

A. vinelandii has served as an experimental model for the study of the differentiation processes to form metabolically dormant cells in Gram-negative bacteria. This work identifies c-di-GMP as a critical regulator for the production of alginates with specific contents of guluronic residues that are able to structure the rigid laminated layers of the cyst envelope. Although allosteric activation of the alginate polymerase complex Alg8-Alg44 by c-di-GMP has long been recognized, our results show a previously unidentified role during the polymer modification step, controlling the expression of extracellular alginate epimerases. Our results also highlight the importance of c-di-GMP in the control of the physical properties of alginate, which ultimately determine the desiccation resistance of the differentiated cell.
KEYWORDS: Azotobacter vinelandii, c-di-GMP, encystment, alginate, MucR, mannuronan C-5 epimerases, cyst capsule
ABSTRACT
The genus Azotobacter, belonging to the Pseudomonadaceae family, is characterized by the formation of cysts, which are metabolically dormant cells produced under adverse conditions and able to resist desiccation. Although this developmental process has served as a model for the study of cell differentiation in Gram-negative bacteria, the molecular basis of its regulation is still poorly understood. Here, we report that the ubiquitous second messenger cyclic dimeric GMP (c-di-GMP) is critical for the formation of cysts in Azotobacter vinelandii. Upon encystment induction, the levels of c-di-GMP increased, reaching a peak within the first 6 h. In the absence of the diguanylate cyclase MucR, however, the levels of this second messenger remained low throughout the developmental process. A. vinelandii cysts are surrounded by two alginate layers with variable proportions of guluronic residues, which are introduced into the final alginate chain by extracellular mannuronic C-5 epimerases of the AlgE1 to AlgE7 family. Unlike in Pseudomonas aeruginosa, MucR was not required for alginate polymerization in A. vinelandii. Conversely, MucR was necessary for the expression of extracellular alginate C-5 epimerases; therefore, the MucR-deficient strain produced cyst-like structures devoid of the alginate capsule and unable to resist desiccation. Expression of mucR was partially dependent on the response regulator AlgR, which binds to two sites in the mucR promoter, enhancing mucR transcription. Together, these results indicate that the developmental process of A. vinelandii is controlled through a signaling module that involves activation by the response regulator AlgR and c-di-GMP accumulation that depends on MucR.
IMPORTANCE A. vinelandii has served as an experimental model for the study of the differentiation processes to form metabolically dormant cells in Gram-negative bacteria. This work identifies c-di-GMP as a critical regulator for the production of alginates with specific contents of guluronic residues that are able to structure the rigid laminated layers of the cyst envelope. Although allosteric activation of the alginate polymerase complex Alg8-Alg44 by c-di-GMP has long been recognized, our results show a previously unidentified role during the polymer modification step, controlling the expression of extracellular alginate epimerases. Our results also highlight the importance of c-di-GMP in the control of the physical properties of alginate, which ultimately determine the desiccation resistance of the differentiated cell.
INTRODUCTION
Cellular differentiation in bacteria is a highly coordinated process involving major metabolic, biochemical, and morphological changes leading to the formation of specialized cells, including spores or cysts, that are able to resist unfavorable conditions. The molecular mechanisms sustaining this process rely on complex and hierarchical signal transduction pathways that ultimately affect gene expression. In both cyst- and spore-forming bacteria, the role of nucleotide-based second messengers such as cyclic dimeric GMP (c-di-GMP), cGMP, and cyclic dimeric AMP (c-di-AMP) have been described as crucial for successful cell development (1–4).
c-di-GMP is a nearly universal second messenger in bacteria. It is synthesized from two GTP molecules by diguanylate cyclases (DGCs) characterized by the presence of a GGDEF domain (the active site or A-site). Some DGCs also contain an allosteric inhibitory c-di-GMP binding site (I-site); hence, they are negatively regulated by their enzymatic product. c-di-GMP degradation is mediated by specific phosphodiesterases (PDEs) containing either EAL or HD-GYP domains (2, 5). In many cases, DGCs and PDEs harbor N-terminal sensory domains that regulate their activities in response to specific signals (6). c-di-GMP coordinates many cellular processes, including motility, biofilm formation, cell cycle progression, and cell development (2).
The genus Azotobacter belongs to the Pseudomonadaceae family and is characterized by the ability of its members to form cysts, which are differentiated cells that are able to resist desiccation, and Azotobacter vinelandii has served as a model for the study of this process (7, 8). During vegetative growth, A. vinelandii is motile because of peritrichous flagella and has a strictly aerobic type of metabolism (9). This bacterium produces two polymers, the exopolysaccharide alginate and the intracellular polyester poly-β-hydroxybutyrate (PHB). Cyst formation can be synchronously induced in the presence of n-butanol or β-hydroxybutyrate and is completed within 3 to 5 days (7). Mature cysts comprise a central body in which granules of PHB accumulate in the cytoplasm. These cells are surrounded by two rigid alginate layers (the exine and intine), which are essential for desiccation resistance. During encystment, bacteria lose the flagella, nitrogen fixation and aerobic respiration are inhibited, phospholipids are replaced by alkyl-resorcinols, and the carbohydrate metabolism is switched to a lipid type of metabolism (7). We recently reported the proteome of the A. vinelandii cyst and showed that the nature of the differentially produced proteins correlated with changes previously described to occur during differentiation. For instance, production of proteins responsible for flagellum assembly or motility were suppressed in the cyst, whereas the production of enzymes for the glyoxylate shunt, the synthesis of alkyl-resorcinols, or the production and modification of alginate increased with respect to the vegetative cell (10). The nature of the regulation of this developmental process is still poorly understood and most likely occurs at multiple levels.
A major component of the capsule layers of the cyst is the polysaccharide alginate, a linear copolymer composed of residues of β-d-mannuronic acid (M) and its C-5 epimer α-l-guluronic acid (G) (8). Bacterial alginates are produced by two genera, Azotobacter and Pseudomonas. The biochemistry and genetics for the biosynthesis of this polymer are very well conserved between Pseudomonas aeruginosa and A. vinelandii. All of the biosynthetic genes, except for algC, are clustered in the chromosome, with the first gene being algD (11, 12). This gene cluster (algD-8-44-K-J-G-X-L-I-V-F-A) encodes proteins necessary for the generation of the alginate monomer, for alginate polymerization, for the periplasmic transfer of the polymer, and for modification and export through the outer membrane. The process of polymerization and cytoplasmic membrane transfer is conducted by Alg8 and Alg44 proteins. The activity of the inner membrane synthase protein Alg8 is posttranslationally regulated by Alg44 (also called the copolymerase), an inner membrane receptor that binds c-di-GMP (11, 13). c-di-GMP binding to the PilZ domain of Alg44 is essential for alginate polymerization in P. aeruginosa (13, 14).
In A. vinelandii, the secreted alginate is originally made as poly-mannuronan but is further modified by a family of extracellular enzymes, the Ca2+-dependent mannuronan C-5 epimerases (AlgE1 to AlgE7) (15). These enzymes are sequence homologous proteins with molecular masses ranging from 57 to 191 kDa and consisting of two different modules, A and R. The R modules contain Ca2+-binding motifs and are necessary for full activity, while the A modules catalyze epimerization, introducing different distributions of G residues in the alginate substrate, including G blocks of various lengths (15). A higher G content confers a greater gelling capacity to the polymer. The exine, the outermost layer of the differentiated A. vinelandii cell, is characteristically rich in GG block sequences; therefore, the alginate AlgE C-5 epimerases are essential for cyst formation. Importantly, AlgE1, AlgE2 and/or AlgE5, AlgE4, and AlgE6 associated with the cyst were detected (16). AlgE1 to AlgE7 are secreted by a type I secretion system composed of proteins EexD, EexE, and EexF. In the absence of this secretion system or AlgE proteins, the alginate is produced almost as a mannuronan polymer and the differentiated cells lack the alginate coat and do not survive drought conditions (16, 17).
Regulation by c-di-GMP of the Alg8-Alg44 alginate polymerase activity in P. aeruginosa depends on the presence of the DGC MucR (18). In P. aeruginosa, expression of mucR and the alginate biosynthetic operon (algD-8-44-K-J-G-X-L-I-V-F-A) is directly regulated by the response regulator AlgR, which binds CCGTTGGTC motif elements in the promoter region of these genes (19, 20). Thus, by affecting the expression of mucR and algD, AlgR can control alginate production by indirectly affecting Alg44 activity and promoting the production of the alginate biosynthetic proteins (19, 20). In contrast, AlgR of A. vinelandii is dispensable for alginate production, because a reduction of only 50% was observed in an algR-deficient strain (21). Inactivation of algR in A. vinelandii impairs the proper organization of the alginate cyst coat, and the resulting cells are unable to resist desiccation (21). It is unclear, however, if the phenotypes associated with the absence of AlgR in A. vinelandii involve the same genetic interactions as observed in P. aeruginosa.
In this work, we analyzed the involvement of c-di-GMP in the encystment process of A. vinelandii. We found that c-di-GMP accumulates during the formation of cysts, and this accumulation was found to be MucR dependent. Although MucR was not required for alginate synthesis, it was necessary for alginate modification, as expression of mannuronan C-5 epimerases was reduced in the ΔmucR mutant. As a consequence, the ΔmucR mutant was unable to produce cysts resistant to desiccation. Together, our results show that alginate production and alginate modification can be separately regulated by c-di-GMP in A. vinelandii.
RESULTS
A c-di-GMP signaling module involving MucR regulates cyst formation in A. vinelandii.
Alginate is an essential component of the cyst envelope. Since in P. aeruginosa alginate is regulated by a c-di-GMP signaling module that relies on the DGC MucR, we explored whether cyst formation in A. vinelandii involves changes in c-di-GMP accumulation. To this end, we quantified the abundance of this second messenger at different time points under encysting-inducing conditions. c-di-GMP accumulation showed an ∼4-fold increase at 6 h, compared to uninduced conditions. After this point, c-di-GMP levels decreased over time (Fig. 1). To determine whether MucR (Avin_49140 [GenBank accession no. ACO81013], also referred to as AVIN_RS22475) was responsible for the c-di-GMP accumulation profile observed during cyst formation, we quantified c-di-GMP levels in a ΔmucR strain. In the ΔmucR strain, c-di-GMP levels decreased under cyst-inducing conditions, relative to vegetative conditions (time zero) and were lower along the developmental process, compared to the wild-type (WT) strain (Fig. 1). These results strongly suggest that MucR is part of a specific c-di-GMP signaling module that operates during cyst formation in A. vinelandii. It is worth pointing out that, under uninduced conditions (time zero, vegetative growth), the levels of c-di-GMP in the ΔmucR strain were higher than those in the WT strain, which could be related to the presence of a PDE activity, as has been reported for MucR in P. aeruginosa (22).
FIG 1.
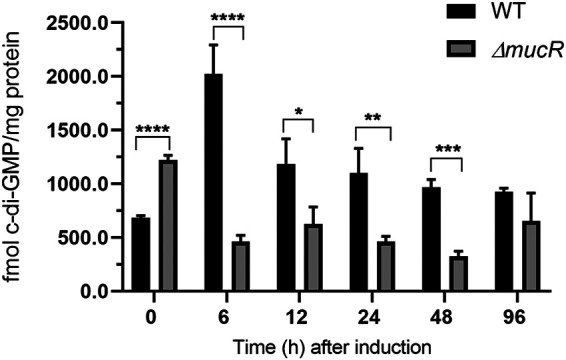
MucR contributes to the c-di-GMP pool under encysting-inducing conditions. c-di-GMP was quantified in the WT strain and in the ΔmucR mutant during encystment induction. Vegetative cells were harvested after 48 h of growth in Burk’s-sucrose medium (0 h), washed three times to remove traces of sucrose, and transferred to Burk’s-butanol medium for the indicated time. The bars for standard deviations from three independent experiments are shown. Significant differences were analyzed by t test. Statistical significance is indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.0001; ****, P < 0.00001.
To further explore the role of c-di-GMP and MucR during the developmental process of A. vinelandii, the ΔmucR mutant was subjected to the desiccation resistance test, which reflects the ability of the encystment-induced cells to form mature cysts. As shown in Fig. 2A, the ΔmucR mutant showed a 50-fold reduced ability to form cysts able to resist a 5-day desiccation period, compared to the WT strain, whereas desiccation resistance was restored in a ΔmucR derivative strain complemented with a WT copy of mucR.
FIG 2.

MucR is necessary for the formation of mature cysts resistant to desiccation but not for the production of alginate. (A) Mature cyst formation assay, based on the percentage of cells resistant to a 5-day period of desiccation. The bars for standard deviations from three independent experiments are shown. Significant differences were analyzed by t test. Statistical significance is indicated. **, P < 0.01; ***, P < 0.0001. (B) Electron micrographs of A. vinelandii cysts after a 5-day period of differentiation. The cells were cultivated on plates of Burk’s-butanol medium for encystment induction. In the WT strain, the central body (CB) and the PHB granules are indicated, as is the cyst capsule, which is composed of two laminated alginate layers, the exine (EX) and the intine (IN). (C) Alginate quantification in A. vinelandii cells grown on plates of Burk’s-butanol medium after 48 h of encystment induction.
Electron microscopy visualization confirmed that, indeed, mature cyst formation was impaired in the ΔmucR strain. In contrast to the cysts formed by the WT strain, the cyst-like structures derived from the ΔmucR strain did not assemble the exine and intine layers and had central bodies lacking any structured capsular material (Fig. 2B). This phenotype is more remarkable than that observed for the ΔalgR mutant, for which an incipient exine layer is formed (21) (Fig. 2B).
The positive role of c-di-GMP during encystment was confirmed using a strain with artificially increased levels of this second messenger. In a companion work (23), we constructed and characterized an A. vinelandii WT derivative strain overproducing the A. vinelandii DGC AvGReg and demonstrated that this strain has elevated levels of c-di-GMP. This strain was subjected to the desiccation resistance test and, as anticipated, the proportion of mature cysts was 43%, almost 3-fold higher than the value for the WT strain (Fig. 2A, DGC+). Together, these results implied that cyst formation in A. vinelandii is regulated by a c-di-GMP signaling module that involves MucR.
MucR is not required for alginate synthesis.
In P. aeruginosa, c-di-GMP and MucR are required for alginate production. Based on this precedent, we hypothesized that the effect of c-di-GMP and MucR on cyst development in A. vinelandii was a result of deficient alginate production. To begin to address this question, we analyzed the ability of the ΔmucR strain to produce alginate under encysting conditions. We observed that the WT strain and the ΔmucR strain accumulated similar amounts of alginate, indicating that in A. vinelandii activation of the Alg8-Alg44 copolymerase complex by c-di-GMP does not rely on MucR (Fig. 2C). On the other hand, complementation of ΔmucR with a WT copy of the mucR gene integrated into the chromosome resulted in increased production of alginate, compared to the WT and ΔmucR strains. The latter result suggests that MucR can promote alginate production, perhaps in a nonspecific manner, by increasing the levels of the c-di-GMP pool.
MucR is critical for the production of mannuronan C-5 epimerases.
Since MucR did not appear to be critical for alginate production in A. vinelandii, we next asked what the molecular basis for the cyst formation phenotype of the ΔmucR strain could be. Mannuronan C-5 epimerases of the AlgE1 to AlgE7 family are critical for the extracellular modification of the alginate chain that is essential for structuring the alginate cyst coats (17), and the ΔmucR mutant made cyst-like structures lacking these alginate coats. Thus, we hypothesized that MucR regulates the expression of algE genes and/or the production of the AlgE mannuronan C-5 epimerases.
As a first approach, the effect of MucR on the transcription of algE genes was evaluated by quantitative PCR (qPCR). During the developmental process, AlgE1, AlgE2, AlgE4, AlgE5, and AlgE6 epimerases are the ones associated to the cyst (24). In a previous work, a single pair of oligonucleotides aligning within a highly conserved region of algE1 to algE6 genes and allowing the simultaneous quantification of their transcript abundance was designed. Therefore, qPCR assays were conducted using this pair of oligonucleotides and total RNA from the WT and ΔmucR strains. We observed transcription of the algE1 to algE6 genes in the WT strain but not in the ΔmucR strain, strongly suggesting that MucR is required for the expression of these genes (Fig. 3A). The complemented ΔmucR strain, which we previously showed produces more alginate, had a 6-fold increase in algE1 to algE6 expression, compared to the WT strain (Fig. 3A).
FIG 3.
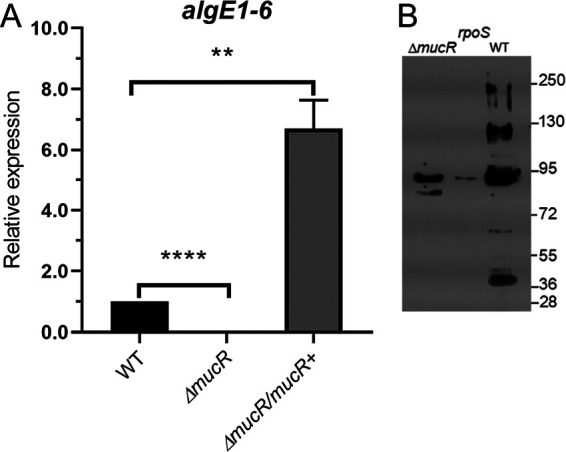
MucR is necessary for the production of extracellular C-5 epimerases. (A) Determination of the relative abundance of algE1 to algE6 mRNA by qPCR in encystment-induced cells at 48 h. The bars for standard deviations from three independent experiments are shown. Significant differences were analyzed by t test. Statistical significance is indicated. **, P < 0.01; ****, P < 0.00001. (B) Detection of AlgE C-5 epimerases associated with the central body of A. vinelandii cells undergoing differentiation in Burk’s-butanol medium for 5 days. Anti-AlgE4 antibody was used as the primary antibody.
We next evaluated whether the mannuronan C-5 epimerases were present in the central body of differentiated cells in both the WT and ΔmucR strains. Five bands detected in Western blots corresponding to AlgE epimerases were detected on the cyst surface of the WT strain, and only one of those was present in the ΔmucR strain (Fig. 3B). Previous studies demonstrated that the AlgE1 to AlgE7 proteins have abnormal migration rates, which prevent their proper identification based only on the expected molecular mass (24, 25). Therefore, the identity of the AlgE1 to AlgE7 epimerases under the control of MucR remains unknown. We included the AlgE epimerase profile of an RpoS-deficient mutant as a control for a previously described positive regulator of algE expression (26).
The mRNA levels of the eexDEF operon, encoding the AlgE1 to AlgE7 type I secretion system, did not change in the ΔmucR strain, compared to the WT strain (0.95 ± 0.16 and 1.0 relative unit, respectively). The gyrA mRNA levels were used as an internal control, allowing normalization of the qRT-PCR data. This result indicated that the absence of AlgE proteins over the cyst surface is not caused by a lack of expression of this secretion machinery.
Together, these data suggest that loss of MucR function results in the loss of expression of the genes coding for the mannuronan C-5 epimerases, likely accounting for the loss of the surface polysaccharide coat.
mucR is directly regulated by the response regulator AlgR in A. vinelandii.
To further explore the signaling module containing MucR in A. vinelandii, we investigated whether the mucR gene is subject to regulation by AlgR in this bacterium. To begin the characterization of the regulatory region of mucR, we first identified the transcription start sites. Two transcripts were identified, and two different transcription start sites were mapped to positions −114 and −157, relative to the ATG translation initiation site of mucR (Fig. 4A). We did not find any conserved −10 and −35 boxes in this region, which could be explained by the need for a transcriptional activator and/or alternative sigma factors (Fig. 4B).
FIG 4.

Identification of the transcription start sites of mucR. (A) Transcription start sites were identified by primer extension with total RNA extracted from the WT (lane 1) and ΔalgR (lane 2) strains grown in Burk´s-sucrose medium for 48 h and a primer complementary to mucR. The cDNA obtained was resolved in a denaturing polyacrylamide gel, along with DNA sequence ladders generated by chemical sequencing of mucR. The transcription start sites (P1 and P2) are indicated. (B) Genomic context of the mucR gene. The mucR flanking genes encode hypothetical conserved proteins. In the lower panel, the location of the two mucR transcription start sites (P1 and P2) identified in panel A are indicated, along with putative AlgR binding sites (BS1 and BS2).
The mucR transcripts could not be detected in a ΔalgR strain, suggesting that AlgR is a positive regulator of this gene (Fig. 4A). The positive effect of AlgR on mucR expression was further confirmed by qPCR assays. The level of mucR transcripts was diminished by 40% in the ΔalgR mutant, compared to the WT strain under vegetative growth conditions. The expression of mucR during encystment-inducing conditions was also under the control of AlgR (Fig. 5A). Based on the qPCR assays, it appears that expression of mucR does not exclusively rely on AlgR. It is possible that an AlgR-independent transcript was not resolved in our primer extension assays and thus will require further experimentation. We next investigated whether transcription of mucR was enhanced under encysting conditions. As shown in Fig. 5B, mucR mRNA levels increased 6-fold, relative to vegetative conditions. This result is somehow expected, considering that we previously reported that the accumulation of AlgR was 22 higher in the cyst, compared to vegetative cells (10).
FIG 5.

Transcriptional regulation of mucR assessed by qPCR analysis. (A) Effect of AlgR on mucR transcription. The mucR mRNA levels in the ΔalgR strain were determined under vegetative (Burk’s-sucrose medium for 48 h) or encysting (Burk’s-butanol medium for 48 h) conditions and were compared to those in the WT strain. (B) mRNA levels of mucR under encysting conditions, relative to those under vegetative conditions. The bars for standard deviations from three independent experiments are shown. Significant differences were analyzed by t test. Statistical significance is indicated. ***, P < 0.0001; ****, P < 0.00001.
To further elucidate the mechanism by which AlgR regulates mucR expression, we analyzed the interaction of this protein with the regulatory region of mucR (PmucR). Two potential AlgR binding sites were found, resembling the previously identified AlgR binding sites in P. aeruginosa (19). The proximal site (BS1 [TCCGTCCGCC]) was located at position −61, whereas the distal site (BS2 [GCCGTCGACG]) was located at position −274, relative to the mucR translation start codon (Fig. 4B and 6A). Binding of AlgR to a DNA fragment of 486 bp containing BS1 and BS2 was tested by electrophoretic mobility shift assays (EMSAs). A DNA fragment carrying the regulatory region of the Avin_05510 gene, encoding a β-ketoacyl synthase, was included as a negative control. AlgR specifically bound the PmucR DNA fragment at concentrations ranging from 0.52 to 1.4 μM (Fig. 6B).
FIG 6.

The response regulator AlgR binds directly to PmucR. (A) Schematic representation of the putative AlgR binding sites (BS1 and BS2) identified in PmucR. The DNA fragments used in panels B (438 bp), C (114 bp), and D (211 bp) are shown. (B, C, and D) EMSAs were performed in order to analyze the AlgR binding to a PmucR fragment carrying BS1 and BS2 (B), BS1 only (C), or BS2 only (D). The DNA fragments were incubated with increasing concentrations of AlgR. As a negative control, a fragment carrying the regulatory region of Avin_05510 was included in each DNA binding reaction. The migration of the DNA fragments was visualized by staining with ethidium bromide.
We next determined whether AlgR could bind the two recognition sites individually. AlgR was able to recognize the two sites separately at concentrations ranging from 0.5 to 1.4 μM (Fig. 6C and D). However, less protein (0.87 μM) was required to fully shift the BS2 site, compared to the amount required to fully shift BS1. It is worth noting that the EMSAs were performed using a nonradioactive method (27, 28); thus, the observed amounts of AlgR needed to shift the PmucR DNA fragments are within the range of the technique (up to 1.5 μM). Altogether, these results clearly confirmed the direct control of mucR by the response regulator AlgR.
DISCUSSION
c-di-GMP has been linked to the regulation of cell differentiation in a variety of bacterial models. In the deltaproteobacterium Myxococcus xanthus, a minimum threshold level of c-di-GMP is required for the formation of spore-filled fruiting bodies (1, 29). During this process, c-di-GMP gradually accumulates up to 20-fold after 48 h of nutrient limitation using the cytoplasmic DGC DmxB. The c-di-GMP receptor involved in this process is the transcriptional regulator EpsI/NIa24, which regulates the expression of an exopolysaccharide essential for fruiting body formation (1). In this work, we show that the developmental process of A. vinelandii is regulated through c-di-GMP signaling and relies on MucR. The absence of MucR reduced the concentration of this second messenger along the entire differentiation process and prevented the proper assembly of the alginate cyst layers.
We hypothesized that the relationship between c-di-GMP and encystment would be at the level of alginate production. This hypothesis was based on two observations. The first is that alginate polymerization is regulated through c-di-GMP in P. aeruginosa by the DGC MucR. The second one is that a putative PDE that we named MucG is involved in alginate production in A. vinelandii. In an accompanying work, we demonstrate that MucG is in fact a PDE that negatively regulates alginate production and its molecular mass (23). A. vinelandii also has an orthologue of MucR; therefore, we speculated that this other DGC could be important for alginate production, affecting cyst formation in this bacterium. We found that, in contrast to what is observed in P. aeruginosa, MucR was not essential for alginate polymerization in A. vinelandii but was necessary for the formation of the alginate coat characteristic of mature cysts. Since structuring of the alginate coat requires the activity of extracellular C-5 epimerases, we evaluated whether the abundance and/or localization of these proteins was altered in the absence of MucR, and we found that MucR regulates the expression of algE1 to algE6 genes and the presence of AlgE epimerases in cysts. In order for MucR to regulate transcription of the algE1 to algE6 genes, it must alter the activity of a c-di-GMP receptor that can act as a transcriptional regulator. No examples in A. vinelandii have been described; however, P. aeruginosa has one of the best characterized c-di-GMP receptors of this class, the flagellar gene master regulator FleQ (30, 31). This receptor activates flagellar gene expression at low c-di-GMP levels and switches to activating pel genes, which are involved in the biosynthesis of the exopolysaccharide Pel, when c-di-GMP levels increase (32). A. vinelandii has a FleQ orthologue that shares 77% identity with FleQ from P. aeruginosa. We identified putative FleQ binding sites in the regulatory region of algE1 to algE7 in A. vinelandii by using Virtual Footprint software (33); therefore, our working hypothesis is that MucR regulates the expression of these genes by modulating the activity of FleQ. Work is under way to investigate the genetic relationship between mucR and fleQ in terms of the regulation of both the alginate coat structuring and the expression of the algE1 to algE7 genes.
Together, our results let us propose that MucR helps to produce alginates with a content of G residues that, in the presence of Ca2+ ions, favors the formation of the rigid gel structure characteristic of the exine and intine layers of the A. vinelandii cysts. The cyst envelope is a complex structure in which alginate is one of the major components; however, it also contains other carbohydrates, proteins, alkyl-resorcinol lipids, and DNA. The formation of this structure is a gradual process of discontinuous layering around the cell that mainly occurs during the first 30 to 48 h of differentiation (8); therefore, the outermost layer, the exine, is the first to be formed, followed by the intine. Alginate C-5 epimerases, such as AlgE1, AlgE2, AlgE6, and AlgE4, which generate a variety of alginate structures present in the cyst coat, are released from the encysting cells primarily between 12 h and 36 h after induction (24). Our results show that MucR affects the overall pool of c-di-GMP, even in early stages of differentiation. In fact, the c-di-GMP peak observed within the first 6 h in the WT strain does not occur in the ΔmucR mutant, suggesting that such an increase in the pool of this second messenger is a prerequisite for the successful formation of a mature cyst. Whether MucR is required only for the modification of the alginate chains contained in the exine and intine layers of the cyst or is also involved in the synthesis of other components of the cyst coat is not known. Given the important physiological and morphological changes occurring during early stages of differentiation, such as the loss of the flagella or the arrest of DNA synthesis, we cannot rule out the existence of additional processes under the control of the c-di-GMP signaling regulatory cascade that depends on MucR.
In the companion paper (23), we demonstrate that in A. vinelandii c-di-GMP favors the synthesis of longer alginate chains in an AvGReg- and MucG-dependent manner. AvGReg is a globin-coupled sensor DGC that is necessary for alginate production, while MucG is a PDE that negatively regulates this biosynthetic pathway; these proteins appear to act at a different stage of the biosynthesis of alginate, compared to MucR. Thus, it appears that MucG, together with the DGC AvGReg, affects alginate polymerization, perhaps by targeting Alg44. On the other hand, MucR, together with a currently unidentified c-di-GMP receptor (perhaps FleQ), affects alginate modification by regulating the transcription of genes encoding C-5 mannuronan epimerases. This regulatory scheme suggests that not only the amount but also the physical properties of this polymer are tightly controlled in response to environmental conditions.
In P. aeruginosa, AlgR is a key transcriptional regulator required for the expression of multiple virulence factors, including alginate (34–37), and modulation of the pool of c-di-GMP via mucR transcription (19). Although AlgR was not essential for alginate production in A. vinelandii, it was required for structuring the alginate cyst coats (21). In the present study, we found that, as in P. aeruginosa, mucR in A. vinelandii is directly regulated by AlgR. AlgR abundance was 22-fold higher in soluble protein extracts from cysts, compared to vegetative cells (10), which explains the higher levels of mucR transcription during cell differentiation, favoring accumulation of c-di-GMP and cell cycle progression. In P. aeruginosa, phosphorylation of AlgR by the histidine kinase FimS (also named AlgZ) leads to the differential regulation of target genes. In its phosphorylated form, AlgR is required for the transcriptional activation/modulation of genes necessary for the synthesis of virulence factors, such as rhamnolipids, twitching motility-related proteins, siderophores, or hydrogen cyanide-producing proteins, among others (34, 36, 38–40). However, AlgR is also capable of activating algD transcription, for the production of alginate, in a phosphorylation-independent manner (37). At present, it is unclear whether AlgR needs to be phosphorylated by FimS for the transcriptional activation of mucR in P. aeruginosa (19). FimS is not present in A. vinelandii (21); therefore, it is possible either that a different histidine kinase or phospho-donor regulates AlgR or that AlgR activates mucR transcription in a nonphosphorylated state.
It is unclear what type of signals are responsible for the increased accumulation of c-di-GMP during encystment. Although A. vinelandii is a strict aerobic bacterium, during encystment it faces significant changes in respiratory activity, accompanied by a reduction in oxygen uptake rates and suppression of the activity of the NADH dehydrogenase (8). Thus, it is likely that this bacterium regulates its metabolism, including the amount of alginate and its physical properties, in response to the redox state and/or oxygen availability. MucR contains three MHYT domains, which are signaling domains predicted to bind diatomic gases such as NO, CO, and O2. In P. aeruginosa, it has been proposed that NO acts as a signaling molecule detected by the MHYT domains of MucR, regulating the activity of the output GGDEF and EAL domains (18, 22, 41). The MHYT domains have also been implicated in detecting the redox state by means of a coordinated Cu2+ ion (42).
We are beginning to recognize another level of complexity in the regulation of alginate biosynthesis in A. vinelandii, where multiple c-di-GMP signaling modules appear to regulate the structure and abundance of this exopolysaccharide of great biotechnological, medical, and ecological significance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains, plasmids, and primers used in this work are listed in Tables 1 and 2. The A. vinelandii WT strain AEIV (also named E strain) (43) was used in this work. Vegetative A. vinelandii growth was conducted in Burk’s nitrogen-free salts medium supplemented with 20 g liter−1 of sucrose (Burk’s-sucrose medium). The composition of the culture medium has been described elsewhere (44). Cultures were grown in 250-ml Erlenmeyer flasks with 50 ml of filling volume at 200 rpm and 30°C. For encystment induction, vegetative A. vinelandii cells (grown for 48 h in 50 ml of Burk’s-sucrose medium) were collected by centrifugation and washed three times with Burk’s buffer (the same mineral medium without a carbon source) to remove traces of sucrose. The cells were resuspended in 1 ml of Burk’s buffer, and 200 μl of this suspension was plated over the surface of solid Burk’s medium supplemented with 0.2% (vol/vol) n-butanol (Burk’s-butanol medium), which induces cell differentiation. The plates were incubated for 5 days to allow completion of the differentiation process. In order to have a synchronous suspension of encystment-induced cells, when indicated the vegetative cells were resuspended in the same volume of fresh liquid Burk’s-butanol medium and incubated for the indicated times.
TABLE 1.
Strains and plasmid used in the present study
| Strain or plasmid | Genotype and relevant characteristicsa | Source or reference |
|---|---|---|
| Bacterial strains | ||
| A. vinelandii | ||
| AEIV (also named E strain) | WT strain | 43 |
| ICM01 | AEIV derivative carrying a ΔmucR::Gm mutation | This work |
| ICM09 | ICMO1 derivative complemented with WT copy of mucR integrated into its native locus in the chromosome; Gmr Kmr | This work |
| CN100 | AEIV derivative carrying a ΔalgR::Km mutation | This work |
| Escherichia coli | ||
| DH5α | supE44 ΔlacU169 hsdR17 recA1 endA1 gyrA96 relA | 45 |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | Invitrogen |
| Plasmids | ||
| pJET1.2/Blunt vector | PCR cloning vector; Apr | Thermo Fisher Scientific |
| pBSL141 | Source of the Gmr cassette | 49 |
| pBSL128 | Source of the Kmr cassette | 49 |
| pET-21a(+) | Expression vector | Novagen |
| pBBR1MCS-2 | Cloning vector; Kmr | 50 |
| pJG99 | pJET1.2/Blunt vector derivative carrying a 2.6-kb fragment containing the mucR gene | This work |
| pJG100 | pJG99 derivative carrying a ΔmucR::Gm mutation; used to construct the mutant ICM01 | This work |
| pJG99-Km | pJG99 derivative carrying a Kmr cassette as a selection marker at a ScaI site of the multiple cloning site; used to construct the complemented strain ICM09 | This work |
| pJG98 | pJET1.2/Blunt vector derivative carrying a ΔalgR::Km construction; linearized with ScaI and used to construct the mutant CN100 | This work |
| pETR-3P | pET-21a(+) vector derivative expressing the AlgR protein carrying a His6 tag at the N terminus | This work |
Gm, gentamicin; Gmr, gentamicin resistance; Apr, ampicillin resistance; Km, kanamycin; Kmr, kanamycin resistance.
TABLE 2.
Sequences of the primers used in this study
| Name | Nucleotide sequence (5′ to 3′) | Template | Used for |
|---|---|---|---|
| mucR-F | CAATGCAGACCAGTCGCGCTC | Chromosomal DNA WT | Plasmid pJG99 |
| mucR-R | CTTCGGCTGCTGTCCTGCCAG | ||
| algR km-1 | CTGTTTCAGGCCGTCGATG | Chromosomal DNA WT | Plasmid pJG98 |
| algR km-2 | CACTCACCAGACGGCTG | ||
| algR km-3 | CAGCCGTCTGGTGAGTGACCTGGGATGAATGTCAGCTAC | pBRIMMCS-2 | |
| algR km-4 | TGCGATGGATGCGCACGAGAAGGCGGCGGTGGAATCG | ||
| algR km-5 | CGTGCGCATCCATCGCA | Chromosomal DNA WT | |
| algR km-6 | GTCTCCAGCTCCTTGAC | ||
| gyrAfw | CCAGCAAGGGCAAGGTCTA | NAa | qPCR, internal control |
| gyrArev | TCGTCCAGCGGCAACAGGT | ||
| FwRT-algE1-6 | CACGAGCAGACCATCAACCTG | NA | qPCR, algE1 to algE6 |
| RvRT-algE1-6 | ATGTTGAAGCCGTGGCGGTCGTTG | ||
| FwRT-eexD | GGTGACTATGGGCTCTCTGG | NA | qPCR, eexD |
| RvRT-eexD | CTCGCCCGGTATACATCTCG | ||
| mucR-Rv-primer | GTGTAGGAGGCGAGGATGG | NA | Primer extension, mucR |
| algRBamP | CAAGGATCCAATGTTCTGATCGTC | Chromosomal DNA WT | pETR-3P |
| algRHindP | CACAAGCTTGGAGGCCAGTTGATT | ||
| mucR-F-2-sites | GAATCCCATCACGCCGACTG | Chromosomal DNA WT | EMSA, BS1 and BS2 sites |
| mucR-R-2-sites | GACGCTCCGTACTGCGTAAG | ||
| mucR-F-site 1 | GCTTCCCCGGCATCCCAATA | Chromosomal DNA WT | EMSA, BS1 site |
| mucR-R-2-sites | GACGCTCCGTACTGCGTAAG | ||
| mucR-F-2-sites | GAATCCCATCACGCCGACTG | Chromosomal DNA WT | EMSA, BS2 site |
| mucR-R-site2 | CTACGCCAAAACGCCGAGAG |
NA, not applicable.
Escherichia coli DH5α cells were grown in lysogeny broth (LB) at 37°C (45, 46). When needed, the final antibiotic concentrations used for A. vinelandii and E. coli were as follows: gentamicin, 1 and 10 μg ml−1; kanamycin (Km), 1 and 10 μg ml−1; ampicillin, not used and 200 μg ml−1, respectively.
DNA manipulation.
All DNA manipulations were performed following standard protocols (47). PCR amplifications were performed using the high-fidelity Phusion DNA polymerase (Thermo Fisher Scientific). Restriction enzymes were purchased from New England Biolabs. Plasmid isolation and DNA purification were performed using kits from Thermo Fisher Scientific, following the instructions of the manufacturer. All constructs were sequenced to verify sequence integrity and fidelity. DNA sequencing was conducted by the Sanger method, using fluorescent dideoxy terminators and a cycle sequencing method in a 3130xl analyzer (Applied Biosystems).
Construction of A. vinelandii mutants.
Construction of the plasmids used to build A. vinelandii mutants is described in Table 1. A. vinelandii transformation was conducted using the natural competent state induced under iron-limited growth conditions, as described previously (44, 48). Competent A. vinelandii cells were transformed with plasmid DNA carrying the desired mutation and previously linearized to ensure double reciprocal recombination and allelic exchange. Transformants were selected on Burk’s-sucrose medium amended with the corresponding antibiotic. Due to the polyploid nature of A. vinelandii, gene inactivation in all of the chromosomal copies was confirmed by PCR analysis.
For the construction of a MucR-deficient mutant, a fragment of 2.6 kb carrying the mucR gene was PCR amplified using the primer pair mucR-F/mucR-R and was ligated to plasmid pJET 1.2/Blunt (Thermo Fisher Scientific). The resulting vector, named pJG99, was excised with BssHI enzyme, deleting an internal mucR fragment of 1.5 kb that was replaced by a gentamicin resistance cassette released with the same enzyme from vector pBSL141 (49). The plasmid generated was named pJG100 and carried the resistance cassette ligated in the same orientation as that of mucR transcription. This plasmid, previously linearized with the ScaI endonuclease, was used to transform AEIV cells, generating strain ICM01 (ΔmucR). To genetically complement mutant ICM01, a WT copy of the mucR gene was introduced into the mucR chromosomal locus. To this end, a HindIII fragment carrying a Km resistance (Kmr) cassette from plasmid pBSL128 (49) was ligated to the polylinker of plasmid pJG99 (mucR+), generating pGJ99-Km. This plasmid, which is unable to replicate in A. vinelandii, was introduced into ICM01 by transformation and Kmr single recombinants were selected; one such transformant, named ICM09, was chosen for further characterization.
A strain carrying a deletion of the entire algR gene, ΔalgR::Km, was constructed and was named CN100. A ΔalgR::Km mutation was constructed by recombinant PCR. The three following PCR products were assembled in the order of appearance by overlapping PCR: (i) 458 bp upstream of algR (from −439 to +26 with respect to the ATG start codon); (ii) 1,112 bp corresponding to the Kmr cassette from plasmid pBBRIMCS-2 (50); and (iii) 536 bp downstream of algR (from +598 to +1117 with respect to the ATG start codon). The Kmr cassette was inserted in the same orientation as algR transcription. The ΔalgR::Km construction was cloned into pJET1.2/Blunt vector, giving rise to plasmid pJG98. Finally, this was linearized with endonuclease ScaI and transformed into WT competent cells, generating mutant CN100.
Nucleotide isolation and c-di-GMP quantification.
c-di-GMP quantification was conducted following a protocol previously described (51). For nucleotide extraction, 47 ml of cell culture induced for encystment was centrifuged at 5,752 × g for 7 min. Cell pellets were resuspended in 1 ml of extraction solution (40% acetonitrile, 40% methanol, 0.1% formic acid, and 19.9% H2O) and incubated for 15 min on ice. After this, samples were centrifuged at 17,949 × g for 15 min at 4°C. The supernatant was transferred to a new Eppendorf tube, dried under vacuum, and lyophilized. The samples were resuspended in 100 μl of ultraperformance liquid chromatography (UPLC)-grade water, and 10 μl of this solution was injected into a liquid chromatography-tandem mass spectrometry system. A c-di-GMP standard curve was determined with the following concentrations: 1.9, 3.9, 7.8, 15.6, 31.3, 62.5, and 125 nM. The determinations were conducted in triplicate. The remaining 3 ml of culture was used to quantify protein content by the Lowry method, to normalize the production of c-di-GMP to cell protein levels (52).
Resistance to desiccation.
Resistance to desiccation was assessed as reported previously (53). In brief, cells of A. vinelandii grown for 48 h in 50 ml of liquid Burk’s-sucrose medium were collected by centrifugation and washed three times with Burk’s buffer to remove traces of sucrose. After this, the cells were resuspended in 1 ml of Burk’s buffer; 200 μl of this suspension was spread on the surface of Burk’s-butanol solid medium and incubated for 5 days at 30°C. After this time, the encystment-induced cells were collected and resuspended in 10 mM MgSO4. Approximately 106 CFU was applied to Millipore membranes of 0.2-μm pore size; when dried, they were incubated for 5 days at 30°C, with only mature cysts being able to survive. After this time, the membranes were soaked for 5 h in Burk’s buffer and the cells were released by vortex mixing. Viable cell counts before and after desiccation were determined in serial dilutions.
Electron microscopy assays.
Visualization of A. vinelandii cysts was performed by following a previously reported protocol with some modifications (54). Cells for which encystment had been induced for 5 days in liquid Burk’s-butanol medium were collected and washed three times with phosphate buffer (pH 7.2) at 4°C. They were then washed with 0.16 M sodium cacodylate buffer and fixed with a paraformaldehyde (4%)-glutaraldehyde (2%) mixture for 1 h at room temperature. After the cells were washed again with 0.16 M sodium cacodylate, they were fixed with 2% osmium tetroxide for 2 h at 4°C. Following fixation, cell suspensions were washed and then dehydrated by passage through a graded ethanol series. After exposure to propylene oxide, samples were embedded in an Epon 812 resin, which was allowed to polymerize for 24 h at 65°C. Ultrathin sections were cut, incubated with uranyl acetate, washed with distilled water, treated with lead citrate, washed again, and observed with a Zeiss transmission electron microscope (model Libra 120, at 80 kV).
Analytical methods.
To determine specific alginate production, the strains were cultivated under encysting conditions on plates of Burk’s-butanol medium for 48 h. The presence of 0.2% n-butanol as the sole carbon source triggers cellular differentiation in about 95% of the population (7, 8), allowing synchronization of the cellular response during encystment. After 48 h, the cells were harvested and washed twice with 10 ml of Burk’s buffer to extract the alginate. After centrifugation, the cells were used to determine the total protein content by the Lowry method (52). The alginate from the supernatants was precipitated with 3 volumes of cold 2-propanol, washed twice with 70% ethanol, dried, and resuspended in 1 ml of distilled water. Alginate was quantitated by the spectrophotometric determination of uronic acids with carbazole (55).
Quantitative real-time reverse transcription.
Total RNA was extracted as described (56) from cells grown in liquid Burk’s-sucrose or Burk’s-butanol medium for 48 h. Genomic DNA contamination was removed with DNase I (Thermo Fisher Scientific). Details of cDNA synthesis and reverse transcription-qPCR amplification conditions have been reported elsewhere (44). The relative levels of algE1 to algE6 and eexD were determined by comparing the quantity of each mRNA under the tested conditions, using gyrA (Avin_15810) mRNA as an internal control. The sequences of the primer pairs used are listed in Table 2. These primers were designed using the Primer3 program (http://bioinfo.ut.ee/primer3) (optimal length of 20 bases and melting temperature of 60°C). Validation of each primer set was conducted by verifying specific single-product amplification by melting curve analyses. Three biological replicates (independent cultures) were performed, with three technical replicates for each one. Similar results were obtained for the transcription of all measured genes in the repetitions. A no-template control reaction was included for each gene. The quantification technique used to analyze the data generated was the 2−ΔΔCT method reported previously (57).
Detection of AlgE-type C-5 epimerases.
Western blot assays to detect AlgE epimerases were performed as described previously, using anti-AlgE4 antibodies (24), with some modifications. Cells of A. vinelandii were cultivated on plates of Burk’s-butanol medium for 5 days; after this time, the proteins associated with the cell were extracted. To this end, cells were collected, washed with 1 ml of 50 mM Tris (pH 7.8), centrifuged for 10 min, resuspended in 0.5 ml of 50 mM Tris-3 mM Na2EDTA, and shaken at 250 rpm for 15 min. The cell suspension was centrifuged, and the supernatant contained the epimerases associated with the surface of the central body. The proteins were blotted as described (24).
Primer extension assay.
The transcription start site of mucR was mapped by primer extension analysis under vegetative conditions. Total RNA was extracted from the A. vinelandii WT strain and from mutant CN100 (ΔalgR::Km) grown in Burk’s-sucrose medium for 48 h. The oligonucleotide used as a primer for the extension was end labeled with [γ-32P]ATP and T4 polynucleotide kinase (Roche). Primer extension was performed at 42°C with avian myeloblastosis virus (AMV) reverse transcriptase (Roche), as indicated by the supplier. The extended cDNA product was analyzed by electrophoresis on a denaturing 6% urea-polyacrylamide gel, in parallel with a DNA sequence ladder produced with the same primer by using a Thermo Sequenase cycle sequencing kit (USB). Plasmid pJG99, carrying the regulatory region of mucR, was used as a DNA template. We were unable to identify the mucR transcription start sites under encysting conditions using this technique, which could be related to the limited amount of total RNA extracted from cells undergoing differentiation.
Expression and purification of AlgR-His protein.
The AlgR protein was purified as a recombinant protein with a 6×His tag at the C terminus (AlgR-His). To produce this recombinant protein, the algR gene was cloned in the expression vector pET-21a(+) (Novagen), generating pETR-3P. The E. coli BL21 strain was transformed with plasmid pETR-3P, and overproduction of AlgR-His was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to cells grown to mid-log phase (A600 = 0.5). After 4 h of induction at 37°C, cells were collected by centrifugation and disrupted by sonication in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole). Lysed cells were centrifuged for 30 min at 20,000 × g at 4°C, and the supernatant was loaded onto an equilibrated column containing the nickel resin Ni-nitrilotriacetic acid–agarose. The column was washed twice with washing buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole), and the protein was eluted with elution buffer I (50 mM NaH2PO4, 300 mM NaCl, 500 mM imidazole). The protein was concentrated by using Microcon YM-10 centrifugal filters (Amicon) and stored in elution buffer II (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). Protein concentrations were determined by the Lowry method using bovine serum albumin as a standard (52). SDS-PAGE of the purified protein revealed the expected molecular mass of approximately 28 kDa.
Electrophoretic mobility shift assays.
EMSAs were conducted using a nonradioactive method reported previously (27, 28). Regions of various lengths upstream of mucR were amplified by PCR, as follows: (i) 438 bp containing the entire regulatory region; (ii) 211 bp containing only AlgR binding site 2; and (iii) 114 bp containing only AlgR binding site 1. Binding reactions were performed by mixing an individual PCR product (100 ng) with increasing concentrations of purified AlgR-His protein in binding buffer (10 mM Tris [pH 8], 50 mM KCl, 1 mM dithiothreitol, 0.5 mM EDTA, 5% glycerol, 10 μg/ml bovine serum albumin), in a total volume of 20 μl. These reaction mixtures were incubated at room temperature for 20 min and then analyzed by electrophoresis on 6% nondenaturing acrylamide gels run with 0.5× Tris-borate-EDTA buffer. The migration of the DNA fragments was visualized by staining with ethidium bromide and excitation under UV illumination.
Statistical analysis.
GraphPad Prism (version 6.0) software (GraphPad Software, La Jolla, CA) was used for statistical analyses. Statistical significance was assessed using two-tailed, unpaired Student's t tests, and P values of ≤0.05 were considered significant.
ACKNOWLEDGMENTS
We thank S. Ainsworth and M. Tabche for computational and technical support and G. Zavala for the acquisition of electron microscopy images. We thank H. Ertesvåg for kindly providing the anti-AlgE4 antibodies and G. Soberón-Chávez and G. O’Toole for critical reading of the manuscript.
I.C.M.-O. is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and has received a CONACyT fellowship (no. 596217). This work was supported by project PAPIIT IN204818 (UNAM) to C.N. and NIH grants GM109259 and AI144395 to C.M.W.
REFERENCES
- 1.Skotnicka D, Smaldone GT, Petters T, Trampari E, Liang J, Kaever V, Malone JG, Singer M, Søgaard-Andersen L. 2016. A minimal threshold of c-di-GMP is essential for fruiting body formation and sporulation in Myxococcus xanthus. PLoS Genet 12:e1006080. doi: 10.1371/journal.pgen.1006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenal U, Reinders A, Lori C. 2017. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol 15:271–284. doi: 10.1038/nrmicro.2016.190. [DOI] [PubMed] [Google Scholar]
- 3.Marden JN, Dong Q, Roychowdhury S, Berleman JE, Bauer CE. 2011. Cyclic GMP controls Rhodospirillum centenum cyst development: cGMP control of encystment. Mol Microbiol 79:600–615. doi: 10.1111/j.1365-2958.2010.07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oppenheimer-Shaanan Y, Wexselblatt E, Katzhendler J, Yavin E, Ben-Yehuda S. 2011. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep 12:594–601. doi: 10.1038/embor.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galperin MY. 2010. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol 13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segura D, Núñez C, Espín G. 2020. Azotobacter cysts In eLS. John Wiley & Sons, Chichester, United Kingdom. doi: 10.1002/9780470015902.a0000295.pub3. [DOI] [Google Scholar]
- 8.Sadoff HL. 1975. Encystment and germination in Azotobacter vinelandii. Bacteriol Rev 39:516–539. doi: 10.1128/MMBR.39.4.516-539.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Setubal JC, dos Santos P, Goldman BS, Ertesvåg H, Espin G, Rubio LM, Valla S, Almeida NF, Balasubramanian D, Cromes L, Curatti L, Du Z, Godsy E, Goodner B, Hellner-Burris K, Hernandez JA, Houmiel K, Imperial J, Kennedy C, Larson TJ, Latreille P, Ligon LS, Lu J, Maerk M, Miller NM, Norton S, O'Carroll IP, Paulsen I, Raulfs EC, Roemer R, Rosser J, Segura D, Slater S, Stricklin SL, Studholme DJ, Sun J, Viana CJ, Wallin E, Wang B, Wheeler C, Zhu H, Dean DR, Dixon R, Wood D. 2009. Genome sequence of Azotobacter vinelandii, an obligate aerobe specialized to support diverse anaerobic metabolic processes. J Bacteriol 191:4534–4545. doi: 10.1128/JB.00504-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury-Paul S, Pando-Robles V, Jiménez-Jacinto V, Segura D, Espín G, Núñez C. 2018. Proteomic analysis revealed proteins induced upon Azotobacter vinelandii encystment. J Proteomics 181:47–59. doi: 10.1016/j.jprot.2018.03.031. [DOI] [PubMed] [Google Scholar]
- 11.Hay ID, Rehman ZU, Moradali MF, Wang Y, Rehm BHA. 2013. Microbial alginate production, modification and its applications. Microb Biotechnol 6:637–650. doi: 10.1111/1751-7915.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urtuvia V, Maturana N, Acevedo F, Peña C, Díaz-Barrera A. 2017. Bacterial alginate production: an overview of its biosynthesis and potential industrial production. World J Microbiol Biotechnol 33:198. doi: 10.1007/s11274-017-2363-x. [DOI] [PubMed] [Google Scholar]
- 13.Whitney JC, Whitfield GB, Marmont LS, Yip P, Neculai AM, Lobsanov YD, Robinson H, Ohman DE, Howell PL. 2015. Dimeric c-di-GMP is required for post-translational regulation of alginate production in Pseudomonas aeruginosa. J Biol Chem 290:12451–12462. doi: 10.1074/jbc.M115.645051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merighi M, Lee VT, Hyodo M, Hayakawa Y, Lory S. 2007. The second messenger bis-(3’-5’)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol Microbiol 65:876–895. doi: 10.1111/j.1365-2958.2007.05817.x. [DOI] [PubMed] [Google Scholar]
- 15.Ertesvåg H. 2015. Alginate-modifying enzymes: biological roles and biotechnological uses. Front Microbiol 6:523. doi: 10.3389/fmicb.2015.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steigedal M, Sletta H, Moreno S, Maerk M, Christensen BE, Bjerkan T, Ellingsen TE, Espìn G, Ertesvåg H, Valla S. 2008. The Azotobacter vinelandii AlgE mannuronan C-5-epimerase family is essential for the in vivo control of alginate monomer composition and for functional cyst formation. Environ Microbiol 10:1760–1770. doi: 10.1111/j.1462-2920.2008.01597.x. [DOI] [PubMed] [Google Scholar]
- 17.Gimmestad M, Steigedal M, Ertesvåg H, Moreno S, Christensen BE, Espín G, Valla S. 2006. Identification and characterization of an Azotobacter vinelandii type I secretion system responsible for export of the AlgE-type mannuronan C-5-epimerases. J Bacteriol 188:5551–5560. doi: 10.1128/JB.00236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay ID, Remminghorst U, Rehm BHA. 2009. MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl Environ Microbiol 75:1110–1120. doi: 10.1128/AEM.02416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong W, Zhao J, Kang H, Zhu M, Zhou T, Deng X, Liang H. 2015. ChIP-seq reveals the global regulator AlgR mediating cyclic di-GMP synthesis in Pseudomonas aeruginosa. Nucleic Acids Res 43:8268–8282. doi: 10.1093/nar/gkv747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohr CD, Leveau JH, Krieg DP, Hibler NS, Deretic V. 1992. AlgR-binding sites within the algD promoter make up a set of inverted repeats separated by a large intervening segment of DNA. J Bacteriol 174:6624–6633. doi: 10.1128/jb.174.20.6624-6633.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Núñez C, Moreno S, Soberón-Chávez G, Espín G. 1999. The Azotobacter vinelandii response regulator AlgR is essential for cyst formation. J Bacteriol 181:141–148. doi: 10.1128/JB.181.1.141-148.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Heine S, Entian M, Sauer K, Frankenberg-Dinkel N. 2013. NO-induced biofilm dispersion in Pseudomonas aeruginosa is mediated by an MHYT domain-coupled phosphodiesterase. J Bacteriol 195:3531–3542. doi: 10.1128/JB.01156-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahumada-Manuel CL, Martínez-Ortíz IC, Hsueh BY, Guzmán J, Waters CM, Zamorano-Sánchez D, Espín G, Núñez C. 2020. Increased c-di-GMP levels lead to the production of alginates of high molecular mass in Azotobacter vinelandii. J Bacteriol 202:e00134-20. doi: 10.1128/JB.00134-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Høidal HK, Svanem BIG, Gimmestad M, Valla S. 2000. Mannuronan C-5 epimerases and cellular differentiation of Azotobacter vinelandii. Environ Microbiol 2:27–38. doi: 10.1046/j.1462-2920.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- 25.Høidal HK, Ertesvåg H, Skjåk-Braek G, Stokke BT, Valla S. 1999. The recombinant Azotobacter vinelandii mannuronan C-5-epimerase AlgE4 epimerizes alginate by a nonrandom attack mechanism. J Biol Chem 274:12316–12322. doi: 10.1074/jbc.274.18.12316. [DOI] [PubMed] [Google Scholar]
- 26.Moreno S, Ertesvåg H, Valla S, Núñez C, Espin G, Cocotl-Yañez M. 2018. RpoS controls the expression and the transport of the AlgE1–7 epimerases in Azotobacter vinelandii. FEMS Microbiol Lett 365:fny210. doi: 10.1093/femsle/fny210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bustamante VH, Martínez LC, Santana FJ, Knodler LA, Steele-Mortimer O, Puente JL. 2008. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc Natl Acad Sci U S A 105:14591–14596. doi: 10.1073/pnas.0801205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martínez-Flores I, Pérez-Morales D, Sánchez-Pérez M, Paredes CC, Collado-Vides J, Salgado H, Bustamante VH. 2016. In silico clustering of Salmonella global gene expression data reveals novel genes co-regulated with the SPI-1 virulence genes through HilD. Sci Rep 6:37858. doi: 10.1038/srep37858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konovalova A, Petters T, Søgaard-Andersen L. 2010. Extracellular biology of Myxococcus xanthus. FEMS Microbiol Rev 34:89–106. doi: 10.1111/j.1574-6976.2009.00194.x. [DOI] [PubMed] [Google Scholar]
- 30.Baraquet C, Murakami K, Parsek MR, Harwood CS. 2012. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res 40:7207–7218. doi: 10.1093/nar/gks384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuyama BY, Krasteva PV, Baraquet C, Harwood CS, Sondermann H, Navarro MVAS. 2016. Mechanistic insights into c-di-GMP–dependent control of the biofilm regulator FleQ from Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 113:E209–E218. doi: 10.1073/pnas.1523148113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baraquet C, Harwood CS. 2016. FleQ DNA binding consensus sequence revealed by studies of FleQ-dependent regulation of biofilm gene expression in Pseudomonas aeruginosa. J Bacteriol 198:178–186. doi: 10.1128/JB.00539-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, Jahn D. 2005. Virtual Footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics 21:4187–4189. doi: 10.1093/bioinformatics/bti635. [DOI] [PubMed] [Google Scholar]
- 34.Okkotsu Y, Tieku P, Fitzsimmons LF, Churchill ME, Schurr MJ. 2013. Pseudomonas aeruginosa AlgR phosphorylation modulates rhamnolipid production and motility. J Bacteriol 195:5499–5515. doi: 10.1128/JB.00726-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lizewski SE, Lundberg DS, Schurr MJ. 2002. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun 70:6083–6093. doi: 10.1128/iai.70.11.6083-6093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitchurch CB, Alm RA, Mattick JS. 1996. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 93:9839–9843. doi: 10.1073/pnas.93.18.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma S, Selvaraj U, Ohman DE, Quarless R, Hassett DJ, Wozniak DJ. 1998. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J Bacteriol 180:956–968. doi: 10.1128/JB.180.4.956-968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Little AS, Okkotsu Y, Reinhart AA, Damron FH, Barbier M, Barrett B, Oglesby-Sherrouse AG, Goldberg JB, Cody WL, Schurr MJ, Vasil ML, Schurr MJ. 2018. Pseudomonas aeruginosa AlgR phosphorylation status differentially regulates pyocyanin and pyoverdine production. mBio 9:e02318-17. doi: 10.1128/mBio.02318-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitchurch CB, Erova TE, Emery JA, Sargent JL, Harris JM, Semmler ABT, Young MD, Mattick JS, Wozniak DJ. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol 184:4544–4554. doi: 10.1128/jb.184.16.4544-4554.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cody WL, Pritchett CL, Jones AK, Carterson AJ, Jackson D, Frisk A, Wolfgang MC, Schurr MJ. 2009. Pseudomonas aeruginosa AlgR controls cyanide production in an AlgZ-dependent manner. J Bacteriol 191:2993–3002. doi: 10.1128/JB.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Hay ID, Rehman ZU, Rehm BHA. 2015. Membrane-anchored MucR mediates nitrate-dependent regulation of alginate production in Pseudomonas aeruginosa. Appl Microbiol Biotechnol 99:7253–7265. doi: 10.1007/s00253-015-6591-4. [DOI] [PubMed] [Google Scholar]
- 42.Galperin MY, Gaidenko TA, Mulkidjanian AY, Nakano M, Price CW. 2001. MHYT, a new integral membrane sensor domain. FEMS Microbiol Lett 205:17–23. doi: 10.1111/j.1574-6968.2001.tb10919.x. [DOI] [PubMed] [Google Scholar]
- 43.Larsen B, Haug A. 1971. Biosynthesis of alginate. Carbohydr Res 17:287–296. doi: 10.1016/s0008-6215(00)82536-7. [DOI] [PubMed] [Google Scholar]
- 44.Ahumada-Manuel CL, Guzmán J, Peña C, Quiroz-Rocha E, Espín G, Núñez C. 2017. The signaling protein MucG negatively affects the production and the molecular mass of alginate in Azotobacter vinelandii. Appl Microbiol Biotechnol 101:1521–1534. doi: 10.1007/s00253-016-7931-8. [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 46.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 47.Green MR, Sambrook J, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 48.Page WJ, Sadoff HL. 1976. Physiological factors affecting transformation of Azotobacter vinelandii. J Bacteriol 125:1080–1087. doi: 10.1128/JB.125.3.1080-1087.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexeyev MF, Shokolenko IN, Croughan TP. 1995. Improved antibiotic-resistance gene cassettes and omega elements for Escherichia coli vector construction and in vitro deletion/insertion mutagenesis. Gene 160:63–67. doi: 10.1016/0378-1119(95)00108-i. [DOI] [PubMed] [Google Scholar]
- 50.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 51.Massie JP, Reynolds EL, Koestler BJ, Cong J-P, Agostoni M, Waters CM. 2012. Quantification of high-specificity cyclic diguanylate signaling. Proc Natl Acad Sci U S A 109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- 53.Moreno S, Nájera R, Guzmán J, Soberón-Chávez G, Espín G. 1998. Role of alternative sigma factor algU in encystment of Azotobacter vinelandii. J Bacteriol 180:2766–2769. doi: 10.1128/JB.180.10.2766-2769.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mejía-Ruíz H, Moreno S, Guzmán J, Nájera R, León R, Soberón-Chávez G, Espín G. 1997. Isolation and characterization of an Azotobacter vinelandii algK mutant. FEMS Microbiol Lett 156:101–106. doi: 10.1111/j.1574-6968.1997.tb12712.x. [DOI] [PubMed] [Google Scholar]
- 55.Knutson CA, Jeanes A. 1968. A new modification of the carbazole analysis: application to heteropolysaccharides. Anal Biochem 24:470–481. doi: 10.1016/0003-2697(68)90154-1. [DOI] [PubMed] [Google Scholar]
- 56.Barry T, Geary S, Hannify S, MacGearailt C, Shalloo M, Heery D, Gannon F, Powell R. 1992. Rapid mini-preparations of total RNA from bacteria. Nucleic Acids Res 20:4940. doi: 10.1093/nar/20.18.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]