

Abstract
Reduction oxidation (REDOX) reaction is crucial in life activities, and its dynamic balance is regulated by ROS. Reactive oxygen species (ROS) is associated with a variety of metabolic diseases involving in multiple cellular signalling in pathologic and physiological signal transduction. ROS are the by-products of numerous enzymatic reactions in various cell compartments, including the cytoplasm, cell membrane, endoplasmic reticulum (ER), mitochondria, and peroxisome. ROS signalling is not only involved in normal physiological processes but also causes metabolic dysfunction and maladaptive responses to inflammatory signals, which depends on the cell type or tissue environment. Excess oxidants are able to alter the normal structure and function of DNA, lipids, and proteins, leading to mutations or oxidative damage. Therefore, excessive oxidative stress is usually regarded as the cause of various pathological conditions, such as cancer, neurodegeneration, cardiovascular diseases (CVDs), diabetes, and kidney diseases. Currently, it has been possible to detect diabetes and other cardiac diseases by detecting derivatives accompanied by oxidative stress in vivo as biomarkers, but there is no effective method to treat these diseases. In consequence, it is essential for us to seek new therapy targeting these diseases through understanding the role of ROS signalling in regulating metabolic activity, inflammatory activation, and cardiac diseases related to metabolic dysfunction. In this review, we summarize the current literature on REDOX and its role in the regulation of cardiac metabolism and inflammation, focusing on ROS, local REDOX signalling pathways, and other mechanisms.
1. Introduction
Oxidative stress can be defined as active oxygen/nitrogen excessive production of ROS, such as oxidant, and lack of antioxidant enzymes. The detoxification of compounds in the cells is usually normal, but when the oxidant emissions are excessive, the cell produces excessive oxidation material to change DNA lipid and protein structure, leading to cell mutation and oxidative damage. Excessive oxidative stress, therefore, is considered as the causes and consequences of a variety of pathological processes, including cancer, neural degeneration, CVDs, diabetes, and kidney diseases [1, 2]. Some studies have found that a balance of oxidative stress is associated with aging [3]. Most kinds of natural or synthetic antioxidants have been evaluated against the oxidative stress-related pathological changes [1, 4, 5]. Besides, ROS is a by-product of many cell compartment enzymatic reactions occurring on the cytoplasm membrane endoplasmic reticulum (ER), or mitochondria, which can control intracellular environment balance and work as the main regulatory factor of cell dysfunction in the pathophysiology. In different cell types or organizational environment, ROS signals may participate in increased inflammatory of incommensurate reaction or lead to metabolic dysfunction-related diseases, such as atherosclerosis, diabetes, and heart stroke [6]. In addition, emerging studies have revealed that a healthy diet plays a critical role in the prevention of CVDs by modulating the oxidative balance [7, 8]. For example, a healthy diet can prevent atherosclerosis by inhibiting the oxidation of low-density lipoprotein (LDL) and reducing the production of ROS [9]; the results of the PREDIMED study [10] show that highly unsaturated fat and antioxidant-rich dietary patterns are useful for reducing the risk of CVDs. Therefore, understanding of ROS signals in regulating metabolic activity and inflammation will promote the discovery of new therapies treating CVDs.
2. ROS Generation
The generation of ROS is involved in a series of complex biochemical reactions [11]. The cascade of ROS generation consists of the following five main pathways (Figure 1):
O2−formation: O2− is produced by the coupling of O2 with electrons (e−) from donors, which is usually considered to be the first ROS cascade reaction. In mammalian cells, e− donors are usually reduced nicotinamide adenine dinucleotide (NADH) or reduced nicotinamide adenine dinucleotide phosphate (NADPH). O2− can be converted into other kinds of ROS by an oxidation reaction.
RNS formation: RNS is a derivative of NO∙, and NO∙ is produced by L-arginine (L-Arg) and catalyzed by NOS. NO∙ can react quickly with O2∙− to form ONOO−. The second-order rate constant between NO∙ and O2∙− is nearly 10 times faster than that of O2∙− catalyzed by superoxide dismutase [12, 13]. However, due to the high intracellular SOD content under physiological condition, O2− was removed before encountering NO∙.
H2O2 formation: H2O2 is produced by O2− mutation catalyzed by superoxide dismutase (SOD). At low pH, a small amount of O2− mutation occurs spontaneously, and some of which can react with reductive transition metals, such as [4Fe-4S]2+. Some oxidases (e.g., NOX4 and DUOX1/2) have dismutase activity and can directly convert O2 into H2O2 instead of O2−.
OH∙ formation: OH∙ can be generated from homolysis fission of ONOOH, and most of OH∙ is formed by metal ions (iron or copper) catalyzed by H2O2 and O2− through the Habor-Weiss reaction. In diseases with iron accumulation (e.g., atherosclerotic lesions [14] or sickle cell patients [11]), OH∙ mediated oxidative stress might be the most pivotal mechanism. OH∙ has strong oxidation ability and short half-life, which is the major cause of biological macromolecule damage by ROS.
L∙/LOO∙ formation: The highly active OH∙ or ONOO− can react with the polyunsaturated fatty acid (PUFA) of the biofilm for lipid peroxidation, in which OH∙ can react directly with lipids to capture a hydrogen atom to form a carbon-centered lipid free radical (L∙). L∙ initiates lipid peroxidation under an aerobic condition and generates lipid peroxide group (LOO∙), which is a medium oxidant that can extract H from nearby lipids to produce lipid hydrogen peroxide (LOOH). Moreover, L∙/LOO∙ can exist in the reaction process of the lipoxygenase-catalyzed polyunsaturated fatty acid formation of molecular oxygen to form hydroperoxide [15, 16].
Figure 1.
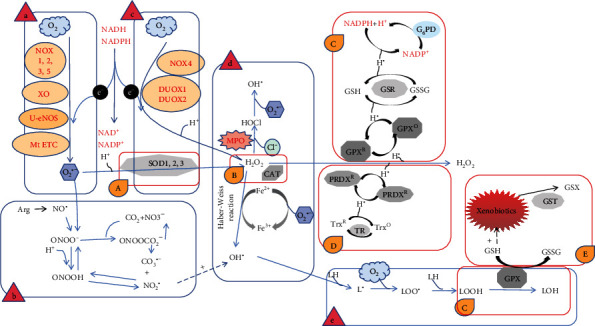
ROS generation and clearance. I. ROS generation: (a) superoxide formation; (b) reactive nitrogen species (RNS) formation; (c) hydrogen peroxide formation; (d) hydroxyl radical formation; (e) lipid radical formation. II. ROS clearance: (A) superoxide dismutation; (B) hydrogen peroxide decomposition; (C) glutathione redox cycle; (D) thioredoxin redox cycle; (E) glutathione-S-transferase detoxification.
3. Dynamics of ROS
In order to maintain the stability of ROS, there are five active oxygen scavenging pathways:
O2− mutated to H2O2 by superoxide dismutase (SOD)
Catalase (CAT) decomposes H2O2 to produce H2O and O2
Glutathione redox cycle: using glutathione as an electron donor, H2O2 and LOOH are decomposed by glutathione peroxidase (GPX).
Thioredoxin reduction cycle: using reduced thioredoxin (TrxR) as electron donor, H2O2 was reduced by redox protein (PRDX) 1-5 to produce H2O.
Exogenous detoxification of glutathione transferase (GST)
4. Oxidative Stress and CVDs
CVD is the leading cause of death worldwide [17], which is a complex pathophysiological disease involved in many factors. The dysdynamics of ROS has been regarded as one of the potential pathogenic factors [18–20]. Increased ROS level is able to lead to decreased availability of nitric oxide and vasoconstriction, which subsequently promotes arterial hypertension [21]. ROS also has negative effects on myocardial calcium treatment, inducing arrhythmias and cardiac remodeling by facilitating hypertrophic signal transduction and apoptosis [22, 23]. In addition, it also promotes the formation of atherosclerotic plaques [24].
4.1. Arterial Hypertension
It is estimated that the global prevalence of hypertension was 1.13 billion in 2015, the risk of which becomes higher with age [25]. A large number of studies have shown that ROS plays an important role in the pathogenesis of hypertension [26–28].
In the vascular system, ROS is mainly produced by vascular endothelial cells, adventitia cells, and smooth muscle cells, primarily induced by NADPH oxidase which produces O2− upon being stimulated by Angiotensin II (Ang-II), Endothelin-1 (ET-1), or urotensin II (U-II). On the other side, increased mechanical forces caused by elevated blood pressure, such as unidirectional laminar flow and oscillatory shear stress, can help to increase the accumulation of ROS. Ca2+ is involved in the regulation of cell contraction, secretion, metabolism, gene expression, and cell survival [29]. The interaction of ROS and Ca2+ plays an important role in the occurrence and development of CVDs [30–32]. Store-operated Ca2+ channel (SOCC) is a ubiquitous Ca2+ influx pathway and is the dominant Ca2+ channel in unexcited cells [33, 34]. ORAI/STIM is a highly selective calcium channel and an important component of SOCC [35, 36]. ORAI/STIM channel is involved in a variety of cardiovascular physiological processes [37]. Oxidative stress can regulate the activity of the ORAI/STIM channel by uncoupling ORAI/STIM complex, regulating the gene expression of ORAI or STIM protein, and oxidizing ORAI or STIM protein [35, 38]. Studies have shown that ROS regulates the ORAI/STIM channel by directly targeting the conserved cysteine residues in ORAI and STIM molecules [39, 40]. ROS can also act as the second messenger in cells to promote the increase of intracellular Ca2+ concentration and lead to vasoconstriction, thus assisting the pathogenesis of hypertension [41]. Ang-II-induced hypertension involves redox-dependent signal cascade activation and NADPH oxidase-induced ROS production [42]. Some common antihypertensive medications, such as Ang-I receptor blockers and angiotensin-converting enzyme (ACE) inhibitors, have been shown to reduce blood pressure partly by inhibiting NADPH oxidase and reducing the ROS production [43].
4.2. Atherosclerosis
Atherosclerosis is one of the main causes of cardiovascular death in developed countries [17]. More and more evidence shows that oxidative stress plays a key role in the formation of atherosclerosis [44, 45]. The activation of proinflammatory signal pathway, the expression of cytokines/chemokines, and the increase of oxidative stress are some of the mechanisms underlying atherosclerosis [20].
ROS is an autophagy trigger factor. Excessive ROS in cells is able to cause oxidative stress, which will further activate autophagy [46, 47]. Autophagy is closely related to the development of atherosclerosis [48, 49]. Excessive autophagy can lead to autophagic cell death [50]. Autophagic death of endothelial cells can damage plaque, form thrombus, and cause atherosclerosis [51]. Therefore, elucidating the specific mechanism of ROS-regulating autophagy may be a feasible way to treat atherosclerosis.
Oxidative stress reduces the expression of prethrombotic antioxidant P-oxidase-2 (PON2) in human atherosclerotic plaques, especially in endothelial cells [52]. Ebert et al. revealed the redox-dependent mechanism of PON2, which involves tissue factor (TF) activity in endothelial cells and prevents systemic coagulation activation and inflammation (Figure 2) [52].
Figure 2.
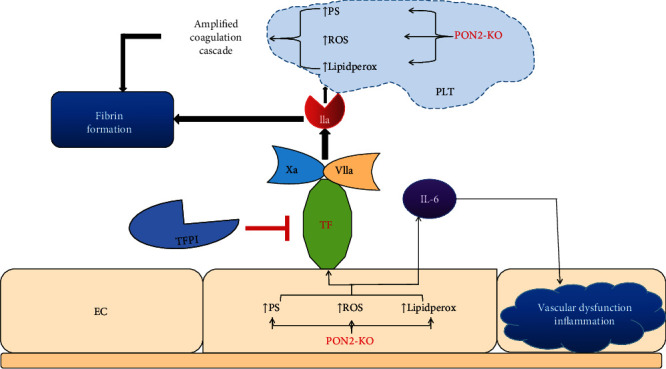
Schematic of EC and platelet-mediated procoagulant and vascular inflammatory processes in pon2−/− environment. EC-mediated systemic inflammation is established by elevated levels of interleukin-6 (IL-6), which may promote vascular inflammation and dysfunction. Knockout of PON2 can lead to the accumulation of phosphatidylserine (PS), ROS, and lipidperox in ECs; result in the production of fibrin through cascade reaction; and ultimately consolidate the function of blood coagulation.
NADPH oxidase is the main source of ROS in atherogenesis, enhancing the production of superoxide and aggravating oxidative stress, leading to the occurrence and development of arterial disease [53, 54]. Gray et al. [55] knocked out the NOX1 and NOX4 genes in streptozotocin (STZ-) induced diabetic ApoE−/− mice and found that loss of NOX1 had a significant antiatherosclerotic effect, which was related to the decreased production of ROS. GPX4 is one of the glutathione peroxidases, which can effectively interact with lipid hydrogen peroxide and catalyze the degradation of peroxides [56]. Mitochondrial GPX4 can avoid ROS damage and maintain intravascular homeostasis by clearing ROS. Overexpression of mitochondrial GPX4 can alleviate myocardial ischemia/reperfusion injury [57]. GPX4 can inhibit ferroptosis by scavenging lipid peroxides and improve the function of the heart [58–60]. GPX4 overexpression inhibits atherosclerosis in ApoE−/− mice [61, 62]. Hyperglycemia can increase the production of ROS, such as O2− and peroxide, through the mitochondrial electron transport chain, and then form a positive feedback effect [63, 64]. For example, PKC can be activated by O2−. Then, activated PKC can promote the production of NADPH oxidase-dependent ROS [65, 66]. O2− in mitochondria can increase the production of intracellular advanced glycation end products (AGEs) in cells [67, 68]. AGEs can add oxygen radical, and the activation of AGEs receptor can cause intracellular oxidative stress, which in turn causes inflammation in endothelial cells [69–71]. Therefore, AGEs eventually lead to atherosclerosis by modifying the extracellular matrix and circulating lipoproteins and activating AGEs receptors [66]. In addition, Zhu et al. showed that AGEs could accelerate vascular calcification through the pathway of hypoxia-inducer/pyruvate dehydrogenase kinase 4 [72].
It is worth mentioning that a recent research found that colchicine, which is a drug widely used in the treatment of nonspecific inflammation, could combine with cholesterol crystal (CC), an important pathological marker for the vulnerability of atherosclerotic plaques [73]. This combination can reduce the intake of cholesterol crystals by endothelial cells, thus attenuating the cellular oxidative stress and endothelial cell prolapse by regulating the AMPK/SIRT1 signaling pathway [74].
4.3. Diabetic Cardiomyopathy
The complications of diabetes mainly include nephropathy, neuropathy, retinopathy, and heart diseases, which are linked with the activation of a series of oxidative stress in the body [75]. ROS can interact with a variety of biological macromolecules, such as DNA, proteins, and lipids [76, 77]. In the case of DNA damage, ROS induces DNA strand breaks and the formation of 8-hydroxydeoxyguanosine, which is a prominent feature of the diabetic heart [78]. The passive stiffness of myocardium is redox dependent, which leads to the increase of cardiac stiffness through actin oxidation and disulfide bond formation [79, 80]. In patients with diabetes, oxidative stress leads to decreased actin phosphorylation by damaging the NO/cGMP/PKG signaling [81], increased cardiomyocyte stiffness, and collagen and AGE deposition [82]. Polyunsaturated fatty acids rich in membrane lipids are easily oxidized by ROS, which is also involved in the formation of atherosclerotic plaques [83]. Lipid oxidation can lead to excessive formation of carbonyl compounds, such as aldehydes, which can accelerate a variety of pathologies [84]. NADPH oxidase is the main source of cardiac ROS, in which NOX2 and NOX4 are the two main subtypes expressed in the heart. It has been found that ROS produced by NOX is a common downstream mediator of various hemodynamic and metabolic pathways. ROS is involved in the occurrence of endothelial dysfunction and the development of diabetic vascular complications during hyperglycemia [85]. Glucose autooxidation, PKC activation, GAPDH inhibition, AGE formation, and polyol pathway activation can in turn exacerbate oxidative stress [86–88]. For example, the activation of the PKC pathway can lead to an increased expression of nuclear factor κB(NF-κB) [65, 89]. NF-κB can increase the expression of inducible nitric oxide synthase and increase the production of nitric oxide. Excessive nitric oxide reacts with peroxynitrates to produce peroxynitrates. Peroxynitrates can induce the formation of mitochondrial permeability transition holes, resulting in the increase of ROS production and the loss of cytochrome C, which exacerbates the development of diabetic cardiomyopathy [65]. Related studies found that in the aorta of STZ-induced diabetic ApoE−/− mice, the levels of NOX2 and NOX4 increased; in db/db mice (type II diabetes model), the expression of NOX1 and NOX4 was upregulated, and their activation resulted in the oxidation of ROS downstream molecules (e.g., tetrahydrobiopterin) and increased inflammatory response, indicating that NOX1, NOX2, and NOX4 are all involved in the pathological process of diabetic cardiomyopathy [90].
Of course, hyperglycemia is not the only pathogenic factor of diabetic cardiomyopathy, and the excessive oxidation of free fatty acids is also not ignored, which will lead to the activation of oxidative stress mitochondria and endoplasmic reticulum stress proinflammatory signals [91–93]. What can be seen is that a large number of changes in the diabetic heart include the overexpression of ROS and abnormal redox status. It is believed that the genes or drugs that target to block the ROS pathway in the future will bring the dawn of cure to patients with diabetic cardiomyopathy.
ROS also participates in the cardiac hypertrophy signaling transduction. In insulin-induced cardiac hypertrophy, the ROS level was upregulated and the levels of catalase were decreased [94]. Hypertrophy agonist Ang-II can increase the ROS levels in cardiomyocytes, and mitochondrial oxidative stress in turn contributes to Ang-II-induced cardiac hypertrophy [95]. Antioxidant administration can inhibit cardiac hypertrophy [96]. Tumor necrosis factor-alpha causes hypertrophy via the generation of ROS in cardiomyocytes [97]. Several hypertrophic stimuli need ROS to trigger cardiac hypertrophy. ROS could be a potential biological target for the novel therapy for maladaptive cardiac hypertrophy.
4.4. Myocardial Infarction (MI)
MI is one of the leading causes of disability and death in patients with CVDs in the world [98]. Programmed cardiomyocyte death, that is, apoptosis or autophagy, is considered to be the cause of MI. Cardiomyocyte apoptosis induced by ROS is controlled by a complex network of signal pathways involving noncoding RNAs [99]. For instance, under anaerobic conditions, mitochondrial fission and apoptosis-related circRNA (MFACR) suppresses the uninterrupted expression of miR-652-3p and MTP18 proteins, which leads to the imbalance of ROS, triggers the accumulation of mitochondrial fragments, and then results in apoptotic cell death [100]. ROS is involved in the toll-like receptor 4 (TLR4) and its downstream molecular pathway in mediating sympathetic activity post-MI within the paraventricular nucleus (PVN) [101]. The activation of TLR4 enhances the sympathetic activity after myocardial infarction by activating the microglia NF-κB and ROS in the paraventricular nucleus of the hypothalamus.
4.5. Heart Failure (HF)
Heart failure (HF) is a progressive disease with an annual mortality rate of about 10%. Although effective treatment has improved the outcome, the prognosis is still poor [102]. Related experiments and clinical studies have shown that the increase of ROS is related to the pathogenesis of HF [103–106]. ROS stimulates myocardial growth, matrix remodeling, and cellular dysfunction by activating various hypertrophic signal kinases and transcription factors. Activation of G protein-coupled receptor (GPCR) can lead to the production of ROS. Some data shows that ROS can directly induce the dissociation and activation of G protein [107–110]. Therefore, ROS may promote the hypertrophic growth signal of neonatal rat ventricular myocytes by directly activating G protein. ROS also stimulates apoptosis signal kinase-1, a redox-sensitive kinase that, when overexpressed, leads to NF-κB-induced hypertrophy [111]. Mitochondrial ROS and mitochondrial matrix calcium ([Ca2+]m) also participate in the pathogenesis of obesity-induced heart failure [112, 113], which may attack cardiomyocytes through the mechanism of free radical injury and combined with inflammatory cytokines (such as TNF-α and IL-6), resulting in the apoptosis of some cardiomyocytes, decreased cardiac function, and compensatory proliferation of cardiomyocytes, finally leading to myocardial hypertrophy [114].
In vitro hydrogen peroxide treatment induces oxidative stress in cardiomyocyte and leads to all kinds of cellular physiological or pathological processes, including necrosis and apoptosis. Wang et al. had identified several de novo pathways that underlie these processes. Several noncoding RNAs play functional roles in these pathways. In the programmed necrosis induced by hydrogen peroxide, long noncoding RNA NRF can combine with miR-873 and regulate the RIPK1/RIPK3 expression [115]. E2F1/miR-30b/Cyclophilin D forms a pathway in regulating hydrogen peroxide-induced necrotic cell death [116]. During hydrogen peroxide induced apoptosis, Wang et al. found that both E2F1/miR-421/Pink signal pathway and miR-361/PHB1 function in regulating mitochondria fission and apoptosis [118, 119]. All these results indicate that functional noncoding RNAs also play important role in a series of hydrogen peroxide-induced cellular responses. And these studies suggest that there might be relationships between functional noncoding RNAs and ROS, which await further study to unveil.
4.6. Atrial Fibrillation (AF)
Atrial fibrillation (AF) is the most common arrhythmia in clinics, and its risk increases with age [119]. Both human and animal data confirm the role of oxidative stress in the pathogenesis of AF [120, 121]. So far, there are some antioxidants that can positively affect the development of AF [122]. Type 2 ryanodine receptor (RyR2) is the main calcium release channel in atrial myocytes. It is a dysfunction caused by oxidative stress which disturbs the intracellular Ca2+ homeostasis that is linked with the pathogenesis of AF [123]. In atrial myocytes, RyR2 is oxidized by mitochondrial-derived ROS, resulting in increased intracellular Ca2+ leakage. It is worth noting that studies have shown that reducing the production of ROS can reduce atrial diastolic Ca2+ leakage, thus hindering the development of AF [124].
4.7. DNA Methylation and CVDs
DNA methylation, in which methyl is added to the C-5' position in the dinucleotide sequence of cytidine-phosphate-guanosine (CpG) to inhibit gene activity by preventing transcription factors from binding to the promoter or by recruiting chromatin modifying enzymes [125]. DNA methylation is catalyzed by three different DNA methyltransferases (DNMTs): DNMT3a and DNMT3b are mainly responsible for the ab initio methylation of embryonic and postpartum tissues, while DNMT1 subsequently maintains methylation [126].
The latest advances in next-generation sequencing technology have provided de novo understanding of DNA methylation. And more and more studies found that there are significant contributions of noncoding RNA in the pathophysiology of HF [127].
Long noncoding RNAs (lncRNAs) can regulate gene expression at the epigenetic level by directly or indirectly regulating the interaction with other molecules [128]. lncRNAs show epigenetic characteristics similar to those of coding genes, such as maternal effects, DNA methylation and histone modification, and posttranscriptional regulation [129].
In a series of causes, abnormal gene expression may be related to specific DNA methylation. The specific knockout of DNA methylase DNMT3b gene in the heart can lead to cardiomyocyte interstitial fibrosis and sarcomere disorder and accelerate the deterioration of systolic function and thinning of the ventricular wall during HF [130]. lncRNA-H19 is closely related to genomic imprinting [131]. It can change the methylation level of DNA by regulating the activity of S-adenosyl methionine(SAM), which plays an important role in cardiovascular diseases [129]. lncRNA-Mhrt can directly interact with histone modifiers to regulate chromatin modification, and its upregulated expression can prevent pathological myocardial hypertrophy [128]. lncRNA upperhand can regulate the expression of the hand2 gene related to cardiac development by allele specificity and cis-regulation [132].
The interaction between lncRNA-Chaer and the catalytic subunit of histone modification complex PRC2 interferes with the targeted genomic site of PRC2, thus inhibiting the methylation of histone H3 lysine 27 residues in the promoter region of cardiac hypertrophy related genes [133]. Inhibition of Chaer can significantly reduce myocardial hypertrophy and dysfunction.
The application of the targeted drugs to interfere with epigenetic dynamics is likely to become a new direction of drug research and development for cardiovascular diseases in the future. For example, trichostatin A, a class I and II histone deacetylase (HDACs) inhibitor, can prevent ischemia-induced left ventricular remodeling by inhibiting the TNF-α transcription and promote angiogenesis and cardiomyocyte survival by enhancing the Akt phosphorylation [134]. HDAC inhibitor sodium butyrate can block NF-κB signal transduction and inflammatory factors and improve myocardial infarction and atherosclerosis [135]. In addition, folic acid, histone deacetylase inhibitor apicidin, peroxisome proliferator-activated receptor-gamma agonist, and valproic acid were found to contribute to the restoration of chromatin modification in cardiac metabolism [136].
5. Diet Participates in the Regulation of ROS
More and more evidences have pointed that diet is closely linked to inflammation. And some researchers have found that it is possible to reduce the incidence of coronary heart disease through controlling diet [137]. If eating high-refined starch, sugar, saturated fatty acids, and trans fatty acids is kept for a long time, it will lead to a lack of natural antioxidants, fibers, and omega-3 fatty acids, which produce excessive proinflammatory cytokines.
In order to explore the relationship between diet and inflammation, Cavicchia et al. [138] proposed the inflammatory diet index (DII) for the first time in 2009, which is a dietary tool derived from the literature to evaluate the overall inflammatory potential of individual diet. DII consists of a variety of dietary ingredients, classified according to proinflammatory and anti-inflammatory components (Table 1). In recent years, DII has been widely used in clinical research, for example, cancer and CVDs [139]. DII can provide new ideas for the diagnosis and treatment of diseases, but related research remains unclear. In the study of diet and CVDs, we need to focus on the huge role of gut microbes and their metabolites in CVDs [140]. There are huge microecosystems in the intestinal tract, in which there are a large number of bacteria, fungi, viruses, protozoa, etc. Its metabolites play an important role in host metabolism, neurodevelopment, energy balance, and immune regulation, as well as the occurrence and development of cardiovascular diseases [141]. For example, intestinal microorganisms can promote vascular dysfunction and hypertension induced by Ang-II through vascular immune cell infiltration and inflammation [142, 143]. In patients with heart failure, the decrease of cardiac output and blood redistribution lead to reduced intestinal perfusion and breakdown of the intestinal barrier, as intestinal microbes and endotoxins enter the bloodstream and increase systemic inflammation, which in turn increases heart failure [144]. The evidence suggests that trimethylamine-N-oxide (TMAO) and short-chain fatty acids (SCFAs), the main metabolites of intestinal microorganisms, are involved in the pathogenesis of cardiovascular diseases [145]. TMAO can induce endothelial dysfunction and monocyte adhesion by activating NF-κB, protein kinase C, and pyran domain of nucleotide-binding oligomerization domain-like receptor family, and increase the expression of vascular endothelial inflammatory factors [146, 147]. At the same time, TMAO can also upregulate scavenger receptors in macrophages, promote the accumulation of cholesterol and formation of foam cells in macrophages, and further promote the formation of vascular plaques [148] and promote the inflammatory reaction of blood vessels through the MAPK and NF-κB pathways [149]. SCFAs play a key role in maintaining intestinal barrier function and play a positive role in cardiac metabolic health [150]. In addition, some probiotics and their fermented products have been proved to inhibit the production of nitrogen oxides in macrophages, reduce the types of reactive oxygen species, increase dietary calcium absorption, and thus reduce blood pressure [151].
Table 1.
Components of the inflammatory dietary index.
| Name | Proinflammatory or anti-inflammatory | Total inflammatory score |
|---|---|---|
| Alcohol (g) | Anti-inflammatory | -0.278 |
| Anthocyanidins (mg) | Anti-inflammatory | -0.131 |
| Beta carotene (μg) | Anti-inflammatory | -0.584 |
| Black/green tea (g) | Anti-inflammatory | -0.536 |
| Caffeine (g) | Anti-inflammatory | -0.110 |
| Carbohydrate (g) | Proinflammatory | 0.097 |
| Cholesterol (mg) | Proinflammatory | 0.110 |
| Energy (kcal) | Proinflammatory | 0.180 |
| Eugenol (mg) | Anti-inflammatory | -0.140 |
| Fiber (g) | Anti-inflammatory | -0.663 |
| Flavan-3-ol (mg) | Anti-inflammatory | -0.415 |
| Flavonols (mg) | Anti-inflammatory | -0.467 |
| Folic acid (μg) | Anti-inflammatory | -0.190 |
| Garlic (g) | Anti-inflammatory | -0.412 |
| Ginger (g) | Anti-inflammatory | -0.453 |
| Iron (mg) | Proinflammatory | 0.032 |
| Isoflavones (mg) | Anti-inflammatory | -0.593 |
| Magnesium (mg) | Anti-inflammatory | -0.484 |
| Monounsaturated fatty acids (g) | Anti-inflammatory | -0.009 |
| Niacin (g) | Anti-inflammatory | -0.246 |
| Omega 3 (g) | Anti-inflammatory | -0.436 |
| Omega 6 (g) | Anti-inflammatory | -0.159 |
| Onion (g) | Anti-inflammatory | -0.301 |
| Oregano/thyme (mg) | Anti-inflammatory | -0.102 |
| Pepper (g) | Anti-inflammatory | -0.131 |
| Polyunsaturated fatty acids (g) | Anti-inflammatory | -0.337 |
| Protein (g) | Proinflammatory | 0.021 |
| Riboflavin (mg) | Anti-inflammatory | -0.068 |
| Rosemary (mg) | Anti-inflammatory | -0.013 |
| Saturated fat (g) | Proinflammatory | 0.373 |
| Selenium (μg) | Anti-inflammatory | -0.191 |
| Thiamine (mg) | Anti-inflammatory | -0.098 |
| Total fat (g) | Proinflammatory | 0.298 |
| Trans fat (g) | Proinflammatory | 0.229 |
| Turmeric (mg) | Anti-inflammatory | -0.785 |
| Vitamin A (RE) | Anti-inflammatory | -0.401 |
| Vitamin B6 (mg) | Anti-inflammatory | -0.365 |
| Vitamin B12 (μg) | Proinflammatory | 0.106 |
| Vitamin C (mg) | Anti-inflammatory | -0.424 |
| Vitamin D (μg) | Anti-inflammatory | -0.446 |
| Vitamin E (mg) | Anti-inflammatory | -0.419 |
| Zinc (mg) | Anti-inflammatory | -0.313 |
A dietary intervention has been shown to reduce the risk of cardiovascular disease events. High-fat and high-sugar diets can lead to abnormal intestinal flora and increase the risk of cardiovascular disease [152]. Increasing the carbohydrate diet can change the composition of Rosella and rectal true bacilli [153]. A diet rich in dietary fiber can promote the growth of beneficial bacteria and inhibit the growth of the conditional pathogenic bacteria [154]. A high-fiber diet can increase acetate-producing microorganisms, lower blood pressure, and improve ventricular remodelling and fibrosis [155]. Another example in rats after partial nephrectomy indicated that the use of curcumin in ginger can retain the ejection fraction and reduce the lipid peroxidation of the heart muscle [156]. Allicin (40 mg/kg/day, orally), which is a component of garlic extract, could reduce hypertension, lipid, and protein oxidation in the heart, meanwhile accelerate the levels of antioxidant enzymes [157]. Supplementation of 800 IU/day vitamin E as an antioxidant can reduce CVD endpoints and myocardial infarction in haemodialysis patients with CVDs [158].
Aloe is an edible plant in daily life [159], which contains a compound called aloe-emodin (AE) [160, 161]. Yu et al. [162] found that in the H2O2-induced apoptosis model of neonatal rat ventricular myocytes, AE can prevent myocardial infarction by upregulating miR-133, inhibiting the ROS production, and inhibiting the caspase-3 apoptosis signal pathway. In addition, AE treatment significantly reversed the H2O2-induced upregulation of Bax/Bcl-2 and loss of mitochondrial membrane potential. Chen et al. [163] established a rat cardiac inflammation model induced by hyperlipidaemia, and then administered AE to study the potential role and mechanism of AE regulating cardiac oxidative stress and inflammation induced by hyperlipaemia. They found that compared with the normal diet (ND) group, the expression levels of proinflammatory cytokines IL-1β, IL-6, and TNF-α were significantly upregulated in the hyperlipaemia group, while the expression levels of IL-1, IL-6, and TNF-α were dramatically decreased in the AE treatment group. In addition, AE also inhibited the expression of vascular cell adhesion molecule-1 (VCAM1) and intercellular adhesion molecule-1 (ICAM-1). In vitro, AE decreased the expression of IL-1β, IL-6, and TNF-α in palmitic acid (PA-) treated H9C2 cells in a dose-dependent manner. Further experiments showed that AE inhibited PA-induced cell death and promoted the production of intracellular ROS [163]. This study indicates that AE may reduce cardiac inflammation induced by hyperlipidaemia/plasminogen activator by inhibiting the TLR4/NF-κB signal pathway, which may be a promising therapeutic strategy for the prevention of myocardial injury.
Intestinal flora and cardiovascular disease is a new research field in the future, but the specific mechanism of the interaction between intestinal flora and body is not clear. Maintaining the homeostasis of intestinal flora and correcting the imbalance of intestinal flora will become a new target for the prevention and treatment of cardiovascular diseases.
6. Exercise Promotes Balance of ROS
Physical activity has long been considered to be beneficial for CVDs. However, the molecular mechanisms by which triggering and sustaining exercise are beneficial for the heart are poorly understood, which is expected for new therapeutic targets. To explore these mechanisms, Moreira et al. identified cardiac gene targets in rat models by using RNA sequencing [164], whose expression could be disrupted in heart failure but was recovered by exercise. Through a series of elaborate validation, they screened 16 targets to assess whether targeted interference with the silencing RNA of these genes can affect the abundance of a CVD biomarker (BNP, B-type natriuretic peptide) in human cardiomyocytes. Among them, the Proline Dehydrogenase (PRODH) expression is reduced in human failing hearts, but rescued by exercise in a rat model of HF. The knockdown of PRODH also resulted in the rise of the BNP expression in human cardiomyocytes.
Compared with the traditional drug treatments, natural methods of improving the collaterals through exercise training seem to be more effective, especially for patients with intermittent claudication. Exercise has a variety of positive effects on the body, but it also has systemic benefits [165, 166]. In general, physical activity has been shown to greatly improve cardiovascular function, and this is partly due to improved bioavailability of NO, increased endogenous antioxidant defence, and decreased expression of the enzyme involved in the ROS production [167].
7. Summary
ROS is not only a natural by-product of metabolic responses in various cell compartments but also a signalling molecule that regulates specific biochemical pathways in normal cell function and survival. However, the dysregulation of ROS signalling or excessive production of nonspecific ROS can affect the pathophysiology of heart diseases. As this review highlights (Figure 3), ROS are particularly important in cellular metabolism and inflammatory signalling. Therefore, it is not surprising that ROS plays an important role in cardiac diseases associated with metabolic disorders and inflammation. The pathogenesis of cardiovascular and metabolic diseases is complex, and understanding tissue-specific REDOX signals is important for us to develop new and novel therapies to treat diseases. Metabolic dysregulation is a major driver of cell dysfunction and disease progression, and exploring the contribution and effect of reactive oxygen species on metabolic processes is an important field of scientific discovery. At the same time, exploring the effects of a healthy diet and exercise on the regulation of oxidative stress, inflammation, and the improvement of cardiac dysfunction will also provide a new direction for the treatment of CVDs.
Figure 3.
Summary exhibiting effects of REDOX in cardiovascular disease. ROS plays an important role in cardiovascular disease and serious heart disease. The pathogenesis of cardiovascular diseases and metabolic diseases is complex, and understanding the tissue-specific REDOX signal is very important for us to develop new methods to treat diseases. The contribution and influence of reactive oxygen species on metabolic processes is an important area of scientific discovery. Exploring the regulatory effects of a healthy diet and exercise on the improvement of oxidative stress, inflammation, and cardiac dysfunction will also provide new directions for the treatment of CVD.
Acknowledgments
This work was supported by the Natural Science Foundation of China (No: 31701733), National Natural Science Foundation of China (No: 81870236), Qingdao Postdoctoral Applied Research Project, and the Major Research Program of the National Natural Science Foundation of China (No: 91849209).
Contributor Information
Yanhan Dong, Email: dongyanhande@163.com.
Luyu Zhou, Email: lyzhoucas@163.com.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Authors' Contributions
Kai Wang and Yanhan Dong contributed equally to this work.
References
- 1.Pisoschi A. M., Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. European Journal of Medicinal Chemistry. 2015;97:55–74. doi: 10.1016/j.ejmech.2015.04.040. [DOI] [PubMed] [Google Scholar]
- 2.Borut P., Dušan Š., Irina M. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxidative Medicine & Cellular Longevity. 2013;2013:1–11. doi: 10.1155/2013/956792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korovila I., Hugo M., Castro J. P., Weber D., Jung T. Proteostasis, oxidative stress and aging. Redox Biology. 2017;13:550–567. doi: 10.1016/j.redox.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flora S. J. S. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxidative Medicine & Cellular Longevity. 2009;2(4):191–206. doi: 10.4161/oxim.2.4.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poprac P., Jomova K., Simunkova M., Kollar V., Rhodes C. J., Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends in Pharmacological Sciences. 2017;38(7):592–607. doi: 10.1016/j.tips.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Forrester S. J., Kikuchi D. S., Hernandes M. S., Xu Q., Griendling K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research. 2018;122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parikh P., McDaniel M. C., Ashen M. D., et al. Diets and cardiovascular disease: an evidence-based assessment. Journal of the American College of Cardiology. 2005;45(9):1379–1387. doi: 10.1016/j.jacc.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 8.Hankey C. R., Leslie W. S. Nutrition and coronary heart disease. Acta Cardiologica. 2001;5:194–201. [Google Scholar]
- 9.Malekmohammad K., Sewell R. D. E., Rafieian-Kopaei M. Antioxidants and atherosclerosis: mechanistic aspects. Biomolecules. 2019;9(8):p. 301. doi: 10.3390/biom9080301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ros E., Martínez-González M. A., Estruch R., et al. Mediterranean diet and cardiovascular health: teachings of the PREDIMED study. Advances in nutrition. 2014;5(3):330S–336S. doi: 10.3945/an.113.005389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Wang X., Cueto R., et al. Biochemical basis and metabolic interplay of redox regulation. Redox biology. 2019;26:p. 101284. doi: 10.1016/j.redox.2019.101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomasian D., Keaney J. F., Jr., Vita J. A. Antioxidants and the bioactivity of endothelium-derived nitric oxide. Cardiovascular Research. 2000;3 doi: 10.1016/s0008-6363(00)00103-6. [DOI] [PubMed] [Google Scholar]
- 13.Forman H. J., Fridovich I. Superoxide Dismutase: A Comparison of Rate Constants. 1973. [DOI] [PubMed]
- 14.Araujo J. A., Romano E. L., Brito B. E., et al. Iron overload augments the development of atherosclerotic lesions in rabbits. Arteriosclerosis, thrombosis, and vascular biology. 1995;15:1172–1180. doi: 10.1161/01.atv.15.8.1172. [DOI] [PubMed] [Google Scholar]
- 15.Nelson M. J., Cowling R. A., Seitz S. P. Structural characterization of alkyl and peroxyl radicals in solutions of purple lipoxygenase. Biochemistry. 2002;33:4966–4973. doi: 10.1021/bi00182a027. [DOI] [PubMed] [Google Scholar]
- 16.Funk M. O., Andre J. C., Otsuki T. Oxygenation of trans polyunsaturated fatty acids by lipoxygenase reveals steric features of the catalytic mechanism. Biochemistry. 2002;26:6880–6884. doi: 10.1021/bi00395a043. [DOI] [PubMed] [Google Scholar]
- 17.Benjamin E. J., Muntner P., Alonso A., et al. Correction to: Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2020;141(2):p. e33. doi: 10.1161/cir.0000000000000746. [DOI] [PubMed] [Google Scholar]
- 18.Samman Tahhan A., Sandesara P. B., Hayek S. S., et al. Association between oxidative stress and atrial fibrillation. Heart rhythm. 2017;14(12):1849–1855. doi: 10.1016/j.hrthm.2017.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baradaran A., Nasri H., Rafieian-Kopaei M. Oxidative stress and hypertension: possibility of hypertension therapy with antioxidants. Journal of Research in Medical Sciences the Official Journal of Isfahan University of Medical Sciences. 2014;19:358–367. [PMC free article] [PubMed] [Google Scholar]
- 20.Kattoor A. J., Pothineni N. V. K., Palagiri D., Mehta J. L. Oxidative stress in atherosclerosis. Current Atherosclerosis Reports. 2017;19(11) doi: 10.1007/s11883-017-0678-6. [DOI] [PubMed] [Google Scholar]
- 21.Huynh D. T. N., Heo K.-S. Therapeutic targets for endothelial dysfunction in vascular diseases. Archives of Pharmacal Research. 2019;42(10):848–861. doi: 10.1007/s12272-019-01180-7. [DOI] [PubMed] [Google Scholar]
- 22.Anwar M. A., Al Disi S. S., Eid A. H. Anti-hypertensive herbs and their mechanisms of action: part II. Frontiers in Pharmacology. 2016;7 doi: 10.3389/fphar.2016.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senoner T., Dichtl W. Oxidative stress in cardiovascular diseases: still a therapeutic target? Nutrients. 2019;11(9):p. 2090. doi: 10.3390/nu11092090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volobueva A., Grechko A., Yet S. F., Sobenin I., Orekhov A. Changes in mitochondrial genome associated with predisposition to atherosclerosis and related disease. Biomolecules. 2020;9 doi: 10.3390/biom9080377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams B. R., Mancia G., Spiering W., et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Journal of Hypertension. 2019;37 [Google Scholar]
- 26.Peleli M., Flacker P., Zhuge Z., et al. Renal denervation attenuates hypertension and renal dysfunction in a model of cardiovascular and renal disease, which is associated with reduced NADPH and xanthine oxidase activity. Redox Biology. 2017;13:522–527. doi: 10.1016/j.redox.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naregal G. V., Devaranavadagi B. B., Patil S. G., Aski B. S. Elevation of oxidative stress and decline in endogenous antioxidant defense in elderly individuals with hypertension. Journal of Clinical and Diagnostic Research : JCDR. 2017;11:BC09–BC12. doi: 10.7860/jcdr/2017/27931.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paravicini T. M., Touyz R. M. Redox signaling in hypertension. Cardiovascular Research. 2006;71(2):247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Fan M., Zhang J., Tsai C. W., et al. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature. 2020;582(7810):129–133. doi: 10.1038/s41586-020-2309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufmann U., Kahlfuss S., Yang J., Ivanova E., Koralov S. B., Feske S. Calcium signaling controls pathogenic Th17 cell-mediated inflammation by regulating mitochondrial function. Cell metabolism. 2019;29(5):1104–1118.e6. doi: 10.1016/j.cmet.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton S., Terentyeva R., Martin B., et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic research in cardiology. 2020;115(4):p. 38. doi: 10.1007/s00395-020-0797-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olgar Y., Tuncay E., Degirmenci S., et al. Ageing-associated increase in SGLT2 disrupts mitochondrial/sarcoplasmic reticulum Ca homeostasis and promotes cardiac dysfunction. Journal of cellular and molecular medicine. 2020;24:8567–8578. doi: 10.1111/jcmm.15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson M. T., Gudlur A., Zhang X., et al. L-type Ca2+channel blockers promote vascular remodeling through activation of STIM proteins. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:202007598–202017380. doi: 10.1073/pnas.2007598117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gavin R., Koo C., Tomlinson M. Tspan18 is a novel regulator of thrombo-inflammation. Medical Microbiology and Immunology. 2020;209:553–564. doi: 10.1007/s00430-020-00678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang F., Pelzl L., Hauser S., Hermann A., Stournaras C., Schöls L. To die or not to die SGK1-sensitive ORAI/STIM in cell survival. Cell Calcium. 2018;74:29–34. doi: 10.1016/j.ceca.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Pelzl L., Sahu I., Ma K., et al. Beta-glycerophosphate-induced ORAI1 expression and store operated Ca2+ entry in megakaryocytes. Scientific reports. 2020;10(1):p. 1728. doi: 10.1038/s41598-020-58384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W., Trebak M. STIM1 and Orai1: novel targets for vascular diseases? Science China. Life Sciences. 2011;54:780–785. doi: 10.1007/s11427-011-4206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papanikolaou M., Lewis A., Butt A. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Structure & Function. 2017;222:2993–3005. doi: 10.1007/s00429-017-1380-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gough N. Focus issue: the ins and outs of ORAI in immune cells. Science signaling. 2016;9(418):p. eg3. doi: 10.1126/scisignal.aaf4957. [DOI] [PubMed] [Google Scholar]
- 40.Tsujikawa H., Yu A. S., Xie J., et al. Identification of key amino acid residues responsible for internal and external pH sensitivity of Orai1/STIM1 channels. Scientific reports. 2015;5(1):p. 16747. doi: 10.1038/srep16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gouni-Berthold I., Giannakidou E., Faust M., Berthold H. K., Krone W. Oxidative stress in hypertension: mechanisms and therapeutic opportunities. Experimental & Clinical Endocrinology & Diabetes. 2015;123:325–335. doi: 10.1055/s-0035-1548765. [DOI] [PubMed] [Google Scholar]
- 42.Touyz R. M. Reactive oxygen species and angiotensin II signaling in vascular cells -- implications in cardiovascular disease. Brazilian journal of medical and biological research. 2004;37(8):1263–1273. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- 43.Lamb F. S., Choi H., Miller M. R., Stark R. J. TNFα and reactive oxygen signaling in vascular smooth muscle cells in hypertension and atherosclerosis. American journal of hypertension. 2020;33:902–913. doi: 10.1093/ajh/hpaa089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perdomo L., Vidal-Gomez X., Soleti R., et al. Large extracellular vesicle-associated Rap1 accumulates in atherosclerotic plaques, correlates with vascular risks and is involved in atherosclerosis. Circulation research. 2020;127 doi: 10.1161/circresaha.120.317086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouerd S., Idris-Khodja N., Trindade M., et al. Endothelium-restricted endothelin-1 overexpression in type-1 diabetes worsens atherosclerosis and immune cell infiltration via Nox1. Cardiovascular research. 2020 doi: 10.1093/cvr/cvaa168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K., Liu X., Liu Q., et al. Hederagenin potentiated cisplatin- and paclitaxel-mediated cytotoxicity by impairing autophagy in lung cancer cells. Cell death & disease. 2020;11(8):p. 611. doi: 10.1038/s41419-020-02880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang B., Ding L., Yao H., Chen Y., Shi J. A. A metal-organic framework (MOF) fenton nanoagent-enabled nanocatalytic cancer therapy in synergy with autophagy inhibition. Advanced materials. 2020;32(12):p. 1907152. doi: 10.1002/adma.201907152. [DOI] [PubMed] [Google Scholar]
- 48.Lu L., Tian J., Luo X., Peng J. Targeting the pathways of regulated necrosis: a potential strategy for alleviation of cardio-cerebrovascular injury. Cellular and molecular life sciences : CMLS. 2020 doi: 10.1007/s00018-020-03587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Song X., Li Z., et al. MicroRNA-103 protects coronary artery endothelial cells against H2O2-Induced oxidative stress via BNIP3-mediated end-stage autophagy and antipyroptosis pathways. Oxidative Medicine and Cellular Longevity. 2020;2020:15. doi: 10.1155/2020/8351342.8351342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perrotta I., Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxidative Medicine and Cellular Longevity. 2015;2015:10. doi: 10.1155/2015/130315.130315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Förstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research. 2017;120(4):713–735. doi: 10.1161/circresaha.116.309326. [DOI] [PubMed] [Google Scholar]
- 52.Ebert J., Wilgenbus P., Teiber J. F., et al. Paraoxonase-2 regulates coagulation activation through endothelial tissue factor. Blood, The Journal of the American Society of Hematology. 2018;131(19):2161–2172. doi: 10.1182/blood-2017-09-807040. [DOI] [PubMed] [Google Scholar]
- 53.Fulton D. J., Barman S. A. Clarity on the isoform-specific roles of NADPH oxidases and NADPH oxidase-4 in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2016;36(4):579–581. doi: 10.1161/atvbaha.116.307096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin-Ventura J. L., Rodrigues-Diez R., Martinez-Lopez D., Salaices M., Blanco-Colio L. M., Briones A. M. Oxidative stress in human atherothrombosis: sources, markers and therapeutic targets. International journal of molecular sciences. 2017;18(11):p. 2315. doi: 10.3390/ijms18112315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gray S. P., Di Marco E., Okabe J., et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/circulationaha.112.132159. [DOI] [PubMed] [Google Scholar]
- 56.Soula M., Weber R. A., Zilka O., et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nature chemical biology. 2020 doi: 10.1038/s41589-020-0613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng Y., Madungwe N., Imam Aliagan A., Tombo N., Bopassa J. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochemical and Biophysical Research Communications. 2019;520:606–611. doi: 10.1016/j.bbrc.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W., Li W., Leng Y., Xiong Y., Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA and Cell Biology. 2020;39:210–225. doi: 10.1089/dna.2019.5097. [DOI] [PubMed] [Google Scholar]
- 59.Guan X., Li X., Yang X., et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life sciences. 2019;235:p. 116795. doi: 10.1016/j.lfs.2019.116795. [DOI] [PubMed] [Google Scholar]
- 60.Yang W. S., SriRamaratnam R., Welsch M. E., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai T., Li M., Liu Y., Qiao Z., Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free radical biology & medicine. 2020;160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026. [DOI] [PubMed] [Google Scholar]
- 62.Guo Z., Ran Q., Roberts L. J., II, et al. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free radical biology & medicine. 2008;44(3):343–352. doi: 10.1016/j.freeradbiomed.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tacey A., Qaradakhi T., Smith C., et al. The effect of an atherogenic diet and acute hyperglycaemia on endothelial function in rabbits is artery specific. Nutrients. 2020;12(7):p. 2108. doi: 10.3390/nu12072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flynn M. C., Kraakman M. J., Tikellis C., et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circulation research. 2020;127(7):877–892. doi: 10.1161/circresaha.120.316653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kowluru R., Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochimica et biophysica acta. 2015;1852(11):2474–2483. doi: 10.1016/j.bbadis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 66.Neviere R., Yu Y., Wang L., Tessier F., Boulanger E. Implication of advanced glycation end products (Ages) and their receptor (Rage) on myocardial contractile and mitochondrial functions. Glycoconjugate Journal. 2016;33:607–617. doi: 10.1007/s10719-016-9679-x. [DOI] [PubMed] [Google Scholar]
- 67.Patel M., Stovall K., Franklin J. The intrinsic apoptotic pathway lies upstream of oxidative stress in multiple organs. Free Radical Biology & Medicine. 2020;158:13–19. doi: 10.1016/j.freeradbiomed.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu C., Cao B., Zhang Q., et al. Inhibition of thioredoxin 2 by intracellular methylglyoxal accumulation leads to mitochondrial dysfunction and apoptosis in INS-1 cells. Endocrine. 2020;68(1):103–115. doi: 10.1007/s12020-020-02191-x. [DOI] [PubMed] [Google Scholar]
- 69.Fang H., Yang K., Tang P., et al. Glycosylation end products mediate damage and apoptosis of periodontal ligament stem cells induced by the JNK-mitochondrial pathway. Aging. 2020;12(13):12850–12868. doi: 10.18632/aging.103304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moldogazieva N. T., Mokhosoev I. M., Mel’nikova T. I., Porozov Y. B., Terentiev A. A. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxidative Medicine and Cellular Longevity. 2019;2019:14. doi: 10.1155/2019/3085756.3085756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chowdhury S., Ghosh S., Das A., Sil P. Ferulic acid protects hyperglycemia-induced kidney damage by regulating oxidative insult, inflammation and autophagy. Frontiers in Pharmacology. 2019;10:p. 27. doi: 10.3389/fphar.2019.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu Y., Ma W. Q., Han X. Q., Wang Y., Wang X., Liu N. F. Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Scientific reports. 2018;8(1):p. 13730. doi: 10.1038/s41598-018-31877-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang M., Lv H., Liu Q., et al. Colchicine alleviates cholesterol crystal-induced endothelial cell pyroptosis through activating AMPK/SIRT1 pathway. Oxidative Medicine and Cellular Longevity. 2020;2020:18. doi: 10.1155/2020/9173530.9173530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim N. H., Kim H. K., Lee J. H., et al. Juglone suppresses LPS-induced inflammatory responses and NLRP3 activation in macrophages. Molecules. 2020;25(13):p. 3104. doi: 10.3390/molecules25133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koulis C., Watson A. M., Gray S. P., Jandeleit-Dahm K. A. Linking RAGE and Nox in diabetic micro- and macrovascular complications. Diabetes & metabolism. 2015;41(4):272–281. doi: 10.1016/j.diabet.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 76.Zhang H., Chen X., Zong B., et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. Journal of cellular and molecular medicine. 2018;22(9):4437–4448. doi: 10.1111/jcmm.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dai B., Li H., Fan J., et al. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovascular diabetology. 2018;17(1):p. 123. doi: 10.1186/s12933-018-0767-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishio S., Teshima Y., Takahashi N., et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. Journal of molecular and cellular cardiology. 2012;52(5):1103–1111. doi: 10.1016/j.yjmcc.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 79.Evangelista I., Nuti R., Picchioni T., Dotta F., Palazzuoli A. Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. International journal of molecular sciences. 2019;20(13):p. 3264. doi: 10.3390/ijms20133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grützner A., Garcia-Manyes S., Kötter S., Badilla C. L., Fernandez J. M., Linke W. A. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophysical journal. 2009;97(3):825–834. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beckendorf L., Linke W. Emerging importance of oxidative stress in regulating striated muscle elasticity. Journal of Muscle Research and Cell Motility. 2015;36:25–36. doi: 10.1007/s10974-014-9392-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Breitkreuz M., Hamdani N. A change of heart: oxidative stress in governing muscle function? Biophysical Reviews. 2015;7:321–341. doi: 10.1007/s12551-015-0175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zeng Y., Xu J., Hua Y., Peng Y., Xu X. MDM2 contributes to oxidized low-density lipoprotein-induced inflammation through modulation of mitochondrial damage in endothelial cells. Atherosclerosis. 2020;305:1–9. doi: 10.1016/j.atherosclerosis.2020.05.020. [DOI] [PubMed] [Google Scholar]
- 84.Kaludercic N., Carpi A., Nagayama T., et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxidants & redox signaling. 2014;20(2):267–280. doi: 10.1089/ars.2012.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gray S. P., Jandeleit-Dahm K. The pathobiology of diabetic vascular complications--cardiovascular and kidney disease. Journal of molecular medicine. 2014;92(5):441–452. doi: 10.1007/s00109-014-1146-1. [DOI] [PubMed] [Google Scholar]
- 86.Nascimento N. R. F., Lessa L. M. A., Kerntopf M. R., et al. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(1):218–223. doi: 10.1073/pnas.0509779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107:1058–1070. doi: 10.1161/circresaha.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 89.Suzuki H., Kayama Y., Sakamoto M., et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes. 2015;64(2):618–630. doi: 10.2337/db13-1896. [DOI] [PubMed] [Google Scholar]
- 90.Di Marco E., Gray S. P., Chew P., et al. Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune-inflammatory responses in diabetic Apoe -/- mice. Diabetologia. 2014;57 doi: 10.1007/s00125-013-3118-3. [DOI] [PubMed] [Google Scholar]
- 91.Tsushima K., Bugger H., Wende A. R., et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circulation research. 2018;122(1):58–73. doi: 10.1161/circresaha.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ertunc M., Hotamisligil G. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. Journal of Lipid Research. 2016;57:2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wende A., Symons J., Abel E. Mechanisms of lipotoxicity in the cardiovascular system. Current Hypertension Reports. 2012;14:517–531. doi: 10.1007/s11906-012-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tan W. Q., Wang K., Lv D. Y., Li P. F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. The Journal of Biological Chemistry. 2008;283:29730–29739. doi: 10.1074/jbc.M805514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dai D. F., Johnson S. C., Villarin J. J., et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circulation research. 2011;108(7):837–846. doi: 10.1161/circresaha.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang Y., Ago T., Zhai P., Abdellatif M., Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circulation research. 2011;108(3):305–313. doi: 10.1161/circresaha.110.228437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakamura K., Fushimi K., Kouchi H., et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98(8):794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 98.Mozaffarian D., Benjamin E. J., Go A. S., et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/cir.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 99.Dong Y., Xu W., Liu C., Liu P., Li P., Wang K. Reactive oxygen species related noncoding RNAs as regulators of cardiovascular diseases. International journal of biological sciences. 2019;15(3):680–687. doi: 10.7150/ijbs.30464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang K., Gan T. Y., Li N., et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell death and differentiation. 2017;24(6):1111–1120. doi: 10.1038/cdd.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Y., Hu H., Yin J., et al. TLR4 participates in sympathetic hyperactivity post-MI in the PVN by regulating NF-κB pathway and ROS production. Redox biology. 2019;24, article 101186 doi: 10.1016/j.redox.2019.101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tanai E., Frantz S. Pathophysiology of heart failure. Comprehensive Physiology. 2016;6:187–214. doi: 10.1002/cphy.c140055. [DOI] [PubMed] [Google Scholar]
- 103.Mallat Z., Philip I., Lebret M., Chatel D., Maclouf J., Tedgui A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation. 1998;97(16):1536–1539. doi: 10.1161/01.cir.97.16.1536. [DOI] [PubMed] [Google Scholar]
- 104.MF H., PK S. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96(7):2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- 105.Kračun D., Klop M., Knirsch A., et al. NADPH oxidases and HIF1 promote cardiac dysfunction and pulmonary hypertension in response to glucocorticoid excess. Redox biology. 2020;34, article 101536 doi: 10.1016/j.redox.2020.101536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cole L. K., Mejia E. M., Sparagna G. C., et al. Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. Journal of molecular and cellular cardiology. 2020;144:24–34. doi: 10.1016/j.yjmcc.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Nishida M., Schey K. L., Takagahara S., et al. Activation mechanism of Gi and Go by reactive oxygen species. Journal of Biological Chemistry. 2002;277(11):9036–9042. doi: 10.1074/jbc.M107392200. [DOI] [PubMed] [Google Scholar]
- 108.Nishida M., Maruyama Y., Tanaka R., Kontani K., Nagao T., Kurose H. Gαi and Gαo are target proteins of reactive oxygen species. Nature. 2000;408(6811):492–495. doi: 10.1038/35044120. [DOI] [PubMed] [Google Scholar]
- 109.Cao J., Liu X., Yang Y., et al. Decylubiquinone suppresses breast cancer growth and metastasis by inhibiting angiogenesis via the ROS/p53/ BAI1 signaling pathway. Angiogenesis. 2020;23(3):325–338. doi: 10.1007/s10456-020-09707-z. [DOI] [PubMed] [Google Scholar]
- 110.Castaldo M., Zollo C., Esposito G., Ammendola R., Cattaneo F. NOX2-dependent reactive oxygen species regulate formyl-peptide receptor 1-mediated TrkA transactivation in SH-SY5Y cells. Oxidative medicine and cellular longevity. 2019;2019:17. doi: 10.1155/2019/2051235.2051235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guzik T. J., Harrison D. G. Endothelial NF-κB As a Mediator of Kidney Damage. 2007. [DOI] [PubMed]
- 112.Niemann B., Li L., Siegler D., et al. CTRP9 mediates protective effects in cardiomyocytes via AMPK- and adiponectin receptor-mediated induction of anti-oxidant response. Cells. 2020;9(5):p. 1229. doi: 10.3390/cells9051229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Das S., Steenbergen C., Murphy E. Does the voltage dependent anion channel modulate cardiac ischemia-reperfusion injury? Biochimica et Biophysica Acta. 2012;1818:1451–1456. doi: 10.1016/j.bbamem.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zeng S. Y., Yang L., Lu H. Q., Yan Q. J., Gao L., Qin X. P. Rutaecarpine prevents hypertensive cardiac hypertrophy involving the inhibition of Nox4-ROS-ADAM17 pathway. Journal of cellular and molecular medicine. 2019;23:4196–4207. doi: 10.1111/jcmm.14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang K., Liu F., Liu C. Y., et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell death and differentiation. 2016;23(8):1394–1405. doi: 10.1038/cdd.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang K., An T., Zhou L. Y., et al. E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell death and differentiation. 2015;22(5):743–754. doi: 10.1038/cdd.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang K., Zhou L. Y., Wang J. X., et al. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nature communications. 2015;6(1):p. 7619. doi: 10.1038/ncomms8619. [DOI] [PubMed] [Google Scholar]
- 118.Wang K., Liu C. Y., Zhang X. J., et al. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell death and differentiation. 2015;22(6):1058–1068. doi: 10.1038/cdd.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen C., Liu L., Yu Y., et al. Association of systolic blood pressure with atrial fibrillation among treated hypertensive patients. Annals of palliative medicine. 2020;9(4):1752–1763. doi: 10.21037/apm-19-649. [DOI] [PubMed] [Google Scholar]
- 120.Huang C. X., Liu Y., Xia W. F., Tang Y. H., Huang H. Oxidative stress: a possible pathogenesis of atrial fibrillation. Medical Hypotheses. 2009;72:466–467. doi: 10.1016/j.mehy.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 121.Korantzopoulos P., Kolettis T. M., Galaris D., Goudevenos J. A. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. International Journal of Cardiology. 2007;115(2):135–143. doi: 10.1016/j.ijcard.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 122.Sakabe M., Fujiki A., Sakamoto T., Nakatani Y., Mizumaki K., Inoue H. Xanthine oxidase inhibition prevents atrial fibrillation in a canine model of atrial pacing-induced left ventricular dysfunction. Journal of Cardiovascular Electrophysiology. 2012;23(10):1130–1135. doi: 10.1111/j.1540-8167.2012.02356.x. [DOI] [PubMed] [Google Scholar]
- 123.Chelu M. G., Sarma S., Sood S., et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. The Journal of clinical investigation. 2009;119:1940–1951. doi: 10.1172/jci37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xie W., Santulli G., Reiken S. R., et al. Mitochondrial oxidative stress promotes atrial fibrillation. Scientific reports. 2015;14 doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kohli R. M., Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xue J. H., Chen G. D., Hao F., et al. A vitamin-C-derived DNA modification catalysed by an algal TET homologue. Nature. 2019;569(7757):581–585. doi: 10.1038/s41586-019-1160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu C. F., Tang W. H. W. Epigenetics in cardiac hypertrophy and heart failure. JACC. Basic to translational science. 2019;4:976–993. doi: 10.1016/j.jacbts.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Han P., Li W., Lin C. H., et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514(7520):102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou J., Yang L., Zhong T., et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nature communications. 2015;6(1) doi: 10.1038/ncomms10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vujic A., Robinson E. L., Ito M., et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. Journal of molecular and cellular cardiology. 2015;82:174–183. doi: 10.1016/j.yjmcc.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 131.Yoshimura H., Matsuda Y., Yamamoto M., Kamiya S., Ishiwata T. Expression and role of long non-coding RNA H19 in carcinogenesis. Frontiers in bioscience. 2018;23(2):614–625. doi: 10.2741/4608. [DOI] [PubMed] [Google Scholar]
- 132.Jonkers I., Monkhorst K., Rentmeester E., Grootegoed J. A., Grosveld F., Gribnau J. Xist RNA is confined to the nuclear territory of the silenced X chromosome throughout the cell cycle. Molecular and cellular biology. 2008;28(18):5583–5594. doi: 10.1128/mcb.02269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Z., Zhang X. J., Ji Y. X., et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nature medicine. 2016;22(10):1131–1139. doi: 10.1038/nm.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang L., Qin X., Zhao Y., et al. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. The Journal of pharmacology and experimental therapeutics. 2012;341(1):285–293. doi: 10.1124/jpet.111.189910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hu X., Zhang K., Xu C., Chen Z., Jiang H. Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion. Experimental and Therapeutic Medicine. 2014;8:229–232. doi: 10.3892/etm.2014.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circulation Research. 2011;108:1002–1016. doi: 10.1161/circresaha.110.226860. [DOI] [PubMed] [Google Scholar]
- 137.Giugliano D., Ceriello A., Esposito K. The effects of diet on inflammation: emphasis on the metabolic syndrome. Journal of the American College of Cardiology. 2006;48:677–685. doi: 10.1016/j.jacc.2006.03.052. [DOI] [PubMed] [Google Scholar]
- 138.Cavicchia P. P., Steck S. E., Hurley T. G., et al. A new dietary inflammatory index predicts interval changes in serum high-sensitivity C-reactive protein. Journal of Nutrition. 2009;139(12):2365–2372. doi: 10.3945/jn.109.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gardener S. L., Rainey-Smith S. R., Martins R. N. Diet and inflammation in Alzheimer's disease and related chronic diseases: a review. Journal of Alzheimer's disease : JAD. 2016;50:301–334. doi: 10.3233/JAD-150765. [DOI] [PubMed] [Google Scholar]
- 140.Zhu Y., Shui X., Liang Z., et al. Gut microbiota metabolites as integral mediators in cardiovascular diseases (review) International journal of molecular medicine. 2020;46(3):936–948. doi: 10.3892/ijmm.2020.4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peng J., Xiao X., Hu M., Zhang X. Interaction between gut microbiome and cardiovascular disease. Life Sciences. 2018;214:153–157. doi: 10.1016/j.lfs.2018.10.063. [DOI] [PubMed] [Google Scholar]
- 142.Jie Z., Xia H., Zhong S. L., et al. The gut microbiome in atherosclerotic cardiovascular disease. Nature communications. 2017;8(1):p. 845. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Karbach S. H., Schönfelder T., Brandão I., et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. Journal of the American Heart Association. 2016;5(9) doi: 10.1161/jaha.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nagatomo Y., Tang W. Intersections between microbiome and heart failure: revisiting the gut hypothesis. Journal of Cardiac Failure. 2015;21:973–980. doi: 10.1016/j.cardfail.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jia Q., Xie Y., Lu C., et al. Endocrine organs of cardiovascular diseases: gut microbiota. Journal of cellular and molecular medicine. 2019;23(4):2314–2323. doi: 10.1111/jcmm.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Robles-Vera I., Toral M., Romero M., et al. Antihypertensive effects of probiotics. Current hypertension reports. 2017;19(4) doi: 10.1007/s11906-017-0723-4. [DOI] [PubMed] [Google Scholar]
- 147.Ma G., Pan B., Chen Y., et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Bioscience reports. 2017;37(2) doi: 10.1042/bsr20160244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhu W., Gregory J. C., Org E., et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yamashita T., Emoto T., Sasaki N., Hirata K. Gut microbiota and coronary artery disease. International Heart Journal. 2016;57:663–671. doi: 10.1536/ihj.16-414. [DOI] [PubMed] [Google Scholar]
- 150.Muralitharan R. R., Marques F. Z. Diet-related gut microbial metabolites and sensing in hypertension. Journal of human hypertension. 2020 doi: 10.1038/s41371-020-0388-3. [DOI] [PubMed] [Google Scholar]
- 151.Daliri E., Lee B., Oh D. Current perspectives on antihypertensive probiotics. Probiotics and antimicrobial proteins. 2017;9:91–101. doi: 10.1007/s12602-016-9241-y. [DOI] [PubMed] [Google Scholar]
- 152.Yang T., Santisteban M. M., Rodriguez V., et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65(6):1331–1340. doi: 10.1161/hypertensionaha.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Duncan S. H., Lobley G. E., Holtrop G., et al. Human colonic microbiota associated with diet, obesity and weight loss. International journal of obesity. 2008;32(11):1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 154.Foye O., Huang I., Chiou C., Walker W., Shi H. Early administration of probiotic Lactobacillus acidophilus and/or prebiotic inulin attenuates pathogen-mediated intestinal inflammation and Smad 7 cell signaling. FEMS Immunology and Medical Microbiology. 2012;65:467–480. doi: 10.1111/j.1574-695X.2012.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marques F. Z., Nelson E., Chu P. Y., et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135(10):964–977. doi: 10.1161/circulationaha.116.024545. [DOI] [PubMed] [Google Scholar]
- 156.Correa F., Buelna-Chontal M., Hernández-Reséndiz S., et al. Curcumin maintains cardiac and mitochondrial function in chronic kidney disease. Free Radical Biology & Medicine. 2013;61:119–129. doi: 10.1016/j.freeradbiomed.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 157.García-Trejo E., Arellano-Buendía A. S., Argüello-García R., et al. Effects of allicin on hypertension and cardiac function in chronic kidney disease. 2016;2016:1–13. doi: 10.1155/2016/3850402.3850402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Boaz M., Smetana S., Weinstein T., et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. The Lancet. 2000;356(9237):1213–1218. doi: 10.1016/S0140-6736(00)02783-5. [DOI] [PubMed] [Google Scholar]
- 159.Kamath N. P., Tandon S., Nayak R., Naidu S., Kamath Y. S. The effect of aloe vera and tea tree oil mouthwashes on the oral health of school children. European Archives of Paediatric Dentistry. 2019;21 doi: 10.1007/s40368-019-00445-5. [DOI] [PubMed] [Google Scholar]
- 160.Elsohly M. A., Waseem G., Bharathi A., Khan I. A. Determination of the anthraquinones aloe-emodin and aloin-A by liquid chromatography with mass spectrometric and diode array detection. Journal of AOAC International. 2019;90(1) [PubMed] [Google Scholar]
- 161.Ji H., Liu Y., He F., An R., Du Z. LC-MS based urinary metabolomics study of the intervention effect of aloe-emodin on hyperlipidemia rats. Journal of Pharmaceutical & Biomedical Analysis. 2018;156:p. 104. doi: 10.1016/j.jpba.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 162.Yu Y., Liu H., Yang D., He F., Du Z. Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacological Research. 2019;146, article 104315 doi: 10.1016/j.phrs.2019.104315. [DOI] [PubMed] [Google Scholar]
- 163.Chen Y., Feng B., Yuan Y., et al. Aloe emodin reduces cardiac inflammation induced by a high-fat diet through the TLR4 signaling pathway. Mediators of Inflammation. 2020;2020:12. doi: 10.1155/2020/6318520.6318520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moreira J. B., Wohlwend M., Fenk S., et al. Exercise reveals proline dehydrogenase as a potential target in heart failure. Progress in cardiovascular diseases. 2019;62:193–202. doi: 10.1016/j.pcad.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 165.Milani R. V., Lavie C. J. The role of exercise training in peripheral arterial disease. Vascular Medicine. 2007;12:351–358. doi: 10.1177/1358863X07083177. [DOI] [PubMed] [Google Scholar]
- 166.Hamburg N. M., Balady G. J. Exercise rehabilitation in peripheral artery disease: functional impact and mechanisms of benefits. Circulation. 2011;123:87–97. doi: 10.1161/CIRCULATIONAHA.109.881888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gliemann L., Nyberg M., Hellsten Y. Nitric oxide and reactive oxygen species in limb vascular function: what is the effect of physical activity? Free Radical Research. 2014;48:71–83. doi: 10.3109/10715762.2013.835045. [DOI] [PubMed] [Google Scholar]