0.5 M NaCl), low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA [pH 8.0], 20 mM Tris-HCl [pH 8.0], 150 mM NaCl), dialysis buffer (0.2% Sarcosyl, 2 mM EDTA [pH 8.0], 50mM Tris-HCl [pH 8.0]), IP wash buffer (0.25M LiCl, 1% NP-40, 1% Deoxycholate, 1 mM EDTA [pH 8.0], 10 mM Tris-HCl [pH 8.0]) and TE (10mM Tris-HCL [pH 8.0], 1mM EDTA [pH 8.0]). IP chromatin was eluted for 30 min at 37°C with elution buffer (1% SDS, 0.1 M NaHCO3). Crosslinks were reversed with 0.4 mg/mL Proteinase K (ThermoFisher) and 0.2 M NaCl at 65°C for 2 hr. DNA was purified using the ChIP DNA Clean & Concentrator Kit (Zymo).
For H3K27ac, H3K18ac, H3K27me3, H3K4me3 and H3K4me1 IPs were performed using the SimpleChIP Enzymatic Chromatin IP Kit per the manufacturers’ instructions. For H3K27ac and H3K18ac the addition of 5 mM sodium butyrate included in Buffer A and ChIP Buffer. Antibodies used were 10 μg H3K27ac (Active Motif, 39133), 5 μg H3K18ac (ab1191, Abcam), 10 μL H3K27me3 (Cell Signaling, 9733), 10 μL H3K4me3 (Cell Signaling, 9751), or 4 μg H3K4me1 (Abcam, ab8895) per IP. DNA was purified as described above.
Construction and Sequencing of ChIP-seq Libraries
Libraries for Input and IP samples were prepared by the VARI Genomics Core from 10 ng of input and IP material when available, and all material when less than 10 ng available, using the KAPA Hyper Prep Kit (v5.16) (Kapa Biosystems). Prior to PCR amplification, end repaired and A-tailed DNA fragments were ligated to Bioo Scientific NEXTflex Adapters (Bioo Scientific). Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies), QuantiFluor® dsDNA System (Promega), and Kapa Illumina Library Quantification qPCR assays (Kapa Biosystems). Individually indexed libraries were pooled. For P300 ChIP in wild-type cells, 100 bp, single end sequencing was performed on an Illumina NovaSeq6000 sequencer using an SP, 100 cycle sequencing kit (Illumina) and each library was sequenced to minimum read depth of 100M reads per input library, 50M reads per IP library. Base calling was done by Illumina NextSeq Control Software (NCS) v2.0. For differential P300 ChIP, 75 bp, paired end sequencing was performed on an Illumina NextSeq 500 sequencer using 150 cycle HO and MO sequencing kits (v2) (Illumina), with all libraries run across 2 flowcells to return a minimum read depth of 80M reads per input library and 40M read per IP library. Base calling was done by NCS v2.0. For differential H3K27ac, H3K18ac and H3K4me1 ChIP-seq IPs, 50 bp, paired end sequencing was performed on an Illumina NovaSeq6000 sequencer using an S1, 100 cycle sequencing kit and each library was sequenced to minimum read depth of 50M reads per IP library. Input samples were sequenced using 100 bp, single end sequencing to a minimum read depth of 100M reads. Base calling was done by Illumina RTA3. For H3K27me3 and H3K4me3 ChIP, 75 bp, single end sequencing was performed on an Illumina NextSeq 500 sequencer using 75 cycle HO sequencing kits (v2), with all libraries run across two flow cells to return a minimum read depth of 80 M reads per input library and 40 M read per IP library. Base calling was done by Illumina NextSeq Control Software (NCS) v2.0. For all experiments, output data was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.9.0.
Cleavage Under Targets and Release Using Nuclease (CUT&RUN)
The CUT&RUN protocol was adapted from established protocols (Skene et al., 2018). BioMag Plus Concanavalin A–coated magnetic beads (Bangs Laboratories) were washed in Binding Buffer (20 mM HEPES-KOH pH 7.9, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2). 72 hr following siRNA transfection, 500,000 12Z cells were harvested and resuspended in Wash Buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1X protease inhibitor cocktail) and washed twice by centrifuge at 600 × g for 3 min, and then added to the concanavalin A bead suspension and mixed on a tube rotator for 10 min at room temp. Cell/bead conjugates were resuspended in 500 μL of Antibody Buffer (Wash Buffer with 0.05% Digitonin and 2 mM EDTA) containing 5 μg of H3K27ac antibody (Active Motif, cat# 39133) or Rabbit IgG (Cell Signaling, cat#2729) and incubated in a tube nutator overnight at 4°C. The following day, cells were washed in Digitonin Buffer (Wash Buffer with 0.05% Digitonin) three times, resuspended in 250 μL of Digitonin Buffer and 12.5 μL of CUTANA pAG-MNase (EpiCypher, cat# 15–1016) was added. Cells were mixed on a nutator at room temp for 1 hr, followed by two washes in Digitonin Buffer and one wash with Low-Salt Rinse Buffer (20 mM HEPES-NaOH pH 7.5, 0.5 mM Spermidine, 1X protease inhibitor cocktail). Tubes were chilled on ice, 1 mL of Calcium Incubation Buffer (3.5 mM HEPES-NaOH pH 7.5, 10 mM CaCl2, 0.05% Digitonin) was added, and tubes were nutated at 4°C. After 2.5 min, beads were bound to magnet, supernatant was removed and 250 μL of EGTA-STOP Buffer (170 mM NaCl, 20 mM EGTA, 0.05% Digitonin, 20 μg/mL RNase A, 20 μg/mL Glycogen, 0.8 pg/ml S. cerevisiae fragmented nucleosomal DNA) was added. Beads were nutated at 37°C for 30 min, followed by centrifuge at 16,000 × g for 5 min at 4°C. DNA was purified using the NucleoSpin Gel and PCR Clean-up Kit (Takara, cat# 740609.50).
Construction and Sequencing of CUT&RUN Libraries
Libraries for CUT&RUN samples were prepared by the Van Andel Genomics Core from 0.5–1 ng of IP material, using the KAPA Hyper Prep Kit (v5.16) (Kapa Biosystems). Prior to PCR amplification, end-repaired and A-tailed DNA fragments were ligated to Bioo Scientific NEXTflex Adapters (Bioo Scientific) at a concentration of 500 nM. Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies, Inc.), QuantiFluor® dsDNA System (Promega Corp.), and Kapa Illumina Library Quantification qPCR assays (Kapa Biosystems). Individually indexed libraries were pooled and 50 bp, paired end sequencing was performed on an Illumina NovaSeq6000 sequencer using an S1, 100 cycle sequencing kit (Illumina Inc.) Each library was sequenced to an average depth of 75M reads. Base calling was done by Illumina RTA3 and output was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.9.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
For standard mRNA gene-level expression analysis, single-end raw reads were trimmed with cutadapt (Martin, 2011) and Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) followed by quality control analysis via FastQC (Andrews, 2010) and MultiQC (Ewels et al., 2016). Trimmed reads were aligned to GRCh38.p12 genome assembly and indexed to GENCODE (Frankish et al., 2019) v28 GFF3 annotation via STAR (Dobin et al., 2013) aligner with flag ‘–quantMode GeneCounts’ for feature counting. Reverse-stranded, gene-level counts were extracted from the STAR output files and constructed into an experimental read count matrix in R. Low count genes were filtered (1 count per sample on average) prior to DESeq2 (Love et al., 2014) count normalization and differential expression analysis. Modeling design matrices were constructed with a single “condition” variable and included an intercept. Calculated differential expression probabilities were corrected for multiple testing by independent hypothesis weighting (IHW) (Ignatiadis et al., 2016) for downstream analysis. Threshold for differential expression significance was set at FDR < 0.0001. Relative expression heatmaps were produced using relative regularized-logarithm (rlog) (Love et al., 2014) counts by subtracting mean rlog counts of the control group. Relative linear expression bar plots were produced from DESeq2 normalized counts table. Previously published GEMM expression data (Wilson et al., 2019) were extracted from (GEO: GSE129784).
Intergenic eRNA and associated differential expression analysis were also analyzed similarly. Briefly, previously published, paired end total RNA sequencing data from 12Z cells treated with siARID1A or non-targeting siRNA control (Wilson et al., 2019) were extracted from (GEO: GSE129782). Reads were trimmed and aligned as described above. Aligned BAMs were inputted to HOMER (Heinz et al., 2010) in order to count integer RNA-seq reads at each of the 18,050 distal, putatively active enhancer elements described in this study. Counted regions were then excluded which overlapped with any genic regions, including introns, using the genes() function of TxDb.Hsapiens.UCSC.hg38.knownGene (Bioconductor Core Team and Bioconductor Package Maintainer, 2016) R package. Expression status of each eRNA locus was then determined by observation of at least 1 count per sample on average, resulting in 3,668 expressed intergenic eRNAs. The filtered eRNA counts table was then normalized and modeled for differential expression analysis by DESeq2 (Love et al., 2014) as described above.
ChIP-seq analysis
Wild-type P300 and differential H3K27ac, H3K18ac, H3K27me3, and H3K4me3 ChIP-seq experiments were analyzed as single-end libraries, while differential P300 and H3K4me1 ChIP-seq were analyzed as paired-end. Raw reads for IPs and inputs were trimmed with cutadapt (Martin, 2011) and Trim Galore! followed by quality control analysis via FastQC (Andrews, 2010) and MultiQC (Ewels et al., 2016). Trimmed reads were aligned to GRCh38.p12 reference genome via Bowtie2 (Langmead and Salzberg, 2012) with flag ‘–very-sensitive’. Aligned reads were sorted and indexed with samtools (Li et al., 2009). For paired-end analyses, only properly-paired read fragments were retained by samtools view with flag ‘-f 3’ followed by sorting and indexing. Specifically for libraries with differential comparisons, molecular complexity was then estimated from duplicate rates by ATACseqQC (Ou et al., 2018) and preseqR (Daley and Smith, 2013), and libraries were subsampled to equivalent molecular complexity within an experimental design based on these estimates with samtools. Picard MarkDuplicates (http://broadinstitute.github.io/picard/) was used to remove PCR duplicates, followed by sorting and indexing. MACS2 (Zhang et al., 2008) was used to call peaks on each ChIP replicate against the respective input control. For P300 IPs, MACS2 called broadPeaks with FDR < 0.05 threshold and otherwise default settings. For H3K4me3, H3K4me1, H3K27ac and H3K18ac IPs, MACS2 called narrowPeaks with FDR < 0.05 threshold and flags ‘–nomodel–extsize 146’ to bypass model building. For H3K27me3, MACS2 called broadPeaks with FDR < 0.05 threshold and flags ‘–nomodel–extsize 146’ to bypass model building. The resulting peaks were repeat-masked by ENCODE blacklist filtering and filtered for non-standard contigs (Amemiya et al., 2019). A naive overlapping peak set, as defined by ENCODE, was constructed by calling peaks on pooled replicates followed by bedtools intersect (Quinlan and Hall, 2010) to select for peaks of at least 50% overlap with each biological replicate.
ChIP-seq differential binding or abundance analysis was performed with csaw (Lun and Smyth, 2016). Briefly, a consensus peak set was constructed for each differential experiment from the union of replicate-intersecting, filtered MACS2 peaks called in each condition. The replicate intersection criteria used here are less stringent than the naive overlap; any partial intersect between ChIP replicates was accepted as a query region tested for differential binding/abundance. ChIP reads were counted in these query regions by csaw, then filtered for low abundance peaks with average logCPM > −3. When comparing ChIP libraries, any global differences in IP efficiency observed between the two conditions were considered a result of technical bias to ensure a highly conservative analysis. As such, we employed a non-linear loess-based normalization to the peak count matrix, as is implemented in csaw (Lun and Smyth, 2016), to assume a symmetrical MA distribution. A design matrix was then constructed from one “condition” variable, without an intercept. The count matrix and loess offsets were then supplied to edgeR (Robinson et al., 2010) for estimating dispersions and fitting the quasi-likelihood generalized linear model for hypothesis testing. Nearby query regions were then merged up to 500 bp apart for a maximum merged region width of 5 kb, and the most significant probability was used to represent the merged region. An FDR < 0.05 threshold was used to define significant differentially bound/abundant regions.
CUT&RUN analysis
Analysis of CUT&RUN data followed a highly similar procedure as paired-end ChIP-seq. Briefly, raw paired-end reads for H3K27ac or IgG CUT&RUN were trimmed and aligned, filtered for only properly-paired reads, then molecular complexity was estimated, and libraries were subsampled to equalize based on complexity estimates. PCR duplicates were removed, and MACS2 was used to call narrowPeaks against the IgG negative control as input, with FDR < 0.05 threshold and flags ‘–nomodel–extsize 146’ to bypass model building. Peaks were then blacklist-filtered, and a naive overlapping peak set was constructed as described above.
Differential H3K27ac CUT&RUN analysis was computed with csaw. In order to promote similarity between the differential H3K27ac CUT&RUN and ChIP-seq experiments, we used the same consensus peak set as defined by our differential H3K27ac ChIP-seq experiment for the CUT&RUN analysis here. Briefly, H3K27ac CUT&RUN reads were counted in these query regions by csaw, then filtered for low abundance peaks with average logCPM > −3. When comparing CUT&RUN libraries, any global differences in CUT&RUN reaction efficiency observed between two conditions were considered a result of technical bias to ensure a highly conservative analysis. As such, we employed a non-linear loess-based normalization to the peak count matrix, as is implemented in csaw, to assume a symmetrical MA distribution. A design matrix was then constructed from one “condition” variable, without an intercept. The count matrix and loess offsets were then supplied to edgeR for estimating dispersions and fitting the quasi-likelihood generalized linear model for hypothesis testing. Nearby query regions were then merged up to 500 bp apart for a maximum merged region width of 5 kilobases, and the most significant probability was used to represent the merged region.
Chromatin state analysis
ChromHMM (Ernst and Kellis, 2017) was used to segment the hg38 genome based on combinatorial chromatin features in control and ARID1A-depleted 12Z cell conditions. Briefly, all seven chromatin features (total RNA, ATAC, H3K4me1, H3K4me3, H3K27me3, H3K27ac, and H3K18ac) were binarized from aligned BAM files, and chromatin features were modeled in both conditions simultaneously through the “concatenated” option. The concatenated option was selected because it creates a unified model for direct comparison between control and ARID1A-depleted conditions used to identify chromatin state changes. Chromatin state models were built from 5 to 25 states, and each model was manually curated based on inferred biological function to select one with balance between unique and overlapping combinatorial features. We selected 18 states as our final model for downstream analysis. State emissions were then user-reordered to group based on inferred biological function. BED files containing coordinates for each chromatin state in each condition were constructed into non-overlapping GenomicRanges objects in R for downstream enrichment analyses, differential chromatin state analysis, and plotting.
Bioinformatics and statistics
For RNA-seq experiments, three biological replicates were analyzed (n = 3). For ChIP-seq and CUT&RUN experiments two independent IPs were used (n = 2) and were compared against a condition-respective input chromatin sample or IgG negative control, respectively. For in vivo experiments, n represents number of mice. For cell-based assays, n represents biological replicates or independent experiments as indicated in the figure legend. Multiple hypothesis tests corrections via FDR were employed when appropriate to reduce type I errors. Presented probability (p) values are representative of the associated statistical tests as indicated in the figure legends. All boxplots presented for genomic analyses are in the style of Tukey without outliers. The ROSE algorithm (Lovén et al., 2013; Whyte et al., 2013) was used to define active super-enhancers from H3K27ac peaks which overlapped with accessibility (ATAC) in control 12Z cells. GeneHancer (Fishilevich et al., 2017) database was used to associate enhancers to genes with a GeneHancer score > 1 threshold. Various HOMER (Heinz et al., 2010) functions were applied to annotate genomic regions of interest, quantify signal and count reads at sites of interest for tag density heatmaps and meta peak plots. Chromatin analyses involving ChIP signal quantification at regions of interest used pooled reads from both IP replicates, per feature. TxDb.Hsapiens.UCSC.hg38.-knownGene (Bioconductor Core Team and Bioconductor Package Maintainer, 2016) was used to define gene promoters for all standard hg38 genes as 3 kilobase regions surrounding the primary TSS. MACS2 (Zhang et al., 2008) was used to produce genome-wide signal log-likelihood ratio (logLR) tracks for IGV (Robinson et al., 2011) visualization. ClusterProfiler (Yu et al., 2012) was used to compute and visualize pathway enrichment from a list of gene symbols with respective gene universes. Hallmark pathways and GO Biological Process gene sets were retrieved from MSigDB (Liberzon et al., 2015). ComplexHeatmap (Gu et al., 2016) was used for hierarchical clustering by Euclidean distance and general heatmap visualization. GenomicRanges (Lawrence et al., 2013) functions were frequently used to intersect and manipulate genomic coordinates e.g., for genome-wide association tests. eulerr (Larsson, 2020) was used to produce proportional Euler diagrams. biomaRt (Durinck et al., 2005; Durinck et al., 2009) was used for all gene nomenclature and ortholog conversions. ggplot2 (Wickham, 2016) was used for certain plotting applications. The statistical computing language R (R Core Team, 2018) was used for many applications throughout this manuscript. Mantel-Cox tests and t tests were performed using GraphPad Prism 8 software.
Supplementary Material
Highlights.
In endometrial epithelia, ARID1A binding is highly associated with super-enhancers
ARID1A loss leads to super-enhancer H3K27 hyperacetylation and accessibility
P300 inhibition in ARID1A mutant cells suppresses invasion and induces anoikis
SERPINE1 (PAI-1) super-enhancer hyperacetylation drives endometrial invasion
ACKNOWLEDGMENTS
We thank Drs. John Risinger, Jeff MacKeigan, Peter Laird, Fredric Manfredsson, and Jae Wook Jeong for helpful discussions. We thank the Van Andel Genomics Core for providing library construction and sequencing facilities and services, and the Van Andel Histology and Pathology Core for histology services. M.R.W. was supported by an American Cancer Society Postdoctoral Fellowship (PF-17-163-02-DDC). P.J.Y. was supported by a Project Grant (PGT-156084) from the Canadian Institutes of Health Research and a Health Professional Investigator Award from the Michael Smith Foundation for Health Research. R.L.C. was supported by an Innovative Translational Grant from the Mary Kay Foundation (026-16), a Liz Tilberis Early Career Award from the Ovarian Cancer Research Fund Alliance (OCRFA) (457446), and the NIH National Institute for Child Health and Human Development (HD099383-01).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108366.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, Wang W, Wright KL, Lane TF, Zacksenhaus E, and Egan SE (2011). Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 71, 2706–2717. [DOI] [PubMed] [Google Scholar]
- Alotaibi FT, Peng B, Klausen C, Lee AF, Abdelkareem AO, Orr NL, Noga H, Bedaiwy MA, and Yong PJ (2019). Plasminogen activator inhibitor-1 (PAI-1) expression in endometriosis. PLOS ONE 14, e0219064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data (Babraham Bioinformatics; ). [Google Scholar]
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, et al. (2017). Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med 376, 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley J, Jülicher A, Hotz B, Mechsner S, and Hotz H (2014). Epithelial to mesenchymal transition (EMT) seems to be regulated differently in endometriosis and the endometrium. Arch. Gynecol. Obstet 289, 871–881. [DOI] [PubMed] [Google Scholar]
- Bedaiwy MA, Falcone T, Mascha EJ, and Casper RF (2006). Genetic polymorphism in the fibrinolytic system and endometriosis. Obstet. Gynecol 108, 162–168. [DOI] [PubMed] [Google Scholar]
- Bilyk O, Coatham M, Jewer M, and Postovit LM (2017). Epithelial-to-Mesenchymal Transition in the Female Reproductive Tract: From Normal Functioning to Disease Pathology. Front. Oncol 7, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bioconductor Core Team, and Bioconductor Package Maintainer (2016). TxDb.Hsapiens.UCSC.hg38.knownGene: Annotation package for TxDb object(s) R package version 3.4.0 (Bioconductor). [Google Scholar]
- Boretto M, Maenhoudt N, Luo X, Hennes A, Boeckx B, Bui B, Heremans R, Perneel L, Kobayashi H, Van Zundert I, et al. (2019). Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol 21, 1041–1051. [DOI] [PubMed] [Google Scholar]
- Borrelli GM, Abrão MS, Taube ET, Darb-Esfahani S, Köhler C, Chiantera V, and Mechsner S (2016). (Partial) Loss of BAF250a (ARID1A) in rectovaginal deep-infiltrating endometriosis, endometriomas and involved pelvic sentinel lymph nodes. Mol. Hum. Reprod 22, 329–337. [DOI] [PubMed] [Google Scholar]
- Boyd KE, and Farnham PJ (1997). Myc versus USF: discrimination at the cad gene is determined by core promoter elements. Mol. Cell. Biol 17, 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Brien GL, and Verrijzer CP (2019). Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev. 33, 936–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruse C, Bergqvist A, Carlström K, Fianu-Jonasson A, Lecander I, and Astedt B (1998). Fibrinolytic factors in endometriotic tissue, endometrium, peritoneal fluid, and plasma from women with endometriosis and in endometrium and peritoneal fluid from healthy women. Fertil. Steril 70, 821–826. [DOI] [PubMed] [Google Scholar]
- Bruse C, Radu D, and Bergqvist A (2004). In situ localization of mRNA for the fibrinolytic factors uPA, PAI-1 and uPAR in endometriotic and endometrial tissue. Mol. Hum. Reprod 10, 159–166. [DOI] [PubMed] [Google Scholar]
- Bulun SE (2009). Endometriosis. N. Engl. J. Med 360, 268–279. [DOI] [PubMed] [Google Scholar]
- Calo E, and Wysocka J (2013). Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al. (2015). Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun 6, 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, and Cairns BR (2009). The biology of chromatin remodeling complexes. Annu. Rev. Biochem 78, 273–304. [DOI] [PubMed] [Google Scholar]
- Daikoku T, Ogawa Y, Terakawa J, Ogawa A, DeFalco T, and Dey SK (2014). Lactoferrin-iCre: a new mouse line to study uterine epithelial gene function. Endocrinology 155, 2718–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley T, and Smith AD (2013). Predicting the molecular complexity of sequencing libraries. Nat. Methods 10, 325–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, and Kadir RA (2012). Endometrial haemostasis and menstruation. Rev. Endocr. Metab. Disord 13, 289–299. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MJ (2004). The urokinase plasminogen activator system: role in malignancy. Curr. Pharm. Des 10, 39–49. [DOI] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2017). Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47 (D1), D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Elliott NJ, Ho J, Sharp A, Shokhirev MN, and Hargreaves DC (2019). Heterozygous Mutations in SMARCA2 Reprogram the Enhancer Landscape by Global Retargeting of SMARCA4. Mol. Cell 75, 891–904.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargett CE, Nguyen HP, and Ye L (2012). Endometrial regeneration and endometrial stem/progenitor cells. Rev. Endocr. Metab. Disord 13, 235–251. [DOI] [PubMed] [Google Scholar]
- Gellersen B, and Brosens JJ (2014). Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr. Rev 35, 851–905. [DOI] [PubMed] [Google Scholar]
- Gilabert-Estellés J, Estellés A, Gilabert J, Castelló R, España F, Falcó C, Romeu A, Chirivella M, Zorio E, and Aznar J (2003). Expression of several components of the plasminogen activator and matrix metalloproteinase systems in endometriosis. Hum. Reprod 18, 1516–1522. [DOI] [PubMed] [Google Scholar]
- Giudice LC, and Kao LC (2004). Endometriosis. Lancet 364, 1789–1799. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, Chen E, Jeng YM, Wang TL, and Shih IeM. (2011). Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol 35, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, and Huber W (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, and Ge K (2011). Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Wang TL, Shih IeM., Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr., Vogelstein B, et al. (2010). Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and Weinberg RA (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, Murray PJ, van Deursen JM, and Brindle PK (2006). Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell. Biol 26, 789–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalique S, Naidoo K, Attygalle AD, Kriplani D, Daley F, Lowe A, Campbell J, Jones T, Hubank M, Fenwick K, et al. (2018). Optimised ARID1A immunohistochemistry is an accurate predictor of ARID1A mutational status in gynaecological cancers. J. Pathol. Clin. Res 4, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Yoo JY, Wang Z, Lydon JP, Khatri S, Hawkins SM, Leach RE, Fazleabas AT, Young SL, Lessey BA, et al. (2015). ARID1A Is Essential for Endometrial Function during Early Pregnancy. PLOS Genet. 11, e1005537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JY, Oh S, and Yoo KH (2017). Functional Enhancers As Master Regulators of Tissue-Specific Gene Regulation and Cancer Development. Mol. Cells 40, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, and Lorch Y (1992). Chromatin structure and transcription. Annu. Rev. Cell Biol 8, 563–587. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ, and Shih IeM. (2016). The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol 186, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lac V, Nazeran TM, Tessier-Cloutier B, Aguirre-Hernandez R, Albert A, Lum A, Khattra J, Praetorius T, Mason M, Chiu D, et al. (2019a). Oncogenic mutations in histologically normal endometrium: the new normal? J. Pathol 249, 173–181. [DOI] [PubMed] [Google Scholar]
- Lac V, Verhoef L, Aguirre-Hernandez R, Nazeran TM, Tessier-Cloutier B, Praetorius T, Orr NL, Noga H, Lum A, Khattra J, et al. (2019b). Iatro-genic endometriosis harbors somatic cancer-driver mutations. Hum. Reprod 34, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan R, Andreu-Vieyra C, Lawrenson K, Duymich CE, Gayther SA, Liang G, and Jones PA (2017). Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett. 401, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer LF, Ward JM, and Archer TK (2019). Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. eLife 8, e45672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J (2020). eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R package version 6.1.0 (R Foundation for Statistical Computing). [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLOS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu ZL, Zhao Z, An Q, Wang L, Yu Y, and Piao DX (2017). ARID1A gene knockdown promotes neuroblastoma migration and invasion. Neo-plasma 64, 367–376. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, and Smyth GK (2016). csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 44, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari A, Gurunath S, Fatima F, and Bhattacharya S (2012). Adenomyosis and subfertility: a systematic review of prevalence, diagnosis, treatment and fertility outcomes. Hum. Reprod. Update 18, 374–392. [DOI] [PubMed] [Google Scholar]
- Mao TL, and Shih IeM. (2013). The roles of ARID1A in gynecologic cancer. J. Gynecol. Oncol 24, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao TL, Ardighieri L, Ayhan A, Kuo KT, Wu CH, Wang TL, and Shih IeM. (2013). Loss of ARID1A expression correlates with stages of tumor progression in uterine endometrioid carcinoma. Am. J. Surg. Pathol 37, 1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareel M, and Leroy A (2003). Clinical, cellular, and molecular aspects of cancer invasion. Physiol. Rev 83, 337–376. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12. [Google Scholar]
- Mehta R, and Shapiro AD (2008). Plasminogen activator inhibitor type 1 deficiency. Haemophilia 14, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Mihm M, Gangooly S, and Muttukrishna S (2011). The normal menstrual cycle in women. Anim. Reprod. Sci 124, 229–236. [DOI] [PubMed] [Google Scholar]
- Montgomery BE, Daum GS, and Dunton CJ (2004). Endometrial hyperplasia: a review. Obstet. Gynecol. Surv 59, 368–378. [DOI] [PubMed] [Google Scholar]
- Morice P, Leary A, Creutzberg C, Abu-Rustum N, and Darai E (2016). Endometrial cancer. Lancet 387, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, Prada JG, Couturier DL, Kumar S, Kishore K, et al. (2020). ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet 52, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, and Zhu LJ (2018). ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics 19, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli P, Giannoni E, and Chiarugi P (2013). Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 1833, 3481–3498. [DOI] [PubMed] [Google Scholar]
- Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, Nagle CM, Doherty JA, Cushing-Haugen KL, Wicklund KG, et al. ; Ovarian Cancer Association Consortium (2012). Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol. 13, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott S, and Lieb JD (2015). What are super-enhancers? Nat. Genet 47, 8–12. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Ramón L, Gilabert-Estellés J, Castelló R, Gilabert J, España F, Romeu A, Chirivella M, Aznar J, and Estellés A (2005). mRNA analysis of several components of the plasminogen activator and matrix metalloproteinase systems in endometriosis using a real-time quantitative RT-PCR assay. Hum. Reprod 20, 272–278. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A, Xanthoulea S, Giacomini E, Delvoux B, Alleva E, and Vigano P (2020). Endometriotic cell culture contamination and authenticity: a source of bias in in vitro research? Hum. Reprod 35, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samartzis EP, Samartzis N, Noske A, Fedier A, Caduff R, Dedes KJ, Fink D, and Imesch P (2012). Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod. Pathol 25, 885–892. [DOI] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, and Dekker J (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, and Nakatani Y (1999). Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem 274, 1189–1192. [DOI] [PubMed] [Google Scholar]
- Sengupta D, Kannan A, Kern M, Moreno MA, Vural E, Stack B Jr., Suen JY, Tackett AJ, and Gao L (2015). Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 10, 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HY (2018). Targeting Super-Enhancers for Disease Treatment and Diagnosis. Mol. Cell 41, 506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, Robaa D, Alhalabi Z, Sippl W, and Jung M (2016). KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem 59, 1249–1270. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Smith HW, and Marshall CJ (2010). Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol 11, 23–36. [DOI] [PubMed] [Google Scholar]
- Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yama-waki K, Adachi S, Takahashi T, Kase H, et al. (2018). Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 24, 1777–1789. [DOI] [PubMed] [Google Scholar]
- Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, Gopal P, Zhu M, Nassour I, Chuang JC, et al. (2017). Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 32, 574–589.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swygert SG, and Peterson CL (2014). Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 1839, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed SM, Kumar M, Ghosh A, Tomasetig F, Ali A, Whan RM, Alterman D, and Tanwar PS (2020). Endometrial Axin2(+) Cells Drive Epithelial Homeostasis, Regeneration, and Cancer following Oncogenic Transformation. Cell Stem Cell 26, 64–80.e13. [DOI] [PubMed] [Google Scholar]
- Teixeira J, Rueda BR, and Pru JK (2008). Uterine stem cells In StemBook, The Stem Cell Research Community (Harvard Stem Cell Institute; ). [PubMed] [Google Scholar]
- Trizzino M, Barbieri E, Petracovici A, Wu S, Welsh SA, Owens TA, Licciulli S, Zhang R, and Gardini A (2018). The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 23, 3933–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 68, 1067–1082.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, Drier Y, Gillespie SM, Archer TC, Wu JN, et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet 49, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner HM, Berg A, Wik E, Birkeland E, Krakstad C, Kusonmano K, Petersen K, Kalland KH, Oyan AM, Akslen LA, et al. (2013). ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod. Pathol 26, 428–434. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. (2010). ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med 363, 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, Steidl C, Wiseman SM, Gascoyne RD, Gilks B, and Huntsman DG (2011). Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J. Pathol 224, 328–333. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, Adams M, Johnson B, Su RW, Joshi NR, et al. (2019). ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun 10, 3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR, Holladay J, and Chandler RL (2020). A mouse model of endometriosis mimicking the natural spread of invasive endometrium. Hum. Reprod 35, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Wang TL, and Shih IeM. (2014). The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther 15, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, Sun YH, Yang PY, and Liu F (2014). Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis 35, 867–876. [DOI] [PubMed] [Google Scholar]
- Yang YM, and Yang WX (2017). Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 8, 41679–41689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Vattai A, Zhang X, Zhu J, Thaler CJ, Mahner S, Jeschke U, and von Schönfeldt V (2017). Role of Plasminogen Activator Inhibitor Type 1 in Pathologies of Female Reproductive Diseases. Int. J. Mol. Sci 18, 1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitvogel A, Baumann R, and Starzinski-Powitz A (2001). Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am. J. Pathol 159, 1839–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, and Roberts TM (2005). The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 102, 18443–18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondervan KT, Becker CM, Koga K, Missmer SA, Taylor RN, and Viganò P (2018). Endometriosis. Nat. Rev. Dis. Primers 4, 9. [DOI] [PubMed] [Google Scholar]
- Zondervan KT, Becker CM, and Missmer SA (2020). Endometriosis. N. Engl. J. Med 382, 1244–1256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data generated in this manuscript is GEO: GSE148474. Previously published datasets analyzed herein are also available at GEO: GSE121198.