

SUMMARY
Pancreatic β-cells couple nutrient metabolism with appropriate insulin secretion. Here, we show that pyruvate kinase (PK), which converts ADP and phosphoenolpyruvate (PEP) into ATP and pyruvate, underlies β-cell sensing of both glycolytic and mitochondrial fuels. Plasma membrane-localized PK is sufficient to close KATP channels and initiate calcium influx. Small-molecule PK activators increase the frequency of ATP/ADP and calcium oscillations and potently amplify insulin secretion. PK restricts respiration by cyclically depriving mitochondria of ADP, which accelerates PEP cycling until membrane depolarization restores ADP and oxidative phosphorylation. Our findings support a compartmentalized model of β-cell metabolism in which PK locally generates the ATP/ADP required for insulin secretion. Oscillatory PK activity allows mitochondria to perform synthetic and oxidative functions without any net impact on glucose oxidation. These findings suggest a potential therapeutic route for diabetes based on PK activation that would not be predicted by the current consensus single-state model of β-cell function.
Keywords: Pyruvate kinase, KATP channel, insulin secretion, β-cell metabolism, oxidative phosphorylation, phosphoenolpyruvate cycle, anaplerosis, metabolic oscillations, biosensor imaging, metabolic flux
Graphical Abstract
eTOC BLURB
Lewandowski et al. demonstrate that pyruvate kinase, rather than oxidative phosphorylation, is the ATP/ADP generator that closes β-cell KATP channels to initiate insulin secretion. Small-molecule activators of pyruvate kinase potently amplify insulin secretion by switching mitochondria from oxidative phosphorylation to anaplerotic phosphoenolpyruvate biosynthesis.
INTRODUCTION
A characteristic feature of pancreatic β-cells is their ability to couple metabolic glucose sensing with appropriate insulin secretion to maintain euglycemia. The most widely-accepted description of the sensing mechanism involves the oxidation of glucose carbons in the mitochondria to generate a proton motive force that, through ATP synthase, sequentially raises the ATP/ADP ratio, closes KATP channels, and activates Ca2+ influx, which triggers insulin granule fusion with the plasma membrane (Nicholls, 2016; Prentki et al., 2013). Many different components of the glucose-sensing apparatus are well-characterized. For example, glucokinase (GK) and KATP channels are genetically linked to insulin secretion through both gain- and loss-of-function mutations in humans (Nichols, 2006). It is also established that metabolism generates signals, including ATP and lipids, that amplify insulin secretion independent of KATP closure (Dyachok et al., 2008; Eliasson et al., 1997; Ferdaoussi et al., 2015; Gembal et al., 1992, 1993; Takahashi et al., 1999; Zhao et al., 2014). Each of these pathways is linked by β-cell intrinsic metabolic oscillations (Bertram et al., 2018; Merrins et al., 2010; Tornheim, 1997). However, several lines of evidence challenge one key aspect of this canonical mechanism – the exclusive coupling of oxidative phosphorylation (OxPhos) to KATP channel closure.
A central tenet of mitochondrial respiratory control is that, in the presence of adequate O2 and substrate, mitochondrial OxPhos is dependent on ADP availability (Chance and Williams, 1955). Workload, defined as ATP hydrolysis, is therefore the principal driver for OxPhos in many cells. The requirement of OxPhos for ADP poses a challenge to the canonical model, since ADP privation is the physiological driver of KATP channel closure (Civelek et al., 1997; Nicholls, 2016). The basis for the canonical model is that, following glucose elevation, substantial oxygen consumption occurs prior to calcium influx (Civelek et al., 1996a, 1996b; Juntti-Berggren et al., 1994; Kennedy et al., 2002). However, consistent with ADP limitation, calcium influx precedes respiration during glucose-stimulated oscillations (Kennedy et al., 2002), and mitochondrial respiration is highest after membrane depolarization, rather than during the triggering phase when KATP channels close (Civelek et al., 1997; Jung et al., 2000).
If the dependence of KATP closure on OxPhos is to be questioned, is there an alternative ATP/ADP generator that is limited by glucose and functions prior to membrane depolarization? One clue may be that anaplerotic flux through pyruvate carboxylase (PC) is more strongly correlated with insulin secretion than oxidative flux through pyruvate dehydrogenase (PDH) (Alves et al., 2015; Fransson et al., 2006; MacDonald et al., 2005; Prentki et al., 2013; Schuit et al., 1997). Glucose carbons that transit through PC generate 40% of cytosolic PEP through the cataplerotic mitochondrial PEP carboxykinase (PCK2) reaction (Stark et al., 2009). This ‘PEP cycle’ has been linked to insulin secretion (Jesinkey et al., 2019; Stark et al., 2009) and provides a mechanism distinct from OxPhos for cytosolic ATP/ADP generation via pyruvate kinase (PK), which is allosterically activated by fructose 1,6-bisphosphate (FBP) prior to membrane depolarization (Merrins et al., 2013, 2016).
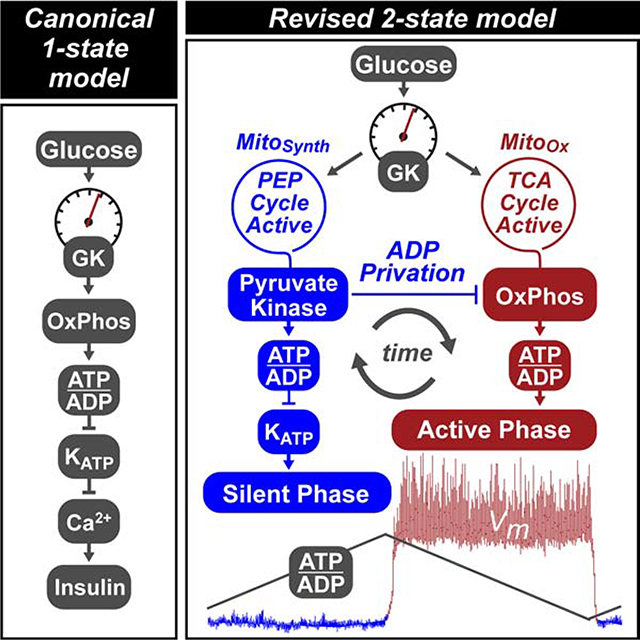
Here, we provide evidence for a revised, more dynamic, model of β-cell metabolism based on the ability of PK to initiate KATP channel closure at the plasma membrane. In this model, cytosolic ADP lowering by PK-driven PEP hydrolysis deprives mitochondria of ADP, at the same time creating antiphase OxPhos oscillations. Such ‘ADP privation’ directs mitochondria to synthesize, rather than oxidize, augmenting the PEP supply for PK. Instead of triggering KATP closure, OxPhos provides the energy to sustain membrane depolarization and insulin secretion. Through a mechanism that would not be predicted by the current canonical linear pathway, pharmacologic activation of PK amplifies the metabolic response without increasing glucose oxidation. Our findings provide a potential new therapeutic strategy for diabetes that does not inappropriately trigger insulin secretion at low glucose.
RESULTS
Membrane-associated pyruvate kinase is sufficient to close KATP channels
The premise for local control of KATP channel closure by glycolysis is established in cardiac myocytes (Weiss and Lamp, 1987; Dhar-Chowdhury et al., 2005; Hong et al., 2011). To test this concept in mouse β-cells, we recorded KATP channels in the inside-out patch clamp configuration to investigate their relationship with membrane-associated PK (Figure 1A). KATP channel opening occurred spontaneously after patch excision, and the channels were reversibly inhibited by 1 mM ATP. A pretest solution containing the KATP-opener ADP (0.5 mM) restored channel activity. When applied to the ADP-containing solution, PEP reduced KATP current or closed the channels completely (Figure 1B), indicating the presence of PK associated with the plasma membrane in mouse islets. PEP reduced the average KATP channel power (reflecting the total number of transported ions) by 80% (Figure 1C), which was due to a 73% decrease in the frequency of events (Figure 1D), as well as a 68% decrease in the fractional time in the open state (Figure 1E). We observed no significant effect of ADP and PEP on current amplitude (Figure 1F). In KATP channels from dispersed human islets, similar trends were observed on channel power, frequency, open time, and amplitude (Figures 1G–K, Figure S1, and Table S1). These data indicate that, even in the presence of high ADP in the bath, endogenous PK is able to locally raise the ATP/ADP ratio to close KATP channels.
Figure 1. Membrane-associated PK is sufficient to close KATP channels.

(A) Experimental setup for excised patch of KATP channels.
(B) Applying the substrates for PK closes KATP channels in mouse islets. ATP, 1 mM ATP; ADP, 0.5 mM ADP + 0.1 mM ATP; PEP, 5 mM phosphoenolpyruvate. Holding potential, −50 mV.
(C-F) Analysis of KATP channel closure in terms of power (C), frequency (D), open time (E), and amplitude (F) in mouse islets.
(G) Applying the substrates for PK closes KATP channels in human islets.
(H-K) Analysis of KATP channel closure in terms of power (H), frequency (I), open time (J), and amplitude (K) from 3 human islet donors.
Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA. See also Figure S1 and Table S1.
PK recruitment potentiates insulin secretion from rodent and human islets
Since GK determines the entry of carbons into glycolysis at elevated glucose (Matschinsky and Ellerman, 1968), the current consensus model predicts that activating distal glycolysis should have no impact on secretion. Nevertheless, given the relationship of PK to KATP channels (Figure 1), we took advantage of the small-molecule PK activator (PKa) TEPP-46 to stabilize the active forms of recruitable PK (Anastasiou et al., 2012). Pancreatic β-cells express three isoforms of PK (M1, M2, and L) (DiGruccio et al., 2016; Mitok et al., 2018) (Figure 2A). While PKM1 is stably active, the recruitable PKM2 and PKL isoforms are allosterically activated by glycolytic FBP (Merrins et al., 2013; Nakatsu et al., 2015). PKa increased the activity of recombinant PKM2 and PKL, while having no effect on PKM1 (Figure 2B). Acute application of PKa (10 μM) to INS1 832/13 cells significantly increased PK activity in cell lysates (Figure 2C), and augmented insulin secretion (Figure 2D). In mouse islets, PKa increased insulin secretion 4-fold by comparison to control (Figure 2E), but did not significantly impact simultaneously measured glucagon secretion (Figure 2F). To control for off-target effects, we tested several alternative PK activators with different chemical scaffolds (Boxer et al., 2010; Jiang et al., 2010; Anastasiou et al., 2012). Six PK activators were capable of enhancing insulin secretion and one was not (Figure S2); although TEPP-46 (referred to as PKa in this manuscript) was not the most effective secretagogue, it is commercially available and was chosen to be used for the rest of the study for future reproducibility.
Figure 2. Pharmacological activation of β-cell PK enhances glucose-stimulated insulin secretion (GSIS) from rodent and human islets.

(A) Schematic depicting PKM2 and PKL activation by glycolytic FBP or PKa.
(B) Activity of recombinant PKM1, PKM2 and PKL in response to PKa (10 μM TEPP-46) (n = 3).
(C) PK activity in INS1 832/13 lysates in response to PKa (n = 3).
(D) GSIS from INS1 832/13 cells in static incubation assays in the absence or presence of PKa (n = 6).
(E-F) Effect of PKa on GSIS (E) and glucagon secretion (F) from perifused mouse islets applied with 10 mM glucose (10G) including during second phase (shaded box) (n = 3).
(G) KCl-stimulated insulin secretion in mouse islets perifused with 2 mM glucose (2G) and 10G in the absence or presence of PKa (n = 3).
(H) Insulin exocytosis, calcium current (ICa), calcium influx (QCa) from mouse islet β-cells with PKa applied via patch pipette (n = 20 cells per treatment).
(I) GSIS from human donor islets in static incubation assays in the absence or presence of PKa. Data points represent the mean of 4 technical replicates for each experiment.
(J) Insulin secretion from human islets (donor H108) perifused with 2.5 mM glucose (2.5G) and 9 mM glucose (9G) in the absence or presence of PKa. Data points represent the mean of 4 technical replicates for each experiment.
Data are shown as mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA (B-D, I), 2-way ANOVA (E-G, J), or Student’s t-test (H). See also Figures S2–3 and Table S1.
PK activation was similarly effective at augmenting depolarization-induced exocytosis. In the control condition, the application of 30 mM KCl (in the presence of diazoxide) elicited biphasic insulin secretion that was more pronounced at elevated glucose (Figure 2G). The subsequent addition of PKa re-initiated biphasic release and further elevated secretion when glucose was present at 10 mM, but not at 2 mM, indicating that the enhancement of secretion by PKa is fuel-dependent. When applied to islet β-cells via patch pipette, PKa increased exocytosis by ~110% in response to 10 step depolarizations, without any change in the evoked calcium current, confirming that PK can amplify insulin secretion independently of KATP channel closure (Figure 2H).
To determine if the effect of PK activation in rodent tissues is relevant to humans, we tested the insulin secretory response to glucose (between 1 and 16.7 mM) in re-aggregated islets from 10 human donors across a spectrum of age and BMI (Figure 2I and Table S1). Insulin secretion was significantly amplified by PKa at glucose concentrations >5 mM, without significant changes at subthreshold glucose concentrations or in the half-maximal effective concentration (EC50) of glucose (vehicle, 10.8 ± 2.5 mM; PKa, 9.1 ± 1.1 mM, n = 10, P > 0.05) (Figure S3). In a dynamic perifusion assay, PKa had no effect on basal or first-phase insulin secretion, and increased second-phase secretion approximately 3.5-fold (Figure 2J). As in mouse islets, these data demonstrate that the actions of PK in human islets are dependent on fuel availability.
PK exerts GK-independent effects on the β-cell triggering and amplifying pathways
GK, unlike the other hexokinases, lacks feedback inhibition and is responsive to glucose across the physiologic range and, consequently, determines the rate of β-cell glucose metabolism (Matschinsky and Ellerman, 1968). Although PK is sufficient to close KATP (Figure 1), the lack of a threshold effect makes it unlikely that PK activation enhances insulin secretion by increasing fuel intake (Figure 2). To test this idea further, we compared PK activation to GK activation in human islets (Figure 3A). PKa primarily increased the amplitude of insulin secretion with only a small effect on the EC50, while GK activation reduced the EC50 for glucose with a lesser effect on the amplitude. When combined, the effects of PK and GK activation were both present, indicating separable mechanisms.
Figure 3. PK amplifies insulin secretion by a distinct mechanism from glucokinase.

(A) Glucose-dose response of insulin secretion in human islets (n = 8) in the absence or presence of GKa and PKa.
(B-E) Representative recordings and quantification of cytosolic calcium and ATP/ADP oscillations (duty cycle and period) in mouse islets. (B) Glucose elevation from 10 mM glucose (10G) to 13 mM glucose (13G) (n = 51). (C) Glucose reduction from 10G to 8 mM glucose (8G) (n = 53). (D) Addition of 500 nM GK activator (GKa) RO-0281675 in 10 mM glucose (n = 37).
(E) Addition of 10 μM PK activator TEPP-46 in 10 mM glucose (n = 62 islets from 4 mice). FuraRed (Ca2+), black scale bar = 0.01 (B, D) or 0.1 (C, E) R430/500; Perceval-HR (ATP/ADP), red scale bar = 0.01 R500/430.
Data are shown as mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA (A) or paired Student’s t-test (B-E). See also Table S1.
To evaluate the β-cell triggering pathway, we performed simultaneous measurements of cytosolic ATP/ADP and calcium oscillations in mouse islets. As the primary glucose-dependent parameter, the calcium duty cycle (fractional time in the active phase of an oscillation) reflects processes that act via KATP (Henquin, 2009). Raising glucose from 10 to 13 mM increased the calcium duty cycle (Figure 3B), while lowering glucose from 10 to 8 mM had the opposite effect (Figure 3C). GK activator (0.5 μM RO-0281675) (Grimsby et al., 2003) increased the duty cycle in a similar fashion to raising glucose (Figure 3D). Failing to match the effects of glucose or GK activator, PKa actually lowered the calcium duty cycle (Figure 3E) despite increasing insulin secretion (Figure 2). At the same time, PKa accelerated ATP/ADP cycling and reduced the time between adjacent depolarizations, as indicated by a marked, 28% reduction in the calcium oscillation period (Figure 3E). Thus, while PK is incapable of matching the effects of GK on glucose uptake, we observed two GK-independent effects: PK modulates the frequency of membrane depolarization and controls the amplitude of insulin secretion.
The GK-independent actions of PK are powered by mitochondrial anaplerosis
An important observation is that PK activators did not stimulate secretion at glucose concentrations < 5mM (Figures 2I and 3A). Even as PK is sufficient to close KATP channels (Figure 1), the insulin secretion and calcium imaging data confirm that, when glucose is the substrate, PK is dependent on glycolytic PEP that is under the metabolic control of GK (Figure 3). But importantly, PEP can arise from both the glycolytic enolase reaction as well as from anaplerosis contributing to PCK2-mediated cataplerosis known as the PEP cycle (Figure 4A) (Jesinkey et al., 2019; Stark et al., 2009). In human islets, the relative PC/PDH flux, which generates the oxaloacetate (OAA) needed by PCK2 to generate PEP, increased in a glucose-dependent manner (Figure 4B). While both PC and PDH fluxes were well-correlated with GSIS, PC was more strongly glucose-responsive than PDH (Figure 4C).
Figure 4. The GK-independent action of PK is powered by mitochondrial anaplerosis.

(A) Cartoon depicting the sources of phosphoenolpyruvate (PEP) from glycolysis and mitochondrial anaplerosis. SAME (monomethyl succinate), PCK2 (mitochondrial phosphoenolpyruvate carboxykinase), Q/L (glutamine/leucine).
(B) Flux of pyruvate carboxylase/pyruvate dehydrogenase (PC/PDH) and insulin secretion in response to various glucose concentrations (donor R082). Significance for PC/PDH is annotated with ** and significance for insulin is annotated with ##. ##P < 0.01, ####P < 0.0001.
(C) Determination of correlation of GSIS with PC and PDH fluxes (donor R082).
(D) Insulin secretion from 2 human islet donors with glucose (1–16.7 mM), 10 mM monomethyl succinate (SAME), and 10 μM PKa as indicated.
(E) Insulin secretion from 2 human islet donors with glucose (1–16.7 mM), 4 mM glutamine plus 10 mM leucine (Q/L), and 10 μM PKa as indicated.
(F) Insulin release from mouse islets in the presence of 2 and 10 mM glucose (2G, 10G), 1 mM leucine (1L), and 10 μM PKa as indicated (n = 8 mice).
(G) Concentration of PEP in INS1 832/13 cells (n = 6) in response to 2.5, 5, 9, 16.7 mM glucose (2.5G, 5G, 9G, 16.7G) in the absence or presence of 4 mM glutamine plus 10 mM leucine (Q/L), 10 mM monomethyl succinate (SAME), and 10 μM PKa.
(H) Representative average β-cell calcium in the absence or presence of PKa and in response to an amino acid ramp at 2.7 mM glucose (2.7G; left; Con, n = 19; PKa, n = 17), 5G (center; Con, n = 20; PKa, n = 19), and 10G (right; Con, n = 14; PKa, n = 13) in mouse islets.
Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 1-way ANOVA (B, D, E), 2-way ANOVA (F,H), or Student’s t-test (G). See also Tables S1–2.
To understand the mechanism by which PEP cycling interacts with PK, we utilized substrates that supply PK with PEP independently of glycolysis. Succinate (or the membrane-permeant succinic acid methyl ester, SAME), while not a direct source of acetyl CoA, has in common with glucose the ability to expand the OAA pool to feed PCK2 (Stark et al., 2009). In human islets, SAME required sub-stimulatory glucose to stimulate insulin secretion but shifted the glucose-sensing curve to the left in a similar manner to GK activators. SAME also displayed an amplifying behavior relative to controls and increased maximal secretion to a similar degree as PK activation (Figure 4D). The combination of SAME and PK activation were also additive and predominantly increased maximal secretion. For SAME to be responsive to PKa, it must enhance the supply of PEP. This fits with the observation, shown in the companion paper, that SAME requires PCK2 to increase insulin secretion (Abulizi et al., 2020), and confirms in human islets that, like glycolysis, mitochondrial anaplerosis-cataplerosis can provide a source of PEP to PK that, in turn, is responsive to PK activation.
At sub-stimulatory glucose concentrations, SAME ± PKa did not stimulate insulin secretion, suggesting a minimal requirement for an oxidizable carbon source such as acetyl CoA (Figure 4D). While glutamine by itself doesn’t stimulate insulin secretion, the combination of glutamine plus leucine (Q/L) provides a direct source of acetyl CoA (through the oxidation of leucine) and a source of anaplerotic α-ketoglutarate (via allosteric activation of glutamate dehydrogenase by leucine), subverting the need for glycolysis. Like SAME, Q/L shifted the glucose-sensing curve to the left and the combination of Q/L and PKa amplified and maximized insulin secretion (Figure 4E). In contrast to SAME, Q/L triggered insulin secretion at sub-stimulatory glucose concentrations that could be amplified by PKa (Figure 4E, shaded box). Interestingly, the augmentation of insulin secretion by Q/L shifted insulin secretion upward and was relatively constant across the whole glucose range, suggesting a fixed acetyl CoA addition by leucine. Consistently, in mouse islets, the synergistic effects of leucine and PKa on insulin secretion were clearly evident at basal and stimulatory glucose concentrations (Figure 4F). Importantly, these data suggest that PK activation does not require GK as long as an ample supply of both oxidizable and anaplerotic carbon are present.
At low glucose concentrations, when PEP cycling is minimal, the steady state PEP concentration is determined simply by its glycolytic supply and PK-mediated clearance. Here, the activity of PKa on its target PK was confirmed by a reduction in the steady state PEP concentration (Figures 4G). The increase in PEP following the addition of SAME or Q/L indicates a PCK2-mediated influx into the PEP pool. This cataplerotic pool was drained by accelerating PEP clearance by PK activation, but only at low glucose, indicating that PEP clearance is matched by supply in the presence of glucose.
We next tested whether calcium influx could be triggered by anaplerotic metabolism in a PK-dependent manner. At low glucose, increasing the mixed amino acid concentrations progressively increased the mean calcium response in mouse islet β-cells, which reached a substantially higher level in the presence of PKa (Figure 4H). As in INS1 832/13 cells (Figure 4G), the effect of PKa on mean calcium progressively diminished as glucose concentrations are raised and glycolytic PEP is generated, suggesting redundancy in the triggering system. These data demonstrate that, like with insulin secretion (Figures 4D–F), mitochondrial PEP works through PK to increase calcium.
PK activation does not alter steady-state metabolism
If interpreted through the lens of the current consensus model, the observation that PK activation significantly increased insulin secretion is expected to be accompanied by a global increase in mitochondrial metabolism. This could occur, for example, if PK activation reduces the concentration of glucose 6-phosphate and disinhibits hexokinase. It was surprising, then, that PK activation did not significantly increase oxygen consumption rates (OCR) in either mouse islets or INS1 832/13 cells (Figures 5A, B). Similarly, at 9 mM glucose, PK activation did not change the concentration of central carbon metabolites, including PEP (Figure 5C). Matching the data in Figure 4G, the lack of a PKa-driven PEP reduction suggests that at higher glucose, glycolysis and PEP cycling are able to keep pace with PK. As metabolite concentrations do not directly correlate with metabolic flux, we used a precision metabolic flux platform, MIMOSA (Mass Isotopomeric MultiOrdinate Spectral Analysis), which has a sensitivity to the impact on metabolism of as little as 0.5 mM glucose (Alves et al., 2015), to directly examine the mitochondrial pathways dependent on pyruvate. In an apparent paradox, despite increasing insulin secretion, independent measures using [U-13C6]-D-glucose for kinetic modeling (ν) or steady state (Φ) calculations, PK did not significantly impact oxidative, PC-dependent anaplerotic, or PCK2-dependent cataplerotic metabolic rates (Figures 5D, E).
Figure 5. PK activation is independent of oxidative mitochondrial metabolism.

(A) Oxygen consumption rate of mouse islets (n = 3) treated with 10 μM PK activator TEPP-46 (PKa, n = 5) or vehicle (Con, n = 4) and the acute addition of electron transport chain inhibitors (Oligo, 1 μM oligomycin; Rot, 1 μM rotenone).
(B) Oxygen consumption rate of INS1 832/13 cells (n = 15) treated with 10 μM PK activator TEPP-46 (PKa) or vehicle (Con) and the acute addition of electron transport chain inhibitors (Oligo, 1 μM oligomycin; Rot, 1 μM rotenone).
(C-E) Impact of PK activation on INS1 832/13 metabolic fluxes. (C) Steady-state concentrations of glycolytic and TCA cycle intermediates. (D) Fractional flux through pyruvate dehydrogenase (VPDH), pyruvate carboxylase (VPC), and phosphoenolpyruvate carboxykinase (VPCK) obtained from the steady-state analysis of enrichments following incubation with [U-13C6]glucose. (E) Absolute fluxes calculated by the mathematical analysis of the time-dependent accumulation of 13C-label into the glycolytic and mitochondrial intermediates (n = 6).
Data are shown as mean ± SEM. Statistics calculated by Student’s t-test. ns, not significant.
By cyclically depriving mitochondria of ADP, PK activation restricts OxPhos
The lack of any evidence that PKa changes steady-state metabolism – despite evidence of increased PK activity and insulin secretion – suggests that the current linear metabolic model is too simplistic. Considering that PKa changes the frequency of metabolic oscillations (instead of the duration) (Figure 3E), then the flux measurements above (Figures 5D, E) would not be sensitive to time-averaged changes in unsynchronized cells at steady state. Consistently, we did observe changes in time-resolved mitochondrial metabolism at a single islet level. PKa effectively reduced the cycling period of NAD(P)H oscillations, which primarily reflect mitochondrial NADH (Patterson et al., 2000), as well as mitochondrial membrane potential (ΔΨm) oscillations (Figure 6A), matching the effects of PKa on ATP/ADP and calcium cycling (Figure 3E). We also observed that PKa increased mitochondrial NAD(P)H fluorescence, and strongly increased ΔΨm, in the presence of both 9 and 2 mM glucose (Figures 6A, B). In particular, PK-induced ΔΨm hyperpolarization would be consistent with decreased OxPhos as would occur in “State 4-like” metabolism when ADP, rather than substrate, is limiting (Figure 6C).
Figure 6. PK requires the mitochondrial PEP cycle to amplify insulin secretion.

(A) Representative recordings and quantification of NAD(P)H fluorescence and mitochondrial membrane potential (ΔΨm) oscillations in mouse islets stimulated by 9 mM glucose (9G) followed by acute application of PKa (Con, n = 36; PKa, n = 43). F/F0 indicates fluorescence normalized to baseline.
(B) Representative recordings and quantification of NAD(P)H fluorescence and mitochondrial membrane potential (ΔΨm) in mouse islets stimulated by 2 mM glucose (2G) followed by acute application of PKa (Con, n = 21; PKa, n = 21). F/F0 indicates fluorescence normalized to baseline.
(C) Cartoon depicting the coordination between PK-mediated ADP lowering, inactivation of the electron transport chain (ETC), and increased mitochondrial GTP (mtGTP) and PEP cycling. PC (pyruvate carboxylase), PDH (pyruvate dehydrogenase), AcCoA (acetyl CoA), OAA (oxaloacetic acid), SCSGTP (GTP-producing isoform of succinyl CoA synthetase), OxPhos (oxidative phosphorylation).
(D) Oxygen consumption of INS1 832/13 cells (n = 6) treated with or without 10 μM FCCP, 100 μM tolbutamide, or 5 μM nimodipine at 2 mM, 5 mM, 7 mM, or 9 mM glucose.
(E) Oxygen consumption of INS1 832/13 cells (n = 6) treated with Plasma Membrane Permeabilizer (Perm), ADP (62.5 μM, 125 μM, or 250 μM), 625 μM PEP, and 4 μM FCCP. Oxygen consumption rate, scale bar = 50 pmol/min.
(F) Oxygen consumption of INS1 832/13 cells (n = 6) treated with Plasma Membrane Permeabilizer (Perm), ADP (125 μM or 500 μM, for 0 μM PEP condition), PEP (625 μM, 313 μM, 156 μM, 78 μM, or 0 μM) and Antimycin A and Rotenone (A/R, 10/5 μM). Oxygen consumption rate, scale bar = 1 pmol/min.
(G) Insulin secretion from PKa- or vehicle-treated control INS1 832/13 cells (Con, n = 12), or INS1 832/13 cells stably overexpressing the ATP and GTP-producing isoforms of succinyl CoA synthetase (SCSATP and SCSGTP, respectively) (n = 6) in response to 9 mM glucose (9G).
Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t-test (B-C), 2-way ANOVA (D), or 1-way ANOVA (G). See also Figure S4.
If β-cell mitochondrial respiration were purely substrate-limited, as predicted by the current canonical model (Nicholls, 2016), then uncoupling should not increase respiration. Instead, ADP limitation was exposed when FCCP uncoupled the proton motive force from ATP synthase, without correlation to the ambient glucose concentration (Figure 6D). Of note, glucose elevation had little effect on oxygen consumption in the presence of physiological glutamine (Alves et al., 2015; Cline et al., 2011). Similar to the ATP/ADP-consuming effect of KCl (Merrins et al., 2016), increasing ATP hydrolysis with tolbutamide increased respiration, while reducing workload (with the further addition of nimodipine) had the opposite effect (Figure 6D). To directly test whether PK itself is capable of limiting OxPhos by ADP privation in situ, respirometry experiments were conducted in permeabilized INS1 832/13 cells in the presence of ADP and succinate (Figure 6E). While ADP dose-dependently stimulated State 3 respiration as would be expected, the further addition of PEP immediately reduced respiration consistent with a shift towards State 4. This inhibition was overcome by replenishment of ADP, which increased respiration to its maximal uncoupled rate (Figure 6E). Considering the observed ADP privation for local control of KATP closure (Figure 1), and the effect of PKa on ΔΨm at low glucose (Figure 6B), we considered that PK may also act locally to restrict OxPhos. Confirming this hypothesis, PEP instantaneously restricted respiration when present at sub-stoichiometric levels relative to ADP (Figure 6F). These data demonstrate the existence of a novel, functionally-defined metabolic compartment in which PK locally controls OxPhos through cytosolic ADP privation.
PK requires the mitochondrial PEP cycle to amplify insulin secretion
The ability of PKa to intermittently restrict ΔΨm and NADH without any net impact on oxidative mitochondrial metabolism (Figures 5 and 6) suggests a mechanism by which PK is able to transiently restrict PDH, and autocatalytically reinforce the anaplerotic fluxes dependent on PC and the cataplerotic PCK2 reactions that refuel the cytosolic PEP pool (Alves et al., 2015; Kibbey et al., 2007) (Figure 6C). PCK2 is dependent on mitochondrial GTP (mtGTP) produced from the GTP-specific isoform of the TCA cycle enzyme succinyl CoA synthetase (SCS). Both the GTP isoform (SCSGTP) and the ATP isoform (SCSATP) are catalytically dependent on a shared α-subunit, so silencing or overexpressing one or the other isoform can modulate mtGTP synthesis and therefore PCK2 activity (Jesinkey et al., 2019; Kibbey et al., 2007). To determine whether PKa requires the PEP cycle to stimulate insulin secretion, we took advantage of INS1 832/13 cells stably overexpressing SCSGTP or SCSATP (Jesinkey et al., 2019). By comparison to the parental INS1 832/13 cells, GSIS was enhanced in SCSGTP cells and blunted in SCSATP cells (Figure 6G). In the SCSATP cells, the effect of PKa to enhance insulin secretion was completely blocked, by comparison to control cells where secretion was increased by 75%. In SCSGTP cells, PKa only modestly increased secretion, in this case because insulin secretion was already near the PKa-stimulated maximum (Figure 6G). At a basal glucose concentration, PKa also slightly increased insulin secretion in SCSGTP cells (Figure S4). These results identify an essential role for the PEP cycle in mediating the effect of PK on insulin secretion.
Evidence for a 2-state model of oscillatory β-cell metabolism
Using molecular biosensors, we characterized the real-time phasic relationship between PK activity, mitochondrial and plasma membrane potentials, KATP conductance, cytosolic calcium, lactate and glutamate, mitochondrial NAD(P)H, and the cytosolic ATP/ADP ratio (Figures 7A–H). In parallel, we used time-resolved stable isotopomer enrichment analysis to measure the oscillatory fluxes of PC relative to PDH (Figure 7I). Based on these measurements, we propose a 2-state model of β-cell metabolism, termed the ‘ADP privation model’ (Figure 7J), describing the relationship of PK activation to β-cell metabolic and electrical oscillations. In this model, cytosolic ADP lowering by PK-driven PEP hydrolysis deprives mitochondria of ADP (ADP privation), at the same time creating antiphase synthetic and OxPhos oscillations (termed ‘MitoSynth’ and ‘MitoOx’, respectively). In reference to the classically defined states of isolated mitochondrial respiration, MitoOx is designated as being ‘state-3-like’ in its metabolism while MitoSynth is ‘state-4-like’. As a conceptual innovation we propose that MitoSynth occurs in intact cells during a physiologic ‘state-4-like’ phase when mitochondria are hyperpolarized and OxPhos is turned off by ADP privation. It is important at the outset to emphasize that the formal designation of ‘state 3’ and ‘state 4’ apply only to isolated mitochondria under a directed biochemical interrogation (Chance and Williams, 1956). Here, the terms MitoSynth and state-4-like or MitoOx and state-3-like are used interchangeably as a frame of reference (and are not intended to imply the unique conditions that may only apply to isolated mitochondria).There is also no inference that in an intact cell or islet that MitoSynth and MitoOx are binary, all-or-none processes. Considering that the TCA cycle is required to produce the mtGTP required by PCK2, it is likely that some combination of oxidative and anaplerotic/cataplerotic metabolism occurs simultaneously.
Figure 7. Evidence for a 2-state model of oscillatory β-cell metabolism.

(A-H) Metabolic and electrical oscillations in islet β-cells stimulated with 10 mM glucose. ATP/ADP, calcium, glutamate, lactate, and PKM2 are cytosolic parameters. NAD(P)H, ΔpHm (SypHer mt), and ΔΨm are mitochondrial parameters. Plasma membrane potential (Vm) and potassium conductance (GK) are electrical parameters. Silent phase, white boxes; active phase, shaded boxes.
(H) Antiphase oscillations in flux through PDH and PC in INS1 832/13 cells following the addition of 9 mM glucose.
(I) Model of oscillatory β-cell metabolism with 2 states (triggering vs. secretory) separated by membrane depolarization. Before depolarization, PK lowers ADP, reducing flux through the ETC (ADP-starved, “State 4-like”) and the TCA cycle while activating the PEP cycle. After PK lowers ADP sufficiently to close KATP channels, workload in the form of ATP hydrolysis restores ADP (e.g. by exocytosis and pumps), increasing flux through the ETC (ADP replete “State 3-like”), the TCA cycle, and glycolysis. Note that recruitable PK isoforms (M2 and L) are active before depolarization, when glycolytic flux is low and the FBP concentration is high.
β-cells have electrically silent and active phases (white versus grey boxes in Figures 7A–H) characterized by the square waves of the plasma membrane potential (Vm). The beginning and ending of each oscillatory phase coincide with the peaks and troughs of ATP/ADP. The metabolic and electrical features described below start with the 1) silent phase with 2) progressive transition into state 4-like respiration with activation of the anaplerotic/cataplerotic PEP cycle 3) to trigger depolarization and 4) activate OxPhos:
1). The electrically-silent phase
The transition into the electrically silent phase begins when ADP levels rise sufficiently to re-open KATP channels and hyperpolarize the plasma membrane as well as close voltage-dependent calcium channels (Figures 7A, B, G, H). The decrease in metabolic demand reduces ATP hydrolysis rates and allows glycolysis and OxPhos to clear calcium and raise ATP/ADP (Figure 7A).
2). MitoSynth: State-4-like respiration is sustained by the PEP cycle
The progressive lowering of ADP by both OxPhos and glycolysis eventually slows the TCA cycle. This is identified by the asymptote of ΔΨm, mitochondrial pH (ΔpHm), and mitochondrial NAD(P)H midway through the silent phase that hallmarks state 4 respiration (Figure 7C, D), as previously observed by Kennedy and colleagues who measured oxygen (Kennedy et al., 2002).
As the TCA cycle stalls, mitochondrial metabolites accumulate so that acetyl CoA can activate the PC-dependent generation of OAA observed by stable isotope patterning (Figure 7I). Through the mass action of OAA synthesis and high mitochondrial NADH, the glutamate-aspartate shuttle slows (as indicated by cytosolic glutamate accumulation) (Figure 7E). The sn-glycerol-3-phosphate shuttle would also be expected to slow in the face of a higher ΔΨm and ΔpHm. In the presence of continuous GK activity but with GAPDH slowing (from increased cytosolic NADH/NAD+), the levels of FBP increase to activate PK allosterically (Figure 7G). With GAPDH slowed, PEP cycling via PCK2 would continue to lower ADP. The lowering of lactate can be explained in the setting of slowed pyruvate dehydrogenation and reduced glycolysis (Figure 7F). A high mitochondrial ATP/ADP ratio reverses SCSATP to provide additional succinyl CoA for enhanced mtGTP synthesis by SCSGTP. PCK2 utilization of OAA and mtGTP supports rapid mitochondrial PEP synthesis to further lower ADP in the cytosol until KATP channels close, marking the end of this phase (Figures 7A, B, G, H). In short, PK-mediated ADP privation during MitoSynth slows OxPhos to divert mitochondrial pyruvate away from oxidation and into PEP synthesis. Such feed forward regulation continues to accelerate PEP cycling until ATP/ADP elevation triggers KATP closure.
3). The electrically-active phase
After membrane depolarization and Ca2+ influx, during the electrically active phase of an oscillation (the shaded boxes in Figures 7A–H), insulin exocytosis occurs. This active phase persists as long as ADP is maintained below the KATP threshold.
4). MitoOx: State 3-like respiration is sustained by the ETC
During this active phase, the sudden surge in metabolic workload (e.g., by Ca2+ pumping and exocytosis, Figure 6D) accelerates ATP hydrolysis (Figure 7A). Importantly, as neither calcium nor the subsequent provision of substrate stimulate mitochondrial respiration (Civelek et al., 1996), it is the elevation of ADP that restores state 3-like OxPhos (Figures 4E, F). Active OxPhos then consumes (depolarizes) ΔΨm, ΔpHm, and mitochondrial NADH (Figure 7C, D), and an active TCA cycle lowers acetyl CoA, transitioning mitochondria from anaplerotic PC to favor oxidative PDH (Figure 7I). Concurrently, the glutamate-aspartate shuttle reactivates with cytosolic glutamate decreasing in the face of a more receptive mitochondrial NADH/NAD+ ratio and diminished PC anaplerosis (Figure 7E), and elevated glycolytic flux lowers the cytosolic NADH/NAD+ ratio as is reflected by the increase in lactate (Figure 7F). As glycolytic intermediates are lowered, the drop in FBP inactivates PKM2 and PKL (Figure 7G). MitoOx, in which calcium and OxPhos synergize, most resembles the β-cell consensus model where the capacity to support the ATP/ADP ratio sustains depolarization in the face of high ATP demand. This electrically-active phase ends as ATP hydrolysis exceeds the capacity of mitochondria to keep ADP low and KATP channels re-open and the plasma membrane repolarizes.
As long as glucose remains elevated, oscillations between these two states persist.
DISCUSSION
These data provide evidence that PEP hydrolysis by PK, rather than OxPhos, provides the energetic push required to raise ATP/ADP beyond the threshold for KATP channel closure. Importantly, PEP can close KATP channels in isolated membrane patches despite the presence of the strong KATP channel-opener ADP, indicating that PK can influence the metabolic microenvironment near the channel. The ability of PK to restrict OxPhos by locally depriving the mitochondria of ADP provides a second example of β-cell metabolic compartmentation in which ADP is the signal carrier. Through this ‘ADP privation’, PK is able to switch mitochondria between synthetic and oxidative functions without impacting glucose oxidation. Finally, the ability of PK activators to amplify insulin secretion solidifies the importance of glycolytic ATP for insulin exocytosis.
The functional linkage between PK and KATP in β-cells is consistent with prior studies in cardiac myocytes, where PK associated with the C-terminus of Kir6.2 closes KATP channels (Weiss and Lamp, 1987; Dhar-Chowdhury et al., 2005). These data do not undermine the previously described metabolic control of glycolysis exerted by GK (Matschinsky and Ellerman, 1968). Real-time imaging of calcium oscillations reaffirmed that the threshold for membrane depolarization is set by GK and observable in the duty cycle (Henquin, 2009). Based on the canonical OxPhos-only model, PK activation should not be able to increase GSIS since GK activity determines the glycolytic and, consequently, mitochondrial flux rates. Mitochondrial anaplerosis has been identified as an important component of metabolic coupling without a clear mechanism (Prentki et al., 2013). Here, we identified a glucose-independent mode of triggering based on mitochondrial PEP biosynthesis that was able to raise mean calcium in the presence of low glucose. Islet perifusion studies further revealed that, independently of initiating membrane depolarization (i.e. in the presence of KCl), the dynamically-regulated, allosterically-recruitable PK appears to play a dominant role in amplifying secretion. This is reinforced by the requirement for an acetyl CoA source for triggering but an anaplerotic carbon source for metabolic amplification in human and mouse islets. Thus, a key finding here is that PK underlies a separate mode of β-cell glucose-sensing downstream of GK that may provide a mechanism dependent upon anaplerosis. While targeting OxPhos in β-cells has not been successful therapeutically, preclinical data suggest the anaplerotic mechanism may be of potential benefit (Abulizi et al., 2020).
Importantly, PK activation did not significantly increase oxidative glucose metabolism, or correspondingly, the duration of the calcium pulses. Instead, it increased the rate of ATP/ADP turnover, and consequently, the frequency of calcium oscillations. Human islet measurements likewise indicated that PK potentiates secretion in a glucose-dependent manner and does not significantly lower the threshold for insulin secretion. The most straightforward explanation for metabolic amplification is that PK provides a source of ATP required for exocytosis (Eliasson et al., 1997; Pizarro-Delgado et al., 2015; Takahashi et al., 1999). As shown previously, ADP and PEP (but not pyruvate) are sufficient for biphasic insulin secretion in membrane-permeabilized islets, including in the presence of rotenone (Pizarro-Delgado et al., 2016). In intact cells, the activity of PK is limiting during periods of high metabolic flux, such as the state induced by KCl or the active phase of calcium oscillations, when FBP levels are low and the recruitable PK isoforms are least active (Merrins et al., 2016). PK activators stabilize the active enzyme and circumvent the control by FBP. Correspondingly, knockdown of PKM2 in MIN6 insulinoma cells reduces insulin secretion (Nakatsu et al., 2015).
Incorporating the data herein, we propose a unifying model for glucose-stimulated insulin secretion termed the ‘ADP privation model’. In this 2-state model, PK-controlled ADP availability switches mitochondria between a biosynthetic state where anaplerosis is high (MitoSynth), and one where OxPhos is high (MitoOx). The importance of metabolic control exerted by ADP stems from the simple fact that ADP must be reduced to low μM concentrations for KATP channels to close (Nicholls, 2016; Tarasov et al., 2006), and that mitochondrial ATP synthase cannot run without ADP (Chance and Williams, 1955). During MitoSynth, the intracellular free energy of PEP favors PK-mediated ADP consumption (ADP privation), that, in effect, turns off OxPhos and converts mitochondria into synthetic organelles. The mitochondrial contribution of PEP boosts PEP production beyond what is achievable by glycolysis alone, enhancing the cytosolic ATP/ADP-generating capacity of PK in the triggering phase. The transition to MitoOx is toggled by the high rate of ATP hydrolysis associated with membrane depolarization, pumps, and vesicle fusion (Nicholls, 2016; Affourtit et al., 2018). ADP restoration increases OxPhos and accelerates TCA cycle and glycolytic fluxes (Civelek et al., 1996). A key advantage of allosteric PK recruitment is the ability to reinforce metabolic oscillations in response to metabolic regulators like FBP (Merrins et al., 2013, 2016). Consequently, the apex of PK activity occurs just prior to membrane depolarization, at the nadir of OxPhos, which resumes following depolarization-initiated ATP hydrolysis. This back and forth between an electrically-silent triggering phase and an electrically-active oxidative secretory phase allow β-cell mitochondria to move between PEP biosynthesis and OxPhos while fuel input remains constant.
Paradoxically, PK recruitment occurs in parallel with the slowing of glycolytic flux. However, this recruitment occurs in the setting of continuous GK flux, when FBP levels rise and flux through PC increases, rerouting pyruvate flux through PCK2 (Jesinkey et al., 2019; Kibbey et al., 2007; Stark et al., 2009). Notably, the expression of PC, generating the OAA necessary for the production of PEP, has been shown to be 80% lower in human islets compared to rodent islets and INS1 832/13 cells (MacDonald et al., 2011). The expression of PC is further reduced islets from type 2 diabetics (MacDonald et al., 2009), which could be a disadvantage when considering PK activation for the treatment of diabetes. However, PC expression does not appear to be a limitation, since PC flux in human islets was found by MIMOSA to be substantial and well-correlated with insulin secretion. Glucose increased flux through PC much more strongly than PDH, in general agreement with prior studies (Alves et al., 2015; Fransson et al., 2006; MacDonald et al., 2005; Prentki et al., 2013). Prior estimations of PEP cycling showed rates could reach ~40% of the total PK flux in islets (Stark et al., 2009). These estimations did not consider that oscillatory metabolism divides OxPhos and anaplerosis into separate phases. As such, mitochondrial PEP cycling could potentially explain nearly all of PK activity during the electrically-silent phase.
The ADP privation model recognizes that the PEP cycle primarily triggers via PK, while OxPhos primarily sustains the active phase. As both processes generate ATP, their individual contributions may not be fully separable in either the time or compartment domains, and both processes are necessary to achieve maximal secretion. However, the timing of OxPhos relative to membrane depolarization (Jung et al., 2000) raises the question of whether it is bioenergetically feasible that mitochondria, even if strategically positioned at the plasma membrane, could lower ADP sufficiently to close KATP channels, as PK was demonstrated to do. Our data are supported by observations of high ATP generation at the plasma membrane relative to the cytosol and mitochondria (Kennedy et al., 1999). It remains to be determined if there is a point at saturating glucose levels where, through either glycolysis and/or the PEP cycle, PK can locally deplete ADP sufficient to close KATP channels, while other mitochondria at sites of active ATP hydrolysis have adequate ADP to conduct OxPhos simultaneously. This work only shows that it is possible for PEP cycle-supported, PK-mediated ADP privation to restrict OxPhos and KATP channel activity.
Although many qualitative β-cell bioenergetics studies have shown that ADP supply and demand are critical for OxPhos and insulin secretion (Ainscow and Rutter, 2002; Doliba et al., 2003; Panten et al., 1986; Sweet et al., 2004; Affourtit et al., 2018), quantitative metabolic control analyses in β-cells have remained lacking (Affourtit et al., 2018; Nicholls, 2016). Since tools to quantify the phosphorylation potential and ATP flux rely on temporal averaging (Affourtit et al., 2018), we currently lack the means to determine how PK-driven ATP/ADP cycling bioenergetically supports increased exocytosis relative to OxPhos. Approaches will need to be developed that can absolutely quantify adenine nucleotides and their sources in a subcompartment-specific and time-resolved way. However, reevaluation of such β-cell bioenergetics may have similar application to other metabolic cycles and pathways that have been proposed (Schuit et al., 1997; Farfari et al., 2000; Joseph et al., 2006; Jensen et al., 2008; Prentki et al., 2013) but are not addressed in this simplified 2-state model.
This new understanding of the native role of PK in β-cell metabolism has broad implications in non-native environments where it has been co-opted, such as in cancer (Dayton et al., 2016; Israelsen et al., 2013). It may be particularly advantageous for cancer or other dividing cells to shut down OxPhos in order for the mitochondria to synthesize, rather than oxidize, building blocks. Time-resolved stable isotope measurements identify that OxPhos and anaplerosis are anti-phase with each other and suggests that mitochondria choose not to oxidize and synthesize simultaneously. In light of this, the phenomenon of PK-associated Warburg metabolism, while often considered for its ATP-generating capacity, may be even more important for its ADP lowering capacity (Harris and Fenton, 2019). Just as in β-cells, ADP privation may shift mitochondrial activity from OxPhos into the biosynthesis of essential nutrients needed to support cell division, or the generation of reactive oxygen species used for signaling (Plecitá-Hlavatá et al., 2020). Of note, cells with more robust PEP cycling have an increased mass of elongated mitochondria while those deficient have fewer more fragmented mitochondria (Jesinkey et al., 2019). The ability of PK isoforms to oscillate may also provide additional advantages not considered here, e.g. intermittently reducing membrane potential to reduce free radical damage that might occur if mitochondria were perpetually kept in a ‘state-4-like’ situation. In this context, activating PK may have some potential therapeutic benefits in certain situations where Warburg metabolism is observed.
Finally, the ability of pharmacologic PK activators to raise insulin secretion has broad conceptual implications for type 2 diabetes therapies. Not only is PK identified as a novel target for diabetes therapy, but we demonstrate that, for a given level of glucose, the secretory pathway can be internally remodeled to tune the glucose responsiveness of healthy and diseased human β-cells. Currently available drugs that increase insulin secretion by stimulating glucose uptake (e.g. GK activators) elevate the metabolic workload on each β-cell (Porat et al., 2011) and can lead to glucotoxic-like damage (Nakamura and Terauchi, 2015). Drugs that increase insulin secretion by directly triggering membrane depolarization such as sulfonylureas, while invaluable for treating some forms of MODY (Kim, 2015), decouple β-cell nutrient sensing with insulin secretion and increase the risk of hypoglycemia. PK-dependent remodeling of the β-cell metabolic pathways could lead to a treatment for diabetes that avoids these pitfalls.
Limitations of Study
To fully test the 2-state model, it will ultimately be important to measure adenine nucleotides and PEP in a subcompartment and time-resolved way. A further limitation is the need to evaluate PKa in vivo. This is addressed by the companion paper (Abulizi et al., 2020).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Merrins (mmerrins@medicine.wisc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate new code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Islet Preparations
C57BL/6J healthy male mice from the Jackson Laboratory were housed 4 per cage at 21–23°C, maintained on a 12 hour light/dark cycle (light on 6:00AM to 6:00PM), with chow diet and water provided ad libitum. All mice used in experiments throughout the study exhibited normal health. At 12–16 weeks old, after at least a one-week acclimation period after importation from the Jackson Laboratory, mice were sacrificed via cervical dislocation and islet isolations were carried out as previously described (Gregg et al., 2016). Islets were cultured overnight in RPMI-1640 supplemented with 10% (v/v) fetal bovine serum (ThermoFisher A31605), 10,000 units/ml penicillin and 10,000 μg/ml streptomycin (Fisher Scientific). All procedures involving animals were approved by the Institutional Animal Care and Use Committees of the University of Wisconsin-Madison and the William S. Middleton Memorial Veterans Hospital, and followed the NIH Guide for the Care and Use of Laboratory Animals (8th ed. The National Academies Press. 2011.).
Human Islet Preparations
Human islets from normal donors were obtained from the University of Alberta Diabetes Institute, the University of Chicago Diabetes Research and Training Center, and the University of Minnesota Schulze Diabetes Institute. The age, sex, body mass index, and %HbA1c of each donor is provided in Table S1. Human islets were cultured in glutamine-free CMRL supplemented with 10 mM niacinamide and 16.7 μM zinc sulfate (Sigma), 1% ITS supplement (Corning), 5 mM sodium pyruvate, 1% Glutamax, 25 mM HEPES (American Bio), 10% HI FBS and antibiotics (10,000 units/ml penicillin and 10 mg/ml streptomycin). All media components were obtained from Invitrogen unless otherwise indicated.
Cell Line Studies
Stocks of the clonal INS-1 832/13 cell line overexpressing the human insulin gene (INS-1) were a gift from C.B. Newgard (Duke University School of Medicine). hSCS-ATP and hSCS-GTP constitutive cell lines (INS-1 832/13 cells transduced to overexpress V5-tagged human ATP- or GTP-specific SCSβ subunit) were prepared previously in R.G. Kibbey’s (Jesinkey et al., 2019). INS1 832/13, hSCS-ATP, and hSCS-GTP cells were cultured as monolayers in RPMI-1640 complete medium as previously described (Kibbey et al., 2007). Cells were incubated at 37°C in 5% CO2, 95% air.
METHOD DETAILS
Cloning and Adenoviral Delivery of Biosensors.
Genetically-encoded sensors for glutamate (Addgene) (GltI253-cpGFP.L1LV/L2NP, Kd = 107 μM) (Marvin et al., 2013), lactate (Addgene) (Laconic/ pcDNA3.1(−)) (San Martín et al., 2013), and mitochondrial pH (Addgene) (SypHer mt) (Poburko et al., 2011) were cloned using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs) into a modified pENTR-SD shuttle vector (Invitrogen) containing the rat insulin promoter (RIP) and rabbit β-globin intron as in a previous study (Merrins et al., 2013). Gateway Clonase II was then used to prepare the full-length adenoviral construct in pAd/PL-DEST (Invitrogen). This procedure was used previously to generate adenovirus for β-cell specific Perceval-HR ATP/ADP biosensors (Merrins et al., 2016) and PKAR PKM2 biosensors (Merrins et al., 2013). For adenoviral delivery of the insulin promoter-driven biosensors, islets were infected immediately post-isolation with high-titer adenovirus for 2 hours at 37°C, then moved to fresh media overnight.
Timelapse Imaging.
For measurements of cytosolic Ca2+, islets were pre-incubated in 2.5 μM FuraRed (Molecular Probes F3020) in islet media for 45 min at 37°C before they were placed in a glass-bottomed imaging chamber (Warner Instruments) mounted on a Nikon Ti-Eclipse inverted microscope equipped with a 20X/0.75NA SuperFluor objective (Nikon Instruments). The chamber was perfused with a standard external solution containing 135 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 20 mM HEPES (pH 7.35). The flow rate was 0.4 mL/min and temperature was maintained at 33°C using solution and chamber heaters (Warner Instruments). Excitation was provided by a SOLA SEII 365 (Lumencor) set to 10% output. Single DiR images utilized a Chroma Cy7 cube (710/75x, T760lpxr, 810/90m). Excitation (x) or emission (m) filters (ET type; Chroma Technology) were used in combination with an FF444/521/608-Di01 dichroic beamsplitter (Semrock) as follows: FuraRed, 430/20x and 500/20x, 630/70m (R430/500); NAD(P)H, 365/20x, 470/24m; Rhodamine-123 and Glutamate, 500/20x, 535/35m; and Perceval-HR and SypHer mt, 430/20x and 500/20x, 535/35m (R500/430). Fluorescence emission was collected with a Hamamatsu ORCA-Flash4.0 V2 Digital CMOS camera every 6 s. A single region of interest was used to quantify the average response of each islet using NIS-Elements (Nikon Instruments) and custom MATLAB software (MathWorks).
Electrophysiology.
β-cell Ca2+ current and exocytosis were measured as in (Merrins and Stuenkel, 2008) with minor changes. Briefly, a Sutter MP-225 micromanipulator was used together with a HEKA EPC10 patch-clamp amplifier (Heka Instruments, Bellmore, NY) in the whole cell patch-clamp configuration to record Ca2+ current from intact islets perfused with standard external solution (above) at 33°C. Pipette tips were filled with an internal solution (in mM: 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2·6H2O, 0.05 EGTA, 5 HEPES, 0.1 cAMP, 3 MgATP; pH 7.15 with CsOH) and 5 μM PKa or vehicle (0.1% DMSO) was added to this as indicated for dialysis into the cell. After membrane rupture, islet β-cells were identified by size (>5.5 pF), and after 1 min Ca2+ current was quantified from a 15 ms depolarization from −70 to 0 mV using a P/4 leak subtraction protocol. After 1 additional min, exocytosis was stimulated by activating VDCCs with a series of ten 500 ms membrane depolarizations from −70 to 0 mV. Capacitance responses (fF) and Ca2+ currents (pA) were normalized to initial cell size (pF). Islet KATP conductance was measured as described previously (Gregg et al., 2016). For single channel patch clamp experiments, mouse or human islets were dispersed with Accutase (Fisher Scientific) and cells were plated on sterilized glass shards. Recordings were performed according to (Krippeit-Drews et al., 2003). Gigaseals were obtained in extra cellular bath solution (in mM): 140 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 0.5 Glucose, 10 HEPES, pH 7.4, adjusted with NaOH and clamped at −50mV before excision into inside-out configuration. Equilibrium solutions with K+ as the charge carrier were used for recording. The bath solution contained (in mM): 130 KCl, 2 CaCl2, 10 EGTA, 0.9 free Mg2+, 10 sucrose, 20 HEPES, pH 7.2 with KOH. The pipette solution contained (in mM): 10 sucrose, 130 KCl, 2 CaCl2, 10 EGTA, 20 HEPES, pH 7.2, adjusted with KOH. Intracellular recording electrodes made of borosilicate glass (Harvard Apparatus, Holliston, MA) with tip resistance of 3 MΩ (Sutter Instruments P-1000) were polished by microforge (Narishige MF-830) to the final tip resistance of 5–10 MΩ. After formation of gigaseal (>2.5 GΩ) and withdrawal of the pipette, excised inside-out configuration was established. A HEKA Instruments EPC10 patch-clamp amplifier was used for registration of current. Data was filtered online at 1kHz with Bessel filter and analyzed offline using Clampfit 10 software (Axon Instruments).
Mouse Islet GSIS.
After incubation, equal numbers of islets were hand-picked and placed into a 12-channel BioRep device (75–100 islets/chamber) containing 2.7 mM glucose KRPH buffer (in mM: 140 NaCl, 4.7 KCl, 1.5 CaCl2, 1 NaH2PO4, 1 MgSO4, 2 NaHCO3, 5 HEPES, 1% fatty acid-free BSA, and 1 mM leucine as indicated; pH 7.4) with 100 μL Bio-Gel P-4 Media (Bio-Rad). Islets were equilibrated for 48 minutes, and then perifused in intervals based on the experimental conditions at 37°C. Insulin and glucagon content and secretion was assessed by Insulin AlphaLISA (Perkin Elmer) with an alpha plate reader (TECAN Spark). After the perifusion, islets from each chamber were collected and incubated at room temperature for 15 minutes in 1 ml 0.1% Triton-X. Islets were then vortexed and frozen. DNA content was determined using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific).
Respirometry.
Islet oxygen consumption rates (OCR in pmoles/min) were measured at 37°C with the Seahorse XF-24 Analyzer, and INS1 832/13 cells with the Seahorse XF-96 Analyzer (Agilent). Islets were recorded in DMEM (Sigma D5030) with addition of 0.02% bovine serum albumin, 2.5 mM glucose, and 5 μM PKa or vehicle (0.1% DMSO) (Figure 5A). 60–70 islets were deposited into each well of XFe24 plate, washed twice with the media, covered with an islet capture screen, and incubated for 1 hour before introduction to the Seahorse instrument. Baseline respiration was measured in 2.5 mM glucose. Islets were then sequentially exposed to 9 mM glucose, 1 μM oligomycin, 1 μM FCCP, and 5 μM rotenone as indicated. Oxygen consumption in INS1 832/13 cells was recorded in similar DMEM as the islets described above with additional 10 mM HEPES and pre-incubated with 2.5 mM glucose and physiologic glutamine (Figures 5B, 6D). Once in the instrument, baseline respiration was measured followed by stimulation with glucose as indicated. Cellular respiration was perturbed following the glucose addition by either 1 μM FCCP or 100 μM tolbutamide followed by 5 μM nimodipine. All cells were then finally exposed to a mixture of 5 μM rotenone and 10 μM antimycin A. For the studies with permeabilized INS1 832/13 cells in the presence of ADP and succinate, the cells were permeabilized using the XF Plasma Membrane Permeabilizer based on the Agilent Technologies’ instructions (Figures 6E, 6F). The 1x Mitochondrial Assay Solution buffer with 10 mM succinate was used. The INS1 832/13 cells were treated with various ADP concentrations (62.5 μM, 125 μM, or 250 μM), 625 μM PEP, and 4 μM FCCP as indicated (Figure 6E). In the experiment for Figure 6F the cells were exposed to 125 μM ADP or 500 μM ADP (for the 0 μM PEP condition), then to various PEP concentrations (0 μM, 78 μM, 156 μM, 313 μM, or 625 μM), next to 1 mM ADP, and finally to antimycin A (10 μM) and rotenone (5 μM).
PK Activity.
The enzymatic assay of pyruvate kinase (EC 2.7.1.40) is from Sigma (Bergmeyer, H.U. et al.), with modifications for a 96-well plate format, and the use of a 1:2 PEP dilution curve starting at 2.5 mM or 5 mM PEP. Unless specified, 3 μM FBP was present. Experiments were carried out at 37°C.
Human Islet Insulin Secretion Studies.
Human donor islets were cultured intact, then dispersed and re-aggregated as pseudo-islets for dynamic insulin secretion studies. Islets were dispersed with Accutase (Fisher Scientific) and the resulting cell suspension was seeded at 5000 cells per well of a 96-well V-bottom plate, lightly centrifuged (200g) and then incubated for 12–24 h at 37°C 5% CO2/95% air. GSIS perifusion studies were performed on an 8-channel BioRep device. The secretion buffer consisted of DMEM (Sigma D5030) supplemented with NaHCO3, 10 mM HEPES, 2 mM glutamine, 0.2% fatty acid-free BSA and 2.5 mM followed by 9 mM glucose in presence of 10 μM TEPP-46 or DMSO (0.1% final) adjusted to the same volume for the vehicle control. Additionally, dynamic GSIS studies were performed 24 h after the islets were plated into the 96 well plates following dispersion and re-aggregation. The human islet plates were washed and incubated at 37°C 5% CO2/95% air in standard KRB (in mM: 115 NaCl, 5 KCl, 24 NaHCO3, 2.2 CaCl2, 1 MgCl2) supplemented with 2.5 mM glucose, 2 mM glutamine, 24 mM HEPES and 0.25% BSA for 1.5 h. After the first incubation, human islet plates were then washed with glucose free KRB and incubated for 2 hours in KRB study media with 1, 2.5, 5, 7, 9, 11.2, 13 or 16.7 mM glucose in presence of 10 μM TEPP-46 or 0.1% DMSO. Supernatants were evaluated by insulin ELISA (ALPCO).
13C-Isotopic Labeling Studies.
INS1 832/13 cells were cultured as monolayers in RPMI-1640 complete medium as previously described (Kibbey et al., 2007). 13C-Isotopic labeling studies were performed in DMEM medium (D5030, Sigma-Aldrich) supplemented with 9 mM glucose, 4 mM glutamine, 0.05 mM pyruvate, and 0.45 mM lactate. Cells were preincubated in this media at 37°C for 2 h until a metabolic steady state was reached at which time unlabeled glucose was replaced with [U-13C6]glucose (Cambridge Isotope Laboratories). Following the addition of 13C-label, cells were quenched at t = 0, 5, 15, 30, 60, 120 and 240 min (n = 6 per time point). Cell quenching and sample preparation was performed as previously described (Alves et al., 2015; Patel et al., 2010).
LC-MS/MS Analysis and Modeling of 13C-labeled Time Courses.
Metabolite concentrations and 13C-enrichments were determined by mass spectrometry using a SCIEX 5500 QTRAP equipped with a SelexION for differential mobility separation (DMS) as described previously (Alves et al., 2015). The integrated analysis of 13C labeling time courses was performed using a mathematical model of the TCA cycle similar to the one described previously (Alves et al., 2015). The model describes the transfer of label through the distal portion of glycolysis and the TCA cycle using [U-13C3]DHAP as a driving function. The label is distributed through all possible isotopomers for PEP, pyruvate, citrate, αKG, glutamate, succinate, malate and OAA and it is used to measure the flux through glycolysis, pyruvate kinase, pyruvate dehydrogenase, pyruvate carboxylase, citrate synthase, reversed isocitrate dehydrogenase and β-oxidation. Isotopomers were grouped in combination pools based on the number and/or position of labeled carbons and used to fit target data. Metabolic modeling and statistical analysis was performed using CWave software, version 4.0 (Mason et al., 2003) running in MATLAB. The flux values is shown as the least-square fit ± the standard deviation of the distribution of uncertainty from 100 Monte-Carlo simulations (Patel et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of experiments can be found in the figure legends. Where applicable mouse and human islets were randomly assigned to groups. Data are expressed as means ± SEM. Statistical significance was determined using one- or two-way ANOVA with Sidak multiple-comparisons test post hoc or Student’s t-test as appropriate. Data were continuous and normally distributed, and consequently analyzed with parametric tests. Differences were considered to be statistically significant at P < 0.05. Statistical calculations were performed with GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| N/A | ||
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| Human islets | University of Alberta Diabetes Institute | N/A |
| Human islets | University of Chicago Diabetes Research and Training Center | N/A |
| Human islets | University of Minnesota Schulze Diabetes Institute | N/A |
| Human islets | Integrated Islet Distribution Program (https://iidp.coh.org/) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TEPP-46 (PKa) | EMD Millipore | 50–548-70001 |
| Glucokinase Activator III, Ro-28–1675 | Fisher Scientific | 50–966-50001 |
| Glucokinase Activator | Merck | 346021 |
| RPMI-1640 cell culture media | Sigma Aldrich | R8758 |
| Fetal bovine serum | Thermo Fisher | A31605 |
| Gateway Clonase II | Fisher Scientific | 11791019 |
| FuraRed | Molecular Probes | F3020 |
| Accutase | Thermo Fisher Scientific | 501129055 |
| Bio-Gel P-4 Media | Bio-Rad | 1504124 |
| DMEM cell culture media | Sigma Aldrich | D5030 |
| Oligomycin, | Sigma Aldrich | 75351 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma Aldrich | C2920 |
| Rotenone | Sigma Aldrich | R8875 |
| Tolbutamide | Fisher Scientific | T0891 |
| Nimodipine | Sigma Aldrich | N149 |
| Antimycin A | Sigma Aldrich | A8674 |
| XF Plasma Membrane Permeabilizer | Agilent Technologies | 102504–100 |
| Phospho(enol)pyruvic acid monopotassium salt (PEP) | Sigma Aldrich | P7127–250mg |
| D-Fructose 1,6-bisphosphate trisodium salt hydrate (FBP) | Sigma Aldrich | F6803 |
| CMRL culture media | GIBCO | N/A |
| 1% Glutamax, | Invitrogen | 35050061 |
| Penicillin-Streptomycin (10,000U/mL) | Fisher Scientific | 15140122 |
| Mono-methyl hydrogen succinate (SAME) | Sigma Aldrich | M81101 |
| [U-13C6]glucose | Cambridge Isotope Laboratories | 110187–42-3 |
| Critical Commercial Assays | ||
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | E5520S |
| Insulin AlphaLISA | Perkin Elmer | AL204F |
| Invitrogen Quant-iT PicoGreen dsDNA Reagent | Thermo Fisher Scientific | P7581 |
| Enzymatic assay of pyruvate kinase | Sigma Aldrich (Bergmeyer, H.U. et al.) | EC 2.7.1.40 |
| Human Insulin ELISA | ALPCO | 80-INSHU-E10 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| Clonal INS-1 832/13 cell line overexpressing the human insulin gene (INS-1) | C.B. Newgard (Duke University School of Medicine) | N/A |
| hSCS-ATP constitutive cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human ATP-specific SCSβ subunit) | (Jesinkey et al., 2019) | N/A |
| hSCS-GTP constitutive cell line (INS-1 832/13 cells transduced to overexpress V5-tagged human GTP-specific SCSβ subunit) | (Jesinkey et al., 2019) | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| GltI253-cpGFP.L1LV/L2NP, Kd = 107 μM | Addgene (Marvin et al., 2013) | 41733 |
| Laconic/pcDNA3.1(−) | Addgene (San Martín et al., 2013) | 44238 |
| SypHer mt | Addgene (Poburko et al., 2011) | 48251 |
| pENTR-SD shuttle vector | Invitrogen | K242020 |
| pAd/PL-DEST Gateway Vector | Invitrogen | V49420 |
| β-cell specific Perceval-HR ATP/ADP biosensor | (Merrins et al., 2016) | N/A |
| β-cell specific PKAR PKM2 biosensor | (Merrins et al., 2013) | N/A |
| Software and Algorithms | ||
| NIS-Elements | Nikon Instruments | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| MATLAB software | Mathworks | https://www.mathworks.com/products/matlab.html |
| Axon pClamp 10 software | Axon Instruments/ Molecular Devices | https://mdc.custhelp.com/app/answers/detail/a_id/18779/~/axon%E2%84%A2pclamp%E2%84%A2–10-electrophysiology-data-acquisition-%26-analysis-software-download |
| CWave software, version 4.0 (Mason et al., 2003) running in MATLAB | (Alves et al., 2015; Jesinkey et al., 2019) | N/A |
| GraphPad Prism 7.0 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| 20X/0.75NA SuperFluor objective | Nikon Instruments | MRF00200 |
| SOLA SEII 365 | Lumencor | N/A |
| Cy7 cube (710/75x, T760lpxr, 810/90m) | Chroma | 49007 |
| FF444/521/608-Di01 dichroic beamsplitter | Semrock | FF444/521/608-Di01–25×36 |
| ORCA-Flash4.0 V2 Digital CMOS camera | Hamamatsu | C11440–22CU |
| HEKA EPC10 patch-clamp amplifier | Heka | N/A |
| SCIEX 5500 QTRAP equipped with a SelexION for differential mobility separation (DMS) | SCIEX | N/A |
| Bio-Rep Perifusion Instrument | Bio-Rep | N/A |
HIGHLIGHTS.
Pyruvate kinase (PK) is a highly compartmentalized β-cell fuel sensor
Membrane-associated PK closes KATP channels and controls calcium influx
By lowering ADP, PK toggles mitochondria between OxPhos and PEP biosynthesis
PK activation increases oscillatory frequency and amplifies insulin secretion
CONTEXT AND SIGNIFICANCE.
The hormone insulin is essential for proper control of blood sugar. Too little insulin results in diabetes, while too much dangerously lowers blood sugar. Insulin-producing β-cells in the pancreas sense blood sugar levels through the metabolism of glucose, which has historically been linked to mitochondrial energy production. However, metabolism of a high-energy glucose byproduct, phosphoenolpyruvate, outside of the mitochondria by an enzyme called pyruvate kinase directly controls the ‘on-off’ switch for insulin secretion by β-cells. Drugs that activate pyruvate kinase redirect metabolic traffic through the mitochondria, enhancing insulin release, and thus may be useful for treating diabetes.
ACKNOWLEDGEMENTS
We thank Sam Stephens for reproducing our PK activator mouse GSIS studies. We thank David Zenisek, Bhupesh Mehta, Colin Nichols, Maria Remedi, Conor McClenaghan, and Christopher Emfinger for assistance with KATP measurements. The Merrins laboratory gratefully acknowledges support from the American Diabetes Association (1-16-IBS-212), the NIH/NIDDK (K01DK101683 and R01DK113103), the NIH/NIA (R21AG050135 and R01AG062328), the Wisconsin Partnership Program, and the Central Society for Clinical and Translational Research. HRV received a postdoctoral fellowship from the American Diabetes Association (1-17-PDF-155), HRF received a postdoctoral fellowship from HRSA (T32HP10010) and NIH/NIA (T32AG000213), SLL received a predoctoral fellowship from NIH/NIDDK (T32DK007665), and we acknowledge Dudley Lamming for contributing support for EP from R01AG062328. The Kibbey laboratory gratefully acknowledges NIH/NIDDK (R01DK092606, R01DK110181 and K08DK080142), CTSA (UL1RR-0024139), and DRC (P30DK045735). CJT acknowledges the support of the Division of Preclinical Innovation, National Center for Advancing Translational Sciences. JEC is funded by the American Diabetes Association (1-18-JDF-017). MEC received a postdoctoral fellowship from NIH/NIDDK (F32DK116542). This work utilized facilities and resources from the William S. Middleton Memorial Veterans Hospital and does not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abulizi A, Stark R, Cardone RL, Lewandowski SL, Zhao X, Alves TC, Thomas C, Kung C, Wang B, Siebel S, et al. (2020). Pharmacologic activation of the mitochondrial phosphoenolpyruvate cycle enhances islet function in vivo. BioRxiv 2020.02.13.947630. [Google Scholar]
- Affourtit C, Alberts B, Barlow J, Carré JE, and Wynne AG (2018). Control of pancreatic β-cell bioenergetics. Biochem. Soc. Trans. 46, 555–564. [DOI] [PubMed] [Google Scholar]
- Ainscow EK, and Rutter GA (2002). Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet beta-cells: evidence for a Ca2+-dependent mechanism. Diabetes 51 Suppl 1, S162–170. [DOI] [PubMed] [Google Scholar]
- Alves TC, Pongratz RL, Zhao X, Yarborough O, Sereda S, Shirihai O, Cline GW, Mason G, and Kibbey RG (2015). Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab. 22, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. (2012). Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeyer HU, Gawehn K, and Grassl M Methods of Enzymatic Analysis. (New York, NY: Academic Press, Inc.), pp. 5109–5510. [Google Scholar]
- Bertram R, Satin LS, and Sherman AS (2018). Closing in on the Mechanisms of Pulsatile Insulin Secretion. Diabetes 67, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer MB, Jiang J, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, et al. (2010). Evaluation of substituted N,N’-diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. J. Med. Chem. 53, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, and Williams GR (1955). Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 217, 383–393. [PubMed] [Google Scholar]
- Chance B, and Williams GR (1956). The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Subj. Biochem. 17, 65–134. [DOI] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Shalosky NJ, Tornheim K, Hansford RG, Prentki M, and Corkey BE (1996a). Regulation of pancreatic beta-cell mitochondrial metabolism: influence of Ca2+, substrate and ADP. Biochem. J 318,615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Kubik K, Schultz V, Tornheim K, and Corkey BE (1996b). Temporal sequence of metabolic and ionic events in glucosestimulated clonal pancreatic beta-cells (HIT). Biochem. J 315, 1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Fusonie GE, Corkey BE, and Tornheim K (1997). Oscillations in oxygen consumption by permeabilized clonal pancreatic beta-cells (HIT) incubated in an oscillatory glycolyzing muscle extract: roles of free Ca2+, substrates, and the ATP/ADP ratio. Diabetes 46, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline GW, Pongratz RL, Zhao X, and Papas KK (2011). Rates of insulin secretion in INS-1 cells are enhanced by coupling to anaplerosis and Kreb’s cycle flux independent of ATP synthesis. Biochem. Biophys. Res. Commun. 415, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton TL, Jacks T, and Vander Heiden MG (2016). PKM2, cancer metabolism, and the road ahead. EMBO Rep. 17, 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, et al. (2005). The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J. Biol. Chem. 280, 38464–38470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, and Huising MO (2016). Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doliba NM, Vatamaniuk MZ, Buettger CW, Qin W, Collins HW, Wehrli SL, Carr RD, and Matschinsky FM (2003). Differential effects of glucose and glyburide on energetics and Na+ levels of betaHC9 cells: nuclear magnetic resonance spectroscopy and respirometry studies. Diabetes 52, 394–402. [DOI] [PubMed] [Google Scholar]
- Dyachok O, Idevall-Hagren O, Sågetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjärvi G, Gylfe E, and Tengholm A (2008). Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab. 8, 26–37. [DOI] [PubMed] [Google Scholar]
- Eliasson L, Renström E, Ding WG, Proks P, and Rorsman P (1997). Rapid ATP-dependent priming of secretory granules precedes Ca(2+)-induced exocytosis in mouse pancreatic B-cells. J. Physiol. 503 (Pt 2), 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfari S, Schulz V, Corkey B, and Prentki M (2000). Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49, 718–726. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest. 125, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson U, Rosengren AH, Schuit FC, Renström E, and Mulder H (2006). Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia 49, 1578–1586. [DOI] [PubMed] [Google Scholar]
- Gembal M, Gilon P, and Henquin JC (1992). Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Invest. 89, 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gembal M, Detimary P, Gilon P, Gao ZY, and Henquin JC (1993). Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Invest. 91, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg T, Poudel C, Schmidt BA, Dhillon RS, Sdao SM, Truchan NA, Baar EL, Fernandez LA, Denu JM, Eliceiri KW, et al. (2016). Pancreatic β-Cells From Mice Offset Age-Associated Mitochondrial Deficiency With Reduced KATP Channel Activity. Diabetes 65, 2700–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsby J, Sarabu R, Corbett WL, Haynes N-E, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, et al. (2003). Allosteric activators of glucokinase: potential role in diabetes therapy. Science 301, 370–373. [DOI] [PubMed] [Google Scholar]
- Harris RA, and Fenton AW (2019). A critical review of the role of M2PYK in the Warburg effect. Biochim. Biophys. Acta Rev. Cancer 1871, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC (2009). Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751. [DOI] [PubMed] [Google Scholar]
- Hong M, Kefaloyianni E, Bao L, Malester B, Delaroche D, Neubert TA, and Coetzee WA (2011). Cardiac ATP-sensitive K+ channel associates with the glycolytic enzyme complex. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 25, 2456–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, et al. (2013). PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, and Newgard CB (2008). Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 295, E1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesinkey SR, Madiraju AK, Alves TC, Yarborough OH, Cardone RL, Zhao X, Parsaei Y, Nasiri AR, Butrico G, Liu X, et al. (2019). Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 28, 759–772.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, et al. (2010). Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 20, 3387–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alárcon C, Rhodes CJ, and Newgard CB (2006). The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 281, 35624–35632. [DOI] [PubMed] [Google Scholar]
- Jung SK, Kauri LM, Qian WJ, and Kennedy RT (2000). Correlated oscillations in glucose consumption, oxygen consumption, and intracellular free Ca(2+) in single islets of Langerhans. J. Biol. Chem. 275, 6642–6650. [DOI] [PubMed] [Google Scholar]
- Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, and Rutter GA (1999). Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J. Biol. Chem. 274, 13281–13291. [DOI] [PubMed] [Google Scholar]
- Kennedy RT, Kauri LM, Dahlgren GM, and Jung S-K (2002). Metabolic oscillations in beta-cells. Diabetes 51 Suppl 1, S152–161. [DOI] [PubMed] [Google Scholar]
- Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, and Shulman GI (2007). Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 5, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-H (2015). Maturity-Onset Diabetes of the Young: What Do Clinicians Need to Know? Diabetes Metab. J 39, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krippeit-Drews P, Bäcker M, Düfer M, and Drews G (2003). Phosphocreatine as a determinant of K(ATP) channel activity in pancreatic beta-cells. Pflugers Arch. 445, 556–562. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, and Kendrick MA (2005). Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 288, E1–15. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Longacre MJ, Langberg E-C, Tibell A, Kendrick MA, Fukao T, and Ostenson C-G (2009). Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52, 1087–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald MJ, Longacre MJ, Stoker SW, Kendrick M, Thonpho A, Brown LJ, Hasan NM, Jitrapakdee S, Fukao T, Hanson MS, et al. (2011). Differences between human and rodent pancreatic islets: low pyruvate carboxylase, atp citrate lyase, and pyruvate carboxylation and high glucose-stimulated acetoacetate in human pancreatic islets. J. Biol. Chem. 286, 18383–18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen T-W, Bargmann CI, et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschinsky FM, and Ellerman JE (1968). Metabolism of glucose in the islets of Langerhans. J. Biol. Chem. 243, 2730–2736. [PubMed] [Google Scholar]
- Merrins MJ, and Stuenkel EL (2008). Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J. Physiol. 586, 5367–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Fendler B, Zhang M, Sherman A, Bertram R, and Satin LS (2010). Metabolic oscillations in pancreatic islets depend on the intracellular Ca2+ level but not Ca2+ oscillations. Biophys. J. 99, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA, and Satin LS (2013). Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 activity. J. Biol. Chem. 288, 33312–33322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Poudel C, McKenna JP, Ha J, Sherman A, Bertram R, and Satin LS (2016). Phase Analysis of Metabolic Oscillations and Membrane Potential in Pancreatic Islet β-Cells. Biophys. J. 110, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitok KA, Freiberger EC, Schueler KL, Rabaglia ME, Stapleton DS, Kwiecien NW, Malec PA, Hebert AS, Broman AT, Kennedy RT, et al. (2018). Islet proteomics reveals genetic variation in dopamine production resulting in altered insulin secretion. J. Biol. Chem. 293, 5860–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, and Terauchi Y (2015). Present status of clinical deployment of glucokinase activators. J. Diabetes Investig. 6, 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu D, Horiuchi Y, Kano F, Noguchi Y, Sugawara T, Takamoto I, Kubota N, Kadowaki T, and Murata M (2015). L-cysteine reversibly inhibits glucose-induced biphasic insulin secretion and ATP production by inactivating PKM2. Proc. Natl. Acad. Sci. U. S. A. 112, E1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG (2016). The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol. Rev. 96, 1385–1447. [DOI] [PubMed] [Google Scholar]
- Nichols CG (2006). KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Panten U, Zünkler BJ, Scheit S, Kirchhoff K, and Lenzen S (1986). Regulation of energy metabolism in pancreatic islets by glucose and tolbutamide. Diabetologia 29, 648–654. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Rothman DL, Behar KL, and Mason GF (2010). Evaluation of cerebral acetate transport and metabolic rates in the rat brain in vivo using 1H-[13C]-NMR. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 30, 1200–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Knobel SM, Arkhammar P, Thastrup O, and Piston DW (2000). Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet beta cells. Proc. Natl. Acad. Sci. U. S. A. 97, 5203–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Delgado J, Deeney JT, Martín-del-Río R, Corkey BE, and Tamarit-Rodriguez J (2015). KCl -Permeabilized Pancreatic Islets: An Experimental Model to Explore the Messenger Role of ATP in the Mechanism of Insulin Secretion. PloS One 10, e0140096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Delgado J, Deeney JT, Corkey BE, and Tamarit-Rodriguez J (2016). Direct Stimulation of Islet Insulin Secretion by Glycolytic and Mitochondrial Metabolites in KCl-Depolarized Islets. PloS One 11, e0166111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecitá-Hlavatá L, Jabůrek M, Holendová B, Tauber J, Pavluch V, Berková Z, Cahová M, Schroeder K, Brandes RP, Siemen D, et al. (2020). Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes. [DOI] [PubMed] [Google Scholar]
- Poburko D, Santo-Domingo J, and Demaurex N (2011). Dynamic Regulation of the Mitochondrial Proton Gradient during Cytosolic Calcium Elevations. J. Biol. Chem. 286, 11672–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, et al. (2011). Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 13, 440–449. [DOI] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, and Madiraju SRM (2013). Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185. [DOI] [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, and Barros LF (2013). A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PloS One 8, e57712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, and Prentki M (1997). Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J. Biol. Chem. 272, 18572–18579. [DOI] [PubMed] [Google Scholar]
- Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shulman GI, and Kibbey RG (2009). Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem. 284, 26578–26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J, and Reems J (2004). Regulation of ATP/ADP in pancreatic islets. Diabetes 53, 401–409. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GC, Miyashita Y, and Kasai H (1999). Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc. Natl. Acad. Sci. U. S. A. 96, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI., Girard CAJ., and Ashcroft FM. (2006). ATP sensitivity of the ATP-sensitive K+ channel in intact and permeabilized pancreatic beta-cells. Diabetes 55, 2446–2454. [DOI] [PubMed] [Google Scholar]
- Tornheim K (1997). Are metabolic oscillations responsible for normal oscillatory insulin secretion? Diabetes 46, 1375–1380. [DOI] [PubMed] [Google Scholar]
- Weiss JN, and Lamp ST (1987). Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science 238, 67–69. [DOI] [PubMed] [Google Scholar]
- Zhao S, Mugabo Y, Iglesias J, Xie L, Delghingaro-Augusto V, Lussier R, Peyot M-L, Joly E, Taïb B, Davis MA, et al. (2014). α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 19, 993–1007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new code.