

Abstract
Background:
Muscle-invasive bladder cancer (MIBC) is a molecularly diverse disease with heterogeneous clinical outcomes. Several molecular classifications have been proposed, but the diversity of their subtype sets impedes their clinical application.
Objective:
To achieve an international consensus on MIBC molecular subtypes that reconciles the published classification schemes.
Design, setting, and participants:
We used 1750 MIBC transcriptomic profiles from 16 published datasets and two additional cohorts.
Outcome measurements and statistical analysis:
We performed a network-based analysis of six independent MIBC classification systems to identify a consensus set of molecular classes. Association with survival was assessed using multivariable Cox models.
Results and limitations:
We report the results of an international effort to reach a consensus on MIBC molecular subtypes. We identified a consensus set of six molecular classes: luminal papillary (24%), luminal nonspecified (8%), luminal unstable (15%), stroma-rich (15%), basal/squamous (35%), and neuroendocrine-like (3%). These consensus classes differ regarding underlying oncogenic mechanisms, infiltration by immune and stromal cells, and histological and clinical characteristics, including outcomes. We provide a single-sample classifier that assigns a consensus class label to a tumor sample’s transcriptome. Limitations of the work are retrospective clinical data collection and a lack of complete information regarding patient treatment.
Conclusions:
This consensus system offers a robust framework that will enable testing and validation of predictive biomarkers in future prospective clinical trials.
Patient summary:
Bladder cancers are heterogeneous at the molecular level, and scientists have proposed several classifications into sets of molecular classes. While these classifications may be useful to stratify patients for prognosis or response to treatment, a consensus classification would facilitate the clinical use of molecular classes. Conducted by multidisciplinary expert teams in the field, this study proposes such a consensus and provides a tool for applying the consensus classification in the clinical setting.
Keywords: Consensus, Molecular taxonomy, Muscle-invasive bladder cancer, Transcriptomic classifier
1. Introduction
Bladder cancer is one of the most frequently diagnosed cancers in North America and Europe. Most bladder cancers are urothelial carcinomas, and are classified as either non-muscle-invasive bladder cancer (NMIBC) or muscle-invasive bladder cancer (MIBC), due to distinct implications for patient management. MIBC is usually diagnosed de novo but may arise from the 10–20% of NMIBC cases that eventually progress. MIBC is a more aggressive disease state, and is associated with a 5-yr survival rate of 60% for patients with localized disease and <10% for patients with distant metastases.
At the molecular level, MIBC is a heterogeneous disease that is characterized by genomic instability and a high mutation rate. Transcriptome profiling facilitates bladder cancer classification into molecular subtypes, for a more precise patient stratification according to prognosis and therapeutic options. A number of teams have reported molecular classifications of bladder cancers. Several expression-based schemes have been proposed, either considering the full spectrum of nonmetastatic bladder cancers [1–6], or focusing separately either on NMIBC [7,8] or on MIBC [9–16]. These classifications have considerably advanced our understanding of bladder cancer biology. Specific genomic alterations are enriched in particular molecular subtypes, including mutations targeting genes involved in cell cycle regulation, chromatin remodeling, and receptor tyrosine kinase signaling. Importantly, several reports have highlighted the clinical significance of molecular stratification of MIBC, by suggesting that responses to chemotherapy and immunotherapy may be enriched in specific MIBC subtypes [12,17–19].
Published MIBC classifications were derived from largely nonoverlapping datasets, using different `s at least for some steps of their respective unsupervised class discovery pipelines (Supplementary Table 1). Nonetheless, they share many characteristics, including subtype-specific molecular features, and a strong overlap has been observed between some subtypes from distinct classification systems [20]. In an initial effort to define features common to all MIBC classifications, Lerner et al. [21] proposed a consensus basal/squamous subtype and reported evidence of a muscle-invasive subtype with urothelial differentiation features. However, the six published classification systems that were considered in their work still differ in the number and relative size of subtypes, and in the use of different subtype names. This diversity has impeded transferring subtypes into clinical practice and highlights that establishing a single consensus set of molecular subtypes would facilitate achieving such a transfer.
2. Patients and methods
2.1. Transcriptomic profiles analyzed
We used 1750 MIBC transcriptomic profiles from 18 datasets to compare six molecular classification schemes and derive a consensus classification. Details of datasets, including their respective normalizations, are given in Supplementary Table 2.
2.2. Consensus classification construction
Transcriptomic classifiers for six published classification systems [9–13,16] were provided and/or validated by the respective teams. These classifiers were merged into an R package (R Foundation for Statistical Computing, Vienna, Austria) that is documented and freely available at https://github.com/cit-bioinfo/BLCAsubtyping.
We applied these classifiers on each of the 18 datasets independently to assign each sample to a subtype in each of the six classification systems. We used a previously validated network-based approach [22] on these subtyping results to identify consensus classes that reconcile the molecular subtypes from the six classification schemes. Briefly, we built a weighted network of subtyping results, using Cohen’s kappa metric to quantify similarities between subtypes from different classification systems, and applied a Markov cluster algorithm to identify robust network substructures corresponding to potential consensus classes. The analysis workflow is summarized in Supplementary Fig. 1 and algorithm details are given in the Supplementary material (Methods). We used a silhouette-based metric to select the most robust consensus solution among those with consensus classes defined by at least three of the six input classification systems.
2.3. Single-sample transcriptomic consensus classifier construction
The network of consensus classes also revealed a core set of consensus samples (see Supplementary material, Methods), that is, tumor samples representative of each consensus class on the basis of their initial subtyping by the six classification systems. We used these core samples (n = 1084) to build a single-sample transcriptomic classifier, as detailed in the Supplementary material (Methods). The classifier was trained on approximately one-third of these samples (n = 403) and achieved 97% mean balanced accuracy on the remaining two-thirds of the core samples (n = 681). This classification tool was implemented as an R package that is documented and freely available at https://github.com/cit-bioinfo/consensusMIBC.
We also offer a stand-alone web application that allows users to classify new samples using the single-sample classifier. By design, it does not store any user data and can be used completely anonymously. The consensusMIBC web application is available at http://cit.ligue-cancer.net:3838/apps/consensusMIBC_web/
2.4. Statistical analyses
We measured associations between consensus classes and categorical variables by Fisher’s exact tests, with Monte-Carlo simulations when necessary. For continuous variables, we evaluated differences by Kruskal-Wallis tests or LIMMA moderated t tests (limma v3.39.1 R package). False discovery rate adjustment of p values was performed to control for multiple testing, for association tests between the consensus classes and either genetic or histological variables, as these types of variables might be interpreted as potential diagnostic or theranostic biomarkers of some consensus classes. We reported unadjusted p values otherwise.
We built a multivariable Cox model integrating consensus classes and clinical risk factors. We used Wald tests to assess survival differences associated with different levels of a given factor included in the Cox model. For each factor level, we computed hazard ratios (HRs) and 95% confidence intervals (CIs). We constructed Kaplan-Meier curves to visualize overall survival stratified by consensus class and used log-rank tests to compare the survival of corresponding patient groups.
All statistical and bioinformatics analyses were performed with R (v3.5.1).
3. Results
3.1. Published molecular classifications of MIBC converge on six classes
We used six published MIBC molecular classifications to define a unified consensus subtyping system, following the approach outlined in Supplementary Fig. 1 and described in the Supplementary material (Methods). We refer to these input classifications as Baylor [16], University of North Carolina (UNC) [10], MD Anderson Cancer Center (MDA) [12], the Cancer Genome Atlas (TCGA) [9], Cartes d’Identité des Tumeurs (CIT)-Curie [11], and Lund [13]. The methods that defined these classifications are summarized in the Supplementary material (Methods) and Supplementary Table 1.
Our analysis converged on six biologically relevant consensus molecular classes, which we labeled as luminal papillary (LumP), luminal nonspecified (LumNS), luminal unstable (LumU), stroma-rich, basal/squamous (Ba/Sq), and neuroendocrine-like (NE-like; Fig. 1). Considerations motivating our choices for these consensus names are detailed in the Supplementary material (Note).
Fig. 1 –
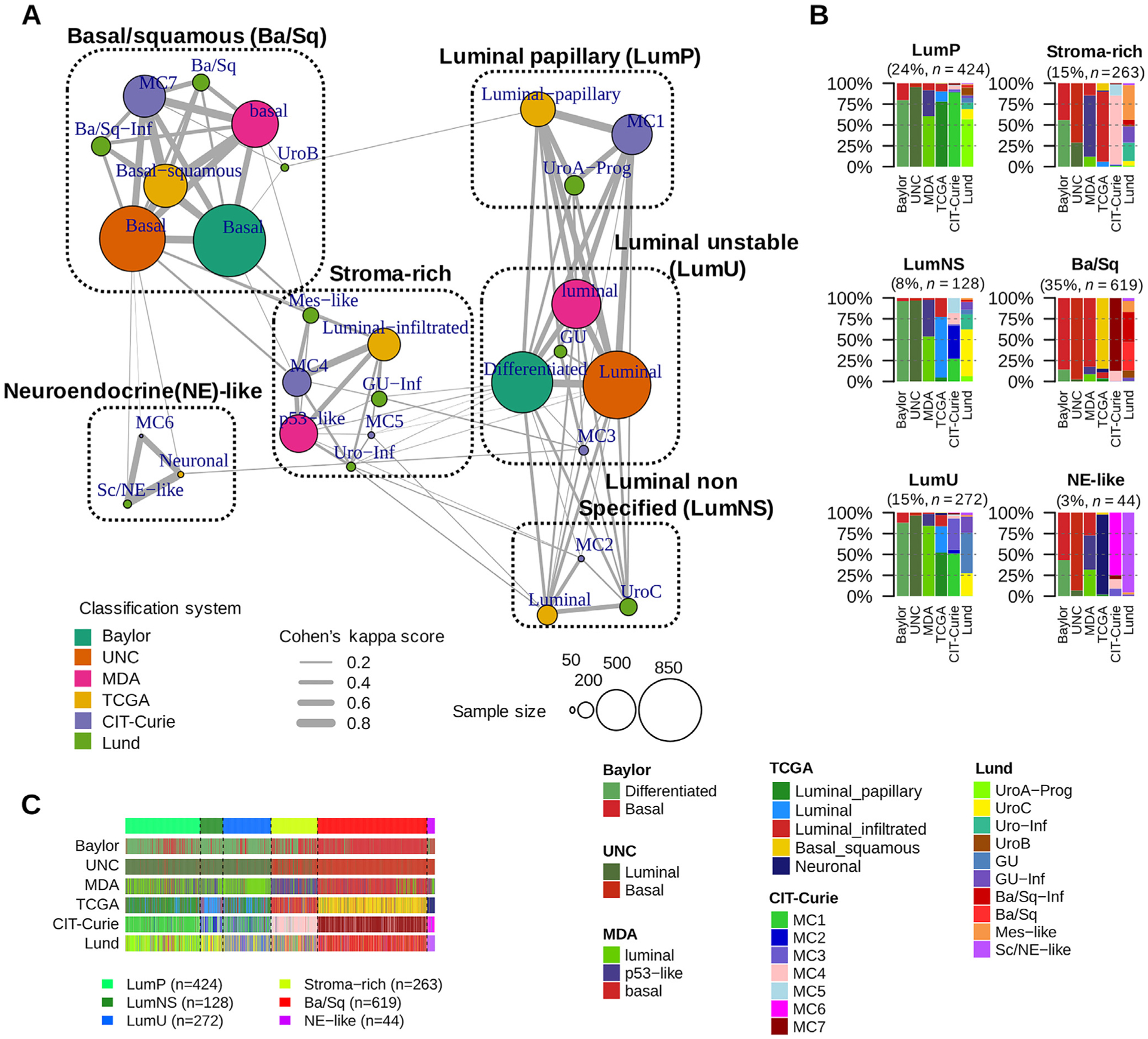
The six consensus classes and their relationships to input molecular subtypes. (A) MCL-clustered network. The six-consensus class solution obtained with MCL clustering on the Cohen’s kappa-weighted network is represented by the six cliques surrounded by black dotted rectangles (see the Supplementary material [Note] for the naming of consensus classes). The circles inside each clique symbolize the input subtypes associated with each consensus class and are colored according to their matching classification system. Circle size is proportional to the number of samples assigned to the subtype. Edge width between subtypes is proportional to the Cohen’s kappa score, which assesses the level of agreement between two classification schemes. (B) Input subtypes repartitioned among each consensus class. Consensus classes were predicted on 1750 MIBC samples using the single-sample classifier described in the Supplementary material (Methods). Here, the samples are grouped by their predicted consensus class labels: LumP, LumNS, LumU, stroma-rich, Ba/Sq, and neuroendocrine (NE)-like. For each consensus class, a bar plot shows the proportion of samples assigned in each input subtype of each input classification system. See also Supplementary Fig. 2 for additional visualization of consensus class distributions across input subtypes and across datasets. (C) Relationship between subtyping results from the six input classification schemes. Samples are ordered by predicted consensus classes. Ba/Sq = basal/squamous; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; MCL = Markov cluster algorithm; MDA = MD Anderson Cancer Center; MIBC = muscle-invasive bladder cancer; TCGA = the Cancer Genome Atlas; UNC = University of North Carolina.
The six molecular classes had variable sample sizes, with Ba/Sq and LumP being the largest (35% and 24% of all samples, respectively). The remaining 41% of samples were LumU (15%), stroma-rich (15%), LumNS (8%), and NE-like (3%) tumors (Fig. 1B). The consensus classification was strongly associated with each of the initial classification systems (Fisher simulated p < 0.001; Fig.1 and Supplementary Fig. 2A).
We compared the consensus classes with the TCGA PanCancer Atlas integrative classification [23] (Supplementary Fig. 2C). We observed associations between the Ba/Sq consensus class and the squamous cell carcinoma C27:Pan-SCC pan-cancer cluster (p < 0.001), and between the stroma-rich class and the stroma-driven C20:Mixed (stromal/immune) pan-cancer cluster (p < 0.001).
3.2. Transcriptomic characterization of the six consensus molecular classes
We used mRNA data from all 1750 samples to characterize consensus classes with molecular gene signatures for bladder cancer pathways and for tumor microenvironment infiltration (Fig. 2A and B, and Supplementary Table 3).
Fig. 2 –
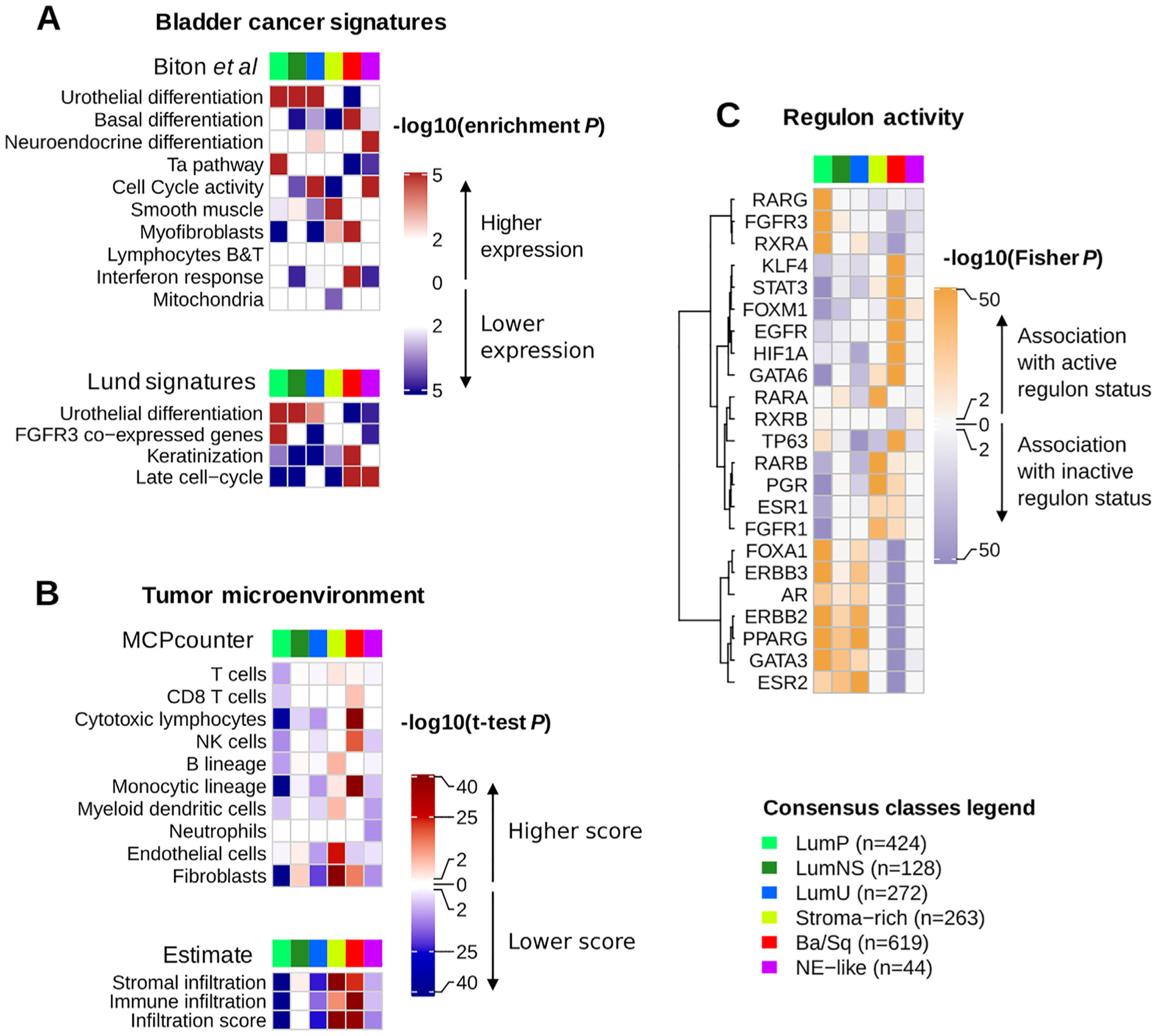
Characterization of tumor and stroma signals using published mRNA signatures and regulon analysis. Descriptions of gene sets and detailed statistics are available in Supplementary Table 3. (A) We performed a gene set analysis (GSA; see the Supplementary material, Methods) in each dataset to test the significance of differential expression of specific bladder cancer-related signatures in each consensus class compared with the others. The heatmaps show Stouffer combined GSA p values over all datasets. The upper panel refers to bladder cancer gene sets extracted from the ICA components described in the study by Biton et al. [25] (see the Supplementary material, Methods). The lower panel displays other bladder cancer-specific signatures retrieved from the literature: urothelial differentiation, keratinization, and late cell-cycle signatures from the study of Eriksson et al. [24], and an FGFR3 coexpressed signature from the study of Sjödahl et al. [4]. (B) We used two mRNA-based computational tools to characterize tumor microenvironments : ESTIMATE (R package, v1.1.0) infers the presence of stromal cells (stromal infiltration) and the infiltration of immune cells (immune infiltration) in a tumor sample using two curated gene signatures described by Yoshihara et al. [26]; MCPcounter (R package, v1.0.13) uses biologically validated transcriptomic markers of specific immune and stromal cell subpopulations to quantify the presence of these populations in a tumor sample [27]. We ran MCPcounter and ESTIMATE independently on each dataset, and used t tests to compare scores for each consensus class relative to the others. The heatmaps show Stouffer combined t test p values over all datasets. (C) We computed discrete regulon status (1 for active regulon status, 0 for undefined status, and −1 for inactive regulon status) in each dataset, as described in the Supplementary material (Methods) and in the work of Robertson et al. [9]. We evaluated the association between each regulon status and each consensus class using Fisher exact tests; the heatmap illustrates the resulting p values.
Ba/Sq = basal/squamous; ICA = independent component analysis; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; NE = neuroendocrine; NK = natural killer.
Differentiation-related mRNA signatures were strongly associated with the consensus classes. Tumors from the three luminal classes overexpressed urothelial differentiation signatures (p < 0.001), including the PPARG/GATA3/FOXA1-related Lund signature [24]. In contrast, Ba/Sq and NE-like tumors, respectively, overexpressed gene signatures associated with basal (p < 0.001) and neuroendocrine differentiation (p < 0.001), respectively.
In addition to their urothelial differentiation status, the three luminal classes exhibited distinct molecular signatures. LumP tumors were characterized by high expression of a noninvasive Ta pathway signature [25] (p < 0.001) and were strongly associated with FGFR3 transcriptional activity, as estimated by an FGFR3 coexpressed gene signature [4] (p < 0.001). LumNS tumors displayed elevated stromal infiltration signatures, mainly fibroblastic, compared with the other luminal tumors (p < 0.001). LumU tumors had a higher cell cycle activity than the other luminal tumors (p < 0.001).
Stroma-rich samples displayed intermediate levels of urothelial differentiation. They were mainly characterized by stromal infiltration as summarized by ESTIMATE stromal scores [26], with overexpression of smooth muscle (p < 0.001), endothelial (p < 0.001), fibroblast (p < 0.001), and myofibroblast (p < 0.001) gene signatures.
Immune infiltration was mainly found within Ba/Sq and stroma-rich tumors, but these two classes were associated with distinct immune cell populations, as estimated by MCPcounter signatures [27]. Ba/Sq tumors were enriched in cytotoxic lymphocytes (p < 0.001) and natural killer cells (p < 0.001), whereas stroma-rich tumors overexpressed T-(p < 0.001) and B-cell (p < 0.001) markers. LumNS tumors were the only luminal type associated with immune infiltration signals; these were mainly for B (p = 0.002) and T (p = 0.004) lymphocytes. We detected no transcriptomic markers of immune infiltration in NE-like tumors. In TCGA samples, an estimation of tumor purity with ABSOLUTE [28] confirmed that stroma-rich and Ba/Sq tumors contained higher levels of nontumor cells (Supplementary Fig. 3).
Analyses of regulatory units (ie, regulons) for 23 regulator genes previously reported as associated with bladder cancer [9] were consistent with the assessed mRNA signatures (Fig. 2C). Luminal tumors, which overexpressed strong urothelial differentiation signals, were associated with active PPARG and GATA3 regulons (p < 0.001 for both comparisons). FGFR3 regulon activity was specifically associated with LumP tumors (p < 0.001), and Ba/Sq tumors showed a strong association with STAT3 regulon activity (p < 0.001), consistent with previous results [16,24]. Additionally, a regulon analysis showed an association of HIF1A regulon activity with Ba/Sq tumors (p < 0.001), suggesting that this class is associated with a hypoxic microenvironment. EGFR regulon activity was specifically associated with Ba/Sq tumors (p < 0.001), consistent with previously reported findings [11].
3.3. Genomic alterations associated with the consensus molecular classes
We used TCGA exome data to identify class-specific mutations (Fig. 3A and Supplementary Table 4) and combined 600 available copy number profiles, grouped by consensus class, to identify class-specific copy number aberrations (CNAs; Supplementary Table 5). In addition, we combined all CNA, gene fusion, and gene mutation data from the 18 cohorts to generate comprehensive profiles of genomic alterations for seven key bladder cancer genes (FGFR3, CDKN2A, PPARG, ERBB2, E2F3, TP53, and RB1) for each consensus class (Fig. 3B).
Fig. 3 –
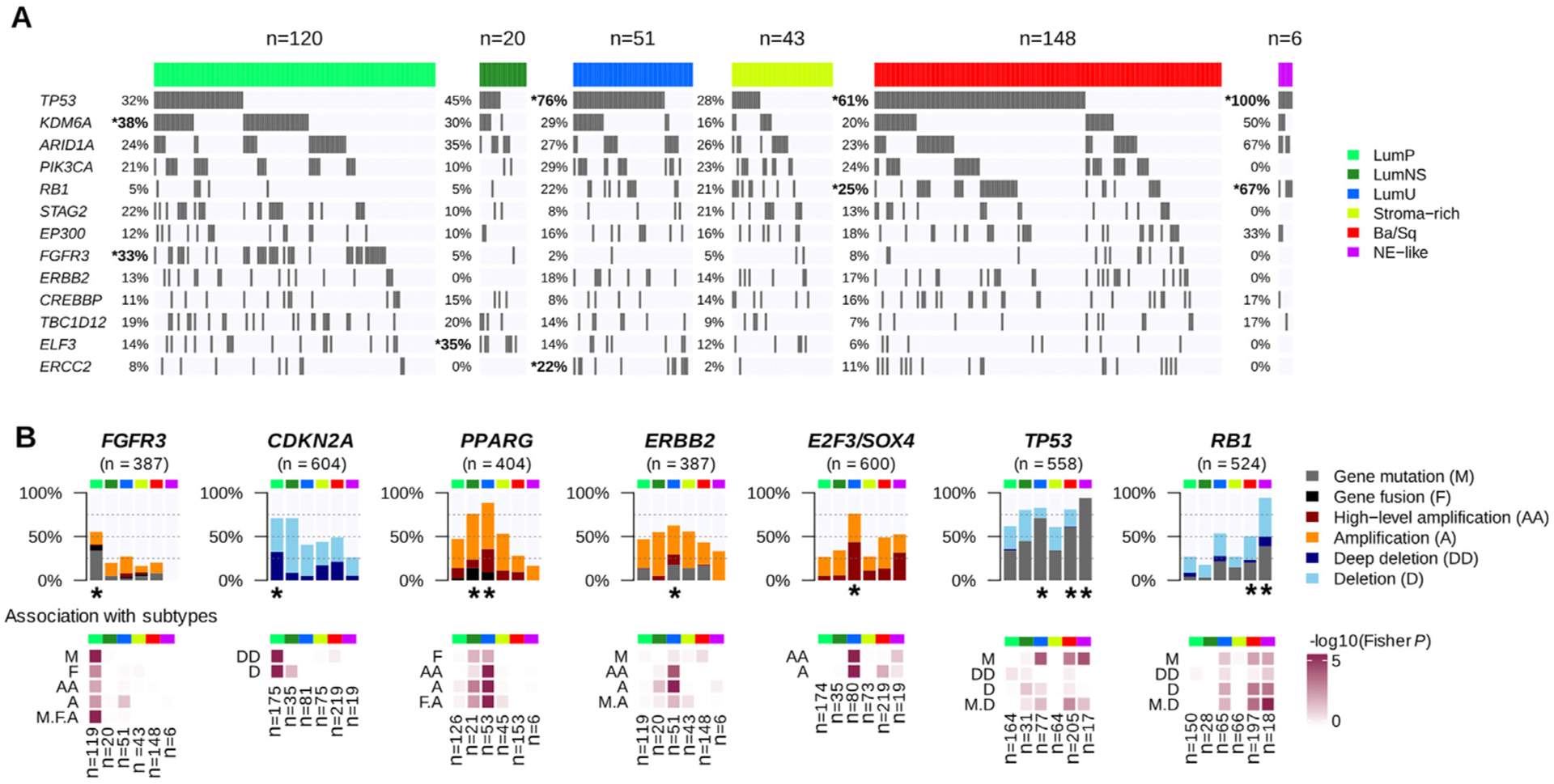
Genomic alterations associated with consensus classes. (A) We used the available exome data from 388 TCGA MIBC samples to study the association between consensus classes and specific gene mutations (see Supplementary Table 4 and Supplementary Fig. 3). The panel displays the 13 genes with MutSig q values <0.02 found in >10% of all tumors. Gene mutations that were significantly enriched in one consensus class are marked by an asterisk. (B) Combined genomic alterations associated with seven bladder cancer-associated genes and statistical association with consensus classes. Upper panels: main alteration types after aggregating CNA profiles (see Supplementary Table 5) from CIT (n = 87), Iyer (n = 58), Sjödahl (n = 29), Stransky (n = 22), and TCGA (n = 404) data; exome profiles (n = 388) and FGFR3 and PPARG fusion data (n = 404) from TCGA data; CDKN2A and RB1 MLPA data from CIT (n = 86 and n = 85, respectively) and Stransky (n = 16 and n = 13, respectively) data; FGFR3 mutation data from MDA (n = 66), CIT (n = 87), Iyer (n = 39), Sjödahl (n = 28), and Stransky (n = 35); TP53 mutation data from MDA (n = 66), CIT (n = 87), Iyer (n = 39), Sjödahl (n = 28), and Stransky (n = 19); and RB1 mutation data from MDA (n = 66), CIT (n = 85), Iyer (n = 39), and Stransky (n = 13). Lower panels: associations between each consensus class, each type of gene alteration, and the combined alterations were evaluated by Fisher’s exact test. Consensus classes significantly enriched with alterations of these candidate genes are marked with a black asterisk.
Ba/Sq = basal/squamous; CIT = Cartes d’Identité des Tumeurs; CNA = copy number aberration; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; MDA = MD Anderson Cancer Center; MIBC = muscle-invasive bladder cancer; NE = neuroendocrine; TCGA = the Cancer Genome Atlas.
LumP tumors were mainly enriched in FGFR3 mutations, with 33% of FGFR3-mutated LumP tumors in the TCGA cohort (p-adjusted <0.001) and summing to 40% by adding FGFR3-targeted sequencing mutation data (n = 255) from additional cohorts (p-adjusted <0.001). LumP tumors also harbored more frequent mutations of KDM6A (38%, p-adjusted = 0.013). Assembling mutations, fusions, and copy number amplifications, FGFR3 genomic alterations were enriched in LumP tumors (55%, p < 0.001). CDKN2A multiplex ligation-dependent probe amplification (MLPA) from 102 tumors and CNA data for 502 tumors revealed homozygous/deep deletions of CDKN2A in 33% of LumP tumors, which was significantly higher than in other classes (p < 0.001). These deletions were consistent with the enrichment of LumP tumors within the TCGA pan-cancer iCluster C7:Mixed (Chr9 del) (p < 0.001), characterized by chromosome 9 deletions (Supplementary Fig. 2).
The LumNS class was mainly characterized by enrichment of mutations in ELF3 (35%, p-adjusted = 0.026), which is an early regulator of urothelial differentiation and is activated by PPARG [29]. PPARG was significantly altered as well, with 76% of LumNS tumors harboring either amplifications or fusions (p = 0.006).
LumU tumors also harbored frequent PPARG alterations (89%, p < 0.001) and high-level amplifications of a 6p22.3 region that contains E2F3 and SOX4 (76%, p < 0.001). ERBB2 amplifications were over-represented in LumU tumors (39%, p < 0.001), but no significant association was found between ERBB2 mutations and any of the consensus classes. In contrast with the other luminal classes, LumU tumors were associated with mutations in TP53 (76%, p-adjusted <0.001) and in ERCC2, which codes for a core nucleotide-excision repair component (22%, p-adjusted = 0.039). More generally, LumU was the most genomically altered class (Supplementary Fig. 3), displaying the highest number of CNAs (p < 0.001) and the highest somatic mutation load (p = 0.009), and including more APOBEC-induced mutations than the other consensus classes (p = 0.01). These features of genomic instability and the association with ERBB2 amplifications were consistent with the enrichment of LumU tumors within the TCGA pan-cancer subtypes C2:BRCA (HER2 amp) (breast tumors characterized by frequent ERBB2 amplifications, p < 0.001) and C13:Mixed (Chr8 del) (enriched in highly aneuploid tumors, p < 0.001, as shown in Supplementary Fig. 2) [23].
For Ba/Sq tumors, as shown previously [30], the most frequently mutated genes based on TCGA exome data were TP53 (61%, p-adjusted = 0.002) and RB1 (25%, p-adjusted = 0.012). Aggregated mutation data, including targeted sequencing data from other cohorts, revealed that 58% (134/232, p = 0.009) and 20% (43/224, p = 0.007) of Ba/Sq tumors contained mutations in TP53 and RB1, respectively; these mutations co-occurred in 14% (32/224) of Ba/Sq cases. Ba/Sq tumors were also strongly associated with genomic deletions of 3p14.2, which occurred in 49% of cases (p = 0.001).
Combining all available data on genomic alterations of TP53 and RB1, we observed strong enrichment of concurrent TP53 and RB1 inactivation in NE-like tumors. TP53 was ubiquitously mutated in these tumors (94%, p-adjusted = 0.030), and co-occurred with RB1 alteration by either mutations or deletions (94%, p-adjusted = 0.029).
3.4. Histological patterns associated with the consensus molecular classes
To characterize the consensus molecular classes histologically, we assembled annotations for urothelial cancer histological variants and specific morphological patterns (Fig. 4 and Supplementary Fig. 4). As expected, Ba/Sq tumors included 79% of tumors in which histological review identified squamous differentiation (126/159, p-adjusted <0.001). However, the Ba/Sq class extended beyond this histological subtype, with only 42% (126/303) of Ba/Sq tumors being associated with squamous differentiation identified by pathologists. Similarly, NE-like tumors were strongly associated with neuroendocrine variant histology, with 72% of histologically reviewed NE-like tumors showing neuroendocrine differentiation (13/18, p-adjusted <0.001), which accounts for 81% of all tumors with such differentiation. LumP tumors were enriched with papillary morphology as compared with other consensus classes (p-adjusted = 0.002). This pattern was observed in 59% (82/139) of histologically reviewed LumP tumors, although it was frequently found in other luminal classes (42% in LumNS and 31% in LumU). LumNS tumors were enriched in micropapillary variant histology (36%, 9/25, p-adjusted = 0.032) and were commonly associated with carcinoma in situ (80%, 4/5, p = 0.005).
Fig. 4 –
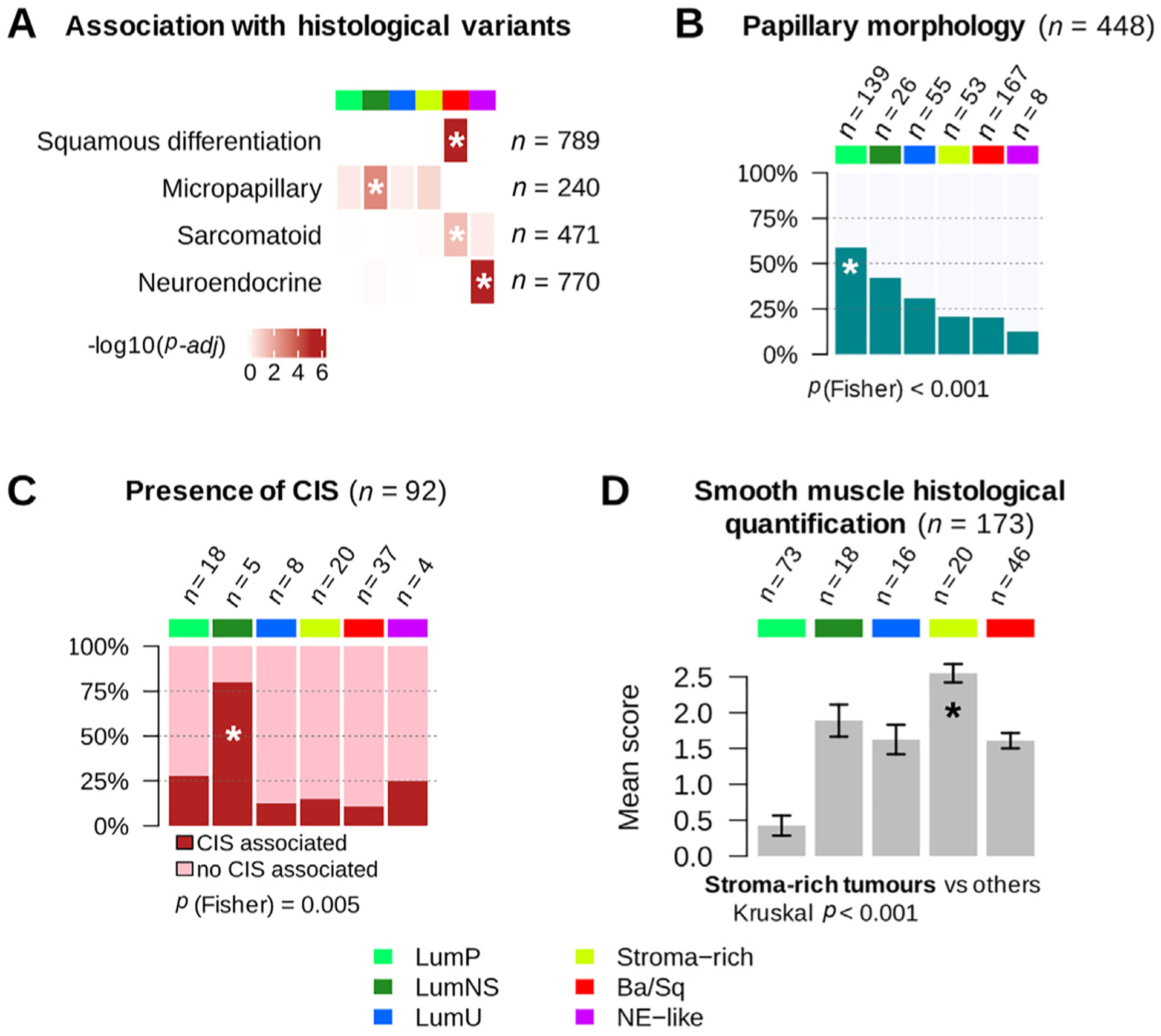
Histopathological associations with consensus classes. (A) Histological variant over-representation within each consensus class. One-sided Fisher exact tests were performed for each class and histological pattern; asterisks indicate a significant association between a consensus class and a histological feature (p < 0.05). Pathological review of histological variants was available for several cohorts: squamous differentiation was evaluated in CIT (n = 75), MDA (n = 46), Sjödahl2012 (n = 23), Sjödahl2017 (n = 239), and TCGA (n = 406) cohorts; neuroendocrine variants were reviewed in CIT (n = 75), MDA (n = 46), Sjödahl2017 (n = 243), and TCGA (n = 406) cohorts; micropapillary variants were reviewed in CIT (n = 75), MDA (n = 46), and TCGA cohorts (n = 118 FFPE tumor slides from TCGA were reviewed by Y.A. and J.F. for this study). Results are displayed on the heatmap as −log10(adj Fisher’s p). Detailed sample counts within each class are given in Supplementary Fig. 4. (B) Occurrence of papillary morphology in tumors from the TCGA cohort (n = 401) and the CIT cohort (n = 47). (C) Proportion of samples with associated CIS within each consensus class in tumors from the CIT cohort (n = 84) and the Dyrskjøt cohort (n = 8). (D) Smooth muscle infiltration from images for 173 tumor slides from the TCGA cohort. Each sample was assigned a semiquantitative score ranging from 0 to 3 (0 = absent, 1 = low, 2 = moderate, and 3 = high) to quantify the presence of large smooth muscle bundles. The bar plot shows means and standard errors for each class.
Ba/Sq = basal/squamous; CIS = carcinoma in situ; CIT = Cartes d’Identité des Tumeurs; FFPE = formalin-fixed paraffin-embedded; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; MDA = MD Anderson Cancer Center; NE = neuroendocrine; TCGA = the Cancer Genome Atlas.
A pathological review of stromal infiltration in slide images corresponding to the TCGA tumor samples confirmed that stroma-rich tumors contained a higher proportion of smooth muscle cells (p < 0.001), consistent with the strong smooth muscle-related mRNA expression characterizing these tumors.
3.5. Association of the consensus molecular classes with clinical characteristics, survival outcomes, and therapeutic opportunities
The consensus classes were associated with gender, stage, and age (Fig. 5A). Ba/Sq tumors were over-represented in females (p < 0.001) and in higher clinical stages (p < 0.001), consistent with published results [4,9–11]. The LumP and LumU consensus classes were enriched in T2 versus T3–4 tumors (p = 0.009 and p < 0.001, respectively) as compared with other classes. Younger patients (<60 yr) were over-represented among LumP tumors (p = 0.001), whereas the LumNS consensus class was enriched with older patients (>80 yr; p = 0.03).
Fig. 5 –
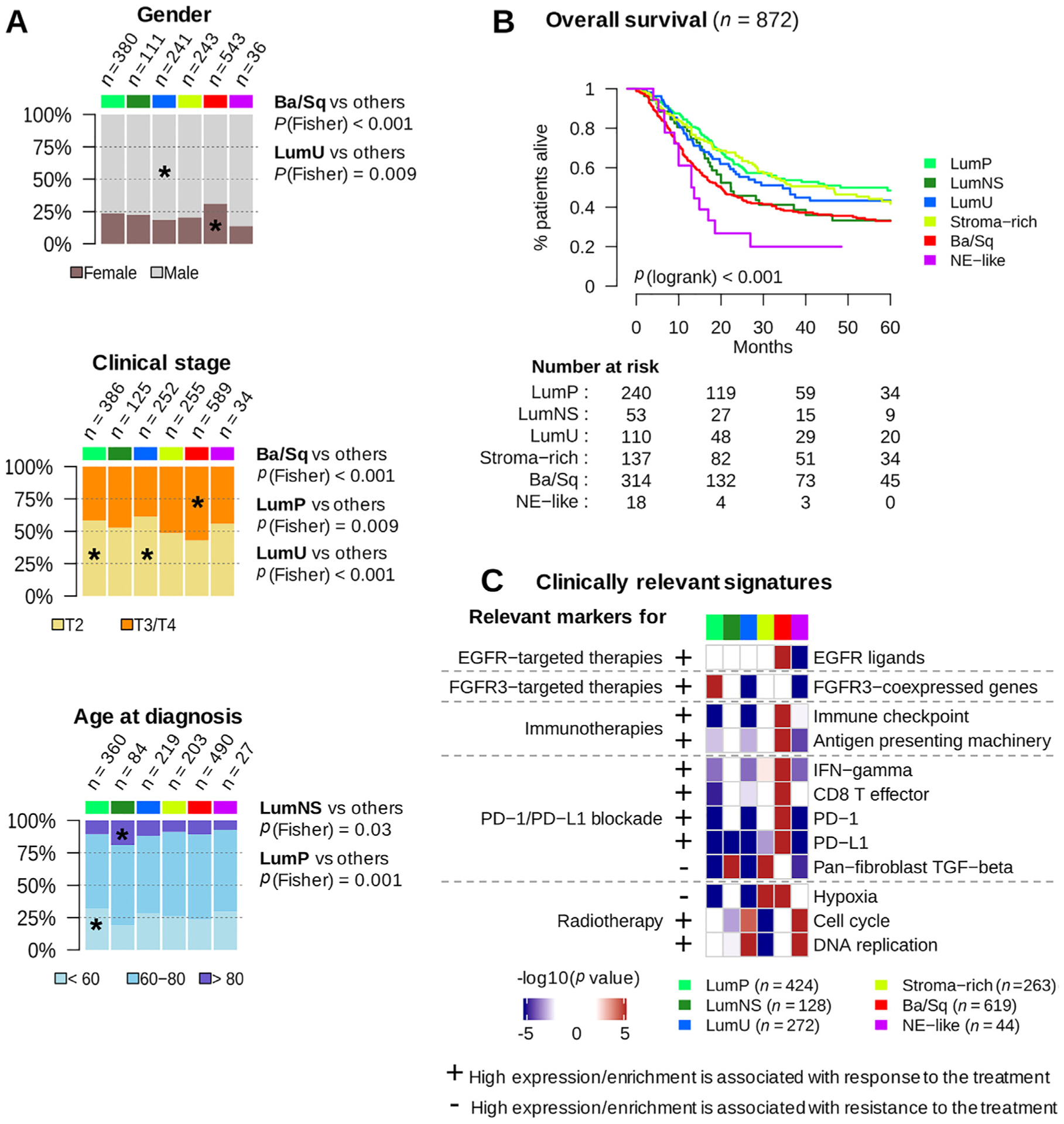
Clinical characteristics and prognostic associations. (A) Association of consensus classes with gender (n = 1554), clinical stage (n = 1641), and age category (n = 1383). (B) Five-year overall survival stratified by consensus class (see also Supplementary Fig. 5). Kaplan-Meier curves were generated from 872 patients with available follow-up data. Patients who had received neoadjuvant chemotherapy were excluded from the survival analysis. Detailed statistics of the multivariable survival analyses is given in Supplementary Table 6. (C) We selected a set of clinically relevant gene signatures (see Supplementary Table 7) and performed a gene set analysis (see the Supplementary material, Methods) in each dataset to test the significance of their differential expression in each consensus class relative to the others. We used one-sided t tests to assess the differential expression of single genes (PD-1 and PD-L1). The heatmaps show Stouffer combined p values over all datasets. Plus/minus annotation of gene sets indicates association of high gene expression levels with response/resistance to the corresponding therapy.
Ba/Sq = basal/squamous; EGFR = epithelial growth factor receptor; FGFR = fibroblast growth factor receptor; IFN = interferon; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; NE = neuroendocrine; TGF = transforming growth factor.
Overall survival was strongly associated with the consensus classes (Fig. 5B, p < 0.001). The association of consensus classes to survival was evaluated in a multivariable Cox model considering tumor stage, node, metastasis, and patient age as covariates (p < 0.001; Supplementary Table 6). The LumP class was used as the reference for class-based survival. Patients with stroma-rich tumors had similar outcomes to patients with LumP tumors (HRStroma-rich/LumP = 0.98, IC95 = [0.65, 1.49]), and their survival was independent of the differentiation status of the tumor sample (Supplementary Fig. 5). Likewise, patients with LumNS tumors had similar prognosis to patients with LumP tumors in this multivariable model, which considers patient age (HRLumNS/LumP = 1.07; CI = [0.63, 1.82]).
The LumU class was associated with poorer patient prognosis (HRLumU/LumP = 1.49, IC = [0.93, 2.39]), although in this setting the difference was modest and not significant.
Ba/Sq tumors were associated with a poor prognosis (HRBaSq/LumP = 1.83, IC = [1.30, 2.58], p < 0.001), consistent with previous studies [11]. Finally, NE-like tumors were associated with the worst prognosis (HRNE-like/LumP = 2.34, IC = [1.09, 5.05], p < 0.03).
We characterized the consensus classes using several clinically relevant mRNA signatures (Fig. 5C and Supplementary Table 7). The FGFR3 signature was strongly and specifically activated in LumP tumors (p < 0.001), suggesting that FGFR3-targeted therapies warrant investigation in patients with tumors of this consensus class. Ba/Sq tumors expressed high levels of EGFR and its ligands (p < 0.001), which may be associated with sensitivity to EGFR-targeted therapies, as suggested by previously reported in vitro and in vivo experiments [11]. Ba/Sq tumors also strongly expressed immune checkpoint markers (p < 0.001) and antigen-presenting machinery genes (p < 0.001), suggesting that such tumors might be more responsive to immunotherapies. Studies integrating mRNA signatures with data on response to anti-PD1/PD-L1 therapies [19,31] have reported associations of anti-PD1/PD-L1 response with high levels of CD8 + T cells, high interferon gamma signals, and low activity of the transforming growth factor-beta pathway. However, considering this combination of factors, no consensus class had an expression profile that clearly suggested either response or resistance to anti-PD1/PD-L1 therapies. In contrast, NE-like and LumU tumors both had profiles associated with a potential response to radiotherapy [32,33], showing elevated cell cycle activity (pNE-like < 0.001 and pLumU < 0.001) and low hypoxia signals (pNE-like = 0.01 and pLumU < 0.001).
Finally, we performed a consensus class-based retrospective analysis of outcomes of patients receiving neoadjuvant chemotherapy (NAC) [12,18] and patients treated with the anti-PD-L1 antibody atezolizumab (IMvigor210) [19] (Supplementary Fig. 6). While outcome was associated with the consensus class for NAC-free patients (Fig. 5B), for NAC-treated patients, we observed no significant association of outcome with the consensus class. Despite this, comparison of survival curves with or without NAC treatment (Supplementary Fig. 6A and Fig. 5B) suggested that patients with Ba/Sq or LumNS tumors may benefit from NAC, while patients with a stroma-rich tumor may not. We observed an enrichment in atezolizumab responders among patients with LumNS (p = 0.05), LumU (p = 0.0044), and NE-like (p = 0.012) tumors. In particular, NE-like tumors may respond to immune checkpoint inhibitors, as suggested by recent results [34].
4. Discussion
While precision genomic medicine promises to transform clinical practice, the diversity of published MIBC classifications has delayed transferring of subtypes into both clinical trials and standard management of bladder cancer patients. In the present study, we generated a stable consensus classification system from existing well-documented molecular subtyping systems, following a procedure similar to that used to identify consensus subtypes in colorectal cancer [22]. The four consensus molecular subtypes identified in that work have helped frame the development of colorectal cancer precision medicine and are now being evaluated in clinical trials [35,36].
Here, we analyzed the relationships among six published classification systems, based on 1750 MIBC transcriptomic profiles. We identified six consensus MIBC molecular classes that reconcile all six classification schemes: LumP, LumU, stroma-rich, LumNS, Ba/Sq, and NE-like. Each consensus class has distinct differentiation patterns, oncogenic mechanisms, tumor microenvironments, and histological and clinical associations, which are summarized in Fig. 6. To facilitate translating the consensus classes to research and clinical settings, we provide the community with an R-based single-sample classifier that assigns a consensus class label to a tumor sample’s transcriptome.
Fig. 6 –
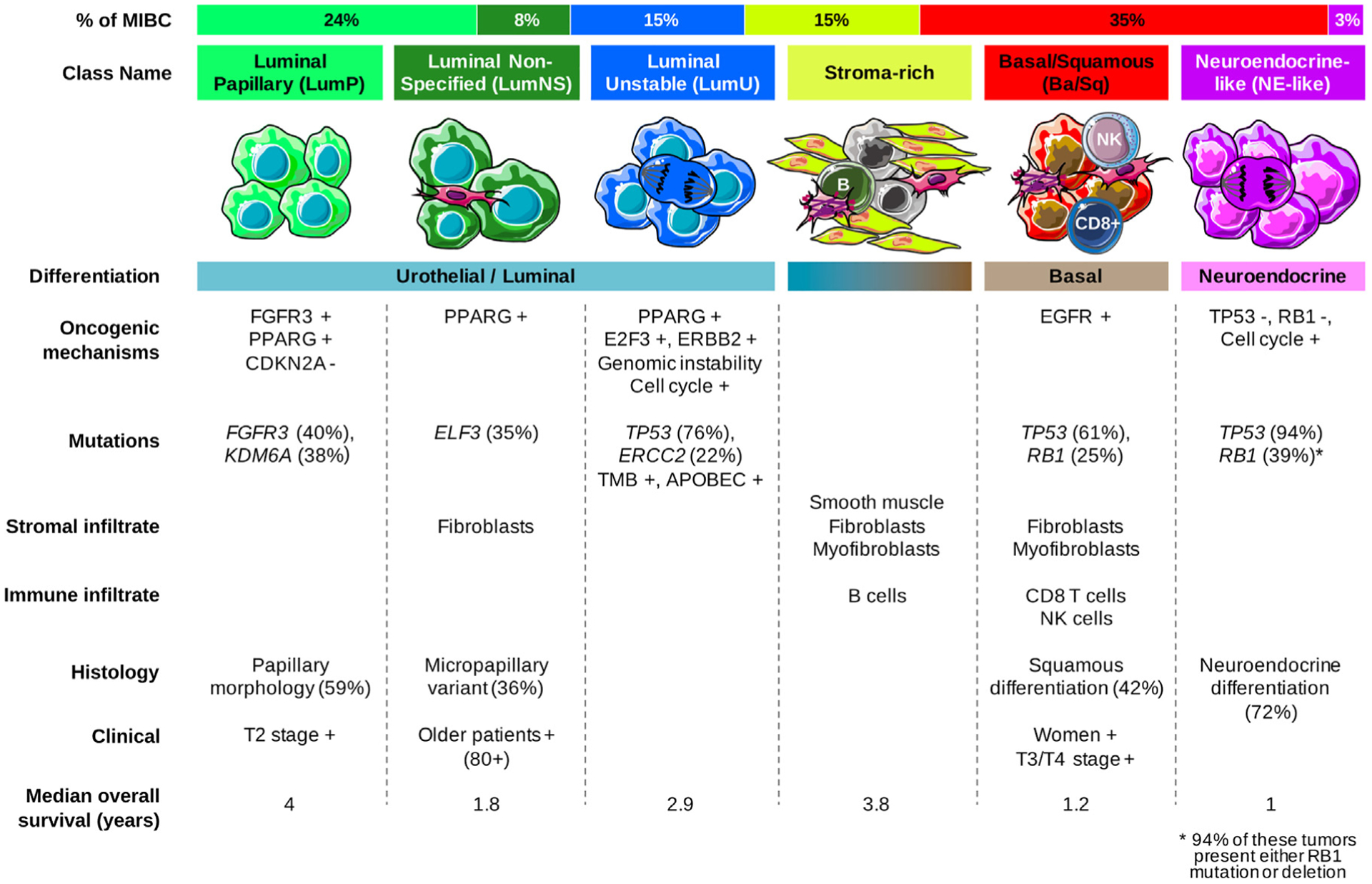
Summary of the main characteristics of the consensus classes. From top to bottom, the following characteristics are presented: proportion of consensus classes in the 1750 tumor samples; consensus class names; schematic graphical representation of tumor cells and their microenvironments (immune cells, fibroblasts, and smooth muscle cells); differentiation-based color scale showing features associated with consensus classes, including a luminal-to-basal gradient and neuroendocrine differentiation; and a table displaying the dominant characteristics such as oncogenic mechanisms, mutations, stromal infiltrate, immune infiltrate, histology, clinical characteristics, and median overall survival.
Ba/Sq = basal/squamous; LumNS = luminal nonspecified; LumP = luminal papillary; LumU = luminal unstable; MIBC = muscle-invasive bladder cancer; NE = neuroendocrine; NK = natural killer.
Some bladder tumors show histological and molecular intratumor heterogeneity [37]. Our consensus subtyping system addresses intertumor heterogeneity and focuses on defining the main molecular subtypes of MIBC. Our transcriptomic classifier will categorize tumors according to the dominant class within the tumor sample analyzed. We recognize that heterogeneous tumor samples may contain multiple subtypes and that some tumor classes are more clearly distinguishable from other tumor classes (eg, Ba/Sq and NE-like tumors). We address how these considerations are likely to interfere with our single-sample classifier by having the classifier report not simply a class label, but also correlation values to the centroids of the six consensus classes, and a separation score that reflects how well a sample is represented by its consensus class. Further studies will be required to assess the impact of intratumor heterogeneity on prognosis and response to treatment.
The consensus classification suggests possible therapeutic implications. Both the high rate of FGFR3 mutations and translocations in LumP tumors, and the FGFR3 activation signatures [4,9] associated with these tumors suggest that they may respond to fibroblast growth factor receptor (FGFR) inhibitors, irrespective of the mutation or translocation status of FGFR3. Novel FGFR inhibitors have been reported to clinically benefit the ~20% of MIBC patients with tumors harboring mutations or translocations in the tyrosine kinase receptor FGFR3 and the ~40% of MIBC patients with tumors overexpressing FGFR3 [38–40].
There is increasing interest in targeting the tumor microenvironment, including the use of immunotherapy strategies. In the USA and most of Europe, PD1 and PD-L1 immune checkpoint inhibition is becoming part of the standard of care for patients with locally advanced or metastatic urothelial cancer who relapse after cisplatin-based chemotherapy or are considered cisplatin ineligible, with a 20% objective response rate. A phase 3 clinical trial has demonstrated the efficacy of targeting tumor vasculature in MIBC using an anti-VEGFR2 inhibitor [41]. The consensus classes are associated with different stromal components, identified by transcriptomic signatures, and likely different response to immunotherapy, as revealed by our analysis of the IMvigor210 data, suggesting that they should be considered for further clinical studies involving immunotherapy or antiangiogenic therapy.
Similarities between MIBC consensus classes and other cancer molecular subtypes reported in the PanCancer Atlas work may also be considered for future basket trials. We showed that such similarities are observed, for example, between Ba/Sq MIBC tumors and squamous cell carcinomas arising in the head and neck, lung, and cervix, which were placed together in the C27 TCGA pan-cancer cluster. LumU tumors and other ERBB2-amplified tumors in breast and gastric cancers were also grouped together in the C2 pan-cancer cluster. More generally, bladder cancer and breast cancer luminal tumors share molecular similarities [10,24]. Indeed, in both cancers, the luminal subtypes rely on GATA3 and FOXA1, two transcription factors that are necessary for luminal differentiation, and on a nuclear receptor (the estrogen receptor (ESR1) in breast cancer or PPARG in bladder cancer) [25]. Intriguingly, in both cancers, there is evidence that the nuclear receptor is involved in differentiation, while also having protumorigenic effects. Such comparisons across tumor types may help transfer treatment information from tumors bearing similar characteristics to bladder cancer and vice versa.
The limitations of our study include cohorts that varied in size, composition, and gene expression technology; retrospective collection of clinical data; and incomplete information regarding patient treatment. Validating our findings, and refining subtype classification as an independent predictor of response or prognosticator of outcome will require prospective studies in which the proposed classes are identified for patients who receive standardized treatments.
5. Conclusions
We emphasize that we report biological rather than clinical classes. We offer a single-sample mRNA classifier (available in an R package and web application) as a research tool for the retrospective and prospective work required to establish how such classes can best be used clinically. The consensus presented here provides a common foundation for the molecular classification of MIBC. Future substratifications may allow defining a system that is more predictive of a response to treatments; in such work, the clinical/strategic issue will be to decide the subtype granularity or resolution [42] that is appropriate for addressing a specific problem. We expect that this consensus classification will help the development of MIBC precision medicine by providing a robust framework to connect clinical findings to molecular contexts and to identify clinically relevant biomarkers for patient management.
Supplementary Material
Acknowledgments:
This work is part of the French national program Cartes d’Identité des Tumeurs (CIT; http://cit.ligue-cancer.net). The results presented in this study are partly based upon data generated by TCGA project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov/. The results presented in this study are partly based on datasets financed by grants from the Swedish Cancer Society (2017/278) and Lund Medical Faculty (ALF). We are grateful to the patients who participated in the studies that made the work reported here possible.
Financial disclosures: Aurélie Kamoun certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Roland Seiler and Peter C. Black share a patent with GenomeDX. Peter C. Black has been provided in-kind research funding by GenomeDX; is an advisory board member of AbbVie, Asieris, AstraZeneca, Astellas, Bayer, Biosyent, BMS, Janssen, Lilly, Merck, Roche, Sanofi, and Urogen; and has participated in clinical trials with Genentech, Janssen, Ferring, Astellas, Sitka, and MDx Health. Joaquim Bellmunt has received research funding from Takeda, Pfizer, Novartis, and Sanofi-Aventis, and has an advisory role for Genentech, MSD, Pfizer, GSK, BMS, Pierre Fabre, Sanofi-Aventis, Astellas, OncoGenex, and Janssen. David J. McConkey has received research funding from AstraZeneca and is a consultant for Rainier Pharmaceuticals. Seth P. Lerner is a consultant for UroGen and Vaxiion; has received support for a clinical trial from Endo, FKD, Urogen, and Viventia; and is an advisory board member of Ferring, miR Scientific, QED Therapeutics, and UroGen. Yves Allory is an advisory board member of Astrazeneca.
Funding/Support and role of the sponsor: None.
Appendix 1. Bladder Cancer Molecular Taxonomy Group and affiliations
Mattias Aine, Division of Molecular Hematology, Department of Laboratory Medicine, Faculty of Medicine, Lund University, Lund, Sweden
Hikmat Al-Ahmadie, Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
Yves Allory, Department of Pathology, Institut Curie, Saint-Cloud, France
Institut Curie, PSL Research University, CNRS, UMR144, Equipe Labellisée Ligue contre le Cancer, Paris, France
Joaquim Bellmunt, Bladder Cancer Center, Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, MA 02215, USA
Isabelle Bernard-Pierrot, Institut Curie, PSL Research University, CNRS, UMR144, Equipe Labellisée Ligue Contre le Cancer, Paris, France
Peter C. Black, Department of Urologic Sciences, University of British Columbia, Vancouver, British Columbia, Canada
Mauro A.A. Castro, Bioinformatics and Systems Biology Laboratory, Federal University of Paraná, Polytechnic Center, Curitiba, Brazil
Keith S. Chan, Cedars-Sinai Samuel Oschin Cancer Institute, Los Angeles, CA 90048, USA
Woonyoung Choi, Johns Hopkins Greenberg Bladder Cancer Institute and Brady Urological Institute, Johns Hopkins University, Baltimore, MD, USA
Bogdan Czerniak, Department of Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Colin P. Dinney, Department of Urology and Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Lars Dyrskjøt, Department of Molecular Medicine, Aarhus University Hospital, Aarhus 8200, Denmark
Pontus Eriksson, Division of Oncology and Pathology, Department of Clinical Sciences, Lund University, Lund, Sweden
Jacqueline Fontugne, Department of Pathology, Institut Curie, Saint-Cloud, France
Institut Curie, PSL Research University, CNRS, UMR144, Equipe Labellisée Ligue Contre le Cancer, Paris, France
Ewan A. Gibb, GenomeDx Biosciences Inc., Vancouver, BC, Canada
Clarice S. Groeneveld, Cartes d’Identité des Tumeurs Program, Ligue Nationale Contre le Cancer, Paris, France
Bioinformatics and Systems Biology Laboratory, Federal University of Paraná, Polytechnic Center, Curitiba, Brazil
Institut Curie, PSL Research University, CNRS, UMR144, Equipe Labellisée Ligue Contre le Cancer, Paris, France
Arndt Hartmann, Institute of Pathology, University Erlangen-Nürnberg, Krankenhausstr 8–10, Erlangen, Germany
Katherine A. Hoadley, Department of Genetics, Department of Medicine, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Mattias Höglund, Division of Oncology and Pathology, Department of Clinical Sciences, Lund University, Lund, Sweden
Aurélie Kamoun, Cartes d’Identité des Tumeurs Program, Ligue Nationale Contre le Cancer, Paris, France
Jordan Kardos, Department of Genetics, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Jaegil Kim, Broad Institute of MIT and Harvard, Cambridge, MA, USA
William Y. Kim, Department of Genetics, Department of Medicine, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
David J. Kwiatkowski, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Thierry Lebret, Department of Urology, University of Versailles-Saint-Quentin-en-Yvelines, Foch Hospital, Suresnes, France
Seth P. Lerner, Scott Department of Urology, Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, USA
Fredrik Liedberg, Department of Translational Medicine, Lund University, Skåne University Hospital, Malmö, Sweden
Núria Malats, Genetic and Molecular Epidemiology Group, Spanish National Cancer Research Centre (CNIO), Madrid, Spain
David J. McConkey, Johns Hopkins Greenberg Bladder Cancer Institute and Brady Urological Institute, Johns Hopkins University, Baltimore, MD, USA
Qianxing Mo, Department of Biostatistics & Bioinformatics, H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL 33612, USA
Thomas Powles, Barts Cancer Institute ECMC, Barts Health and the Royal Free NHS Trust, Queen Mary University of London, London, UK
François Radvanyi, Institut Curie, PSL Research University, CNRS, UMR144, Equipe Labellisée Ligue Contre le Cancer, Paris, France
Francisco X. Real, Epithelial Carcinogenesis Group, Spanish National Cancer Research Centre (CNIO), Madrid, Spain
Aurélien de Reyniès, Cartes d’Identité des Tumeurs Program, Ligue Nationale Contre le Cancer, Paris, France
A. Gordon Robertson, Canada’s Michael Smith Genome Sciences Center, BC Cancer Agency, Vancouver, Canada
Arlene Siefker-Radtke, Department of Genitourinary Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Nanor Sirab, Department of Pathology, Institut Curie Hospital Group, Paris, France
Roland Seiler, Department of Urology, Bern University Hospital, Switzerland
Gottfrid Sjödahl, Division of Urological Research, Department of Translational Medicine, Lund University, Skåne University Hospital Malmö, Sweden
Ann Taber, Department of Molecular Medicine, Aarhus University Hospital, Aarhus 8200, Denmark
John Weinstein, Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Alexandre Zlotta, Department of Surgery, Division of Urology, University of Toronto, Mount Sinai Hospital and University Health Network, Toronto, ON, Canada
Appendix 2. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.eururo.2019.09.006.
References
- [1].Blaveri E, Simko JP, Korkola JE, et al. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res 2005;11:4044–55. [DOI] [PubMed] [Google Scholar]
- [2].Dyrskjøt L, Thykjaer T, Kruhøffer M, et al. Identifying distinct classes of bladder carcinoma using microarrays. Nat Genet 2003;33:90–6. [DOI] [PubMed] [Google Scholar]
- [3].Lindgren D, Frigyesi A, Gudjonsson S, et al. Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer Res 2010;70:3463–72. [DOI] [PubMed] [Google Scholar]
- [4].Sjödahl G, Lauss M, Lövgren K, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res 2012;18:3377–86. [DOI] [PubMed] [Google Scholar]
- [5].Tan TZ, Rouanne M, Tan KT, Huang RY-J, Thiery J-P. Molecular subtypes of urothelial bladder cancer: results from a meta-cohort analysis of 2411 tumors. Eur Urol 2019;75:423–32. [DOI] [PubMed] [Google Scholar]
- [6].Volkmer J-P, Sahoo D, Chin RK, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci U S A 2012;109:2078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hedegaard J, Lamy P, Nordentoft I, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell 2016;30:27–42. [DOI] [PubMed] [Google Scholar]
- [8].Hurst CD, Alder O, Platt FM, et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 2017;32, 701–15.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017;171, 540–56.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Damrauer JS, Hoadley KA, Chism DD, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A 2014;111:3110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rebouissou S, Bernard-Pierrot I, de Reyniès A, et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med 2014;6:244ra91. [DOI] [PubMed] [Google Scholar]
- [12].Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014;25:152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marzouka N, Eriksson P, Rovira C, Liedberg F, Sjödahl G, Höglund M. A validation and extended description of the Lund taxonomy for urothelial carcinoma using the TCGA cohort. Sci Rep 2018;8:3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sjödahl G, Eriksson P, Liedberg F, Höglund M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol 2017;242:113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mo Q, Nikolos F, Chen F, et al. Prognostic power of a tumor differentiation gene signature for bladder urothelial carcinomas. J Natl Cancer Inst 2018;110:448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seiler R, Ashab HAD, Erho N, et al. Impact of molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol 2017;72:544–54. [DOI] [PubMed] [Google Scholar]
- [19].Mariathasan S, Turley SJ, Nickles D, et al. TGFb attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aine M, Eriksson P, Liedberg F, Sjödahl G, Höglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep 2015;5:10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lerner SP, McConkey DJ, Hoadley KA, et al. Bladder cancer molecular taxonomy: summary from a consensus meeting. Bladder Cancer Amst Neth 2016;2:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018;173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Eriksson P, Aine M, Veerla S, Liedberg F, Sjödahl G, Höglund M. Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Med Genomics 2015;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Biton A, Bernard-Pierrot I, Lou Y, et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep 2014;9:1235–45. [DOI] [PubMed] [Google Scholar]
- [26].Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Böck M, Hinley J, Schmitt C, Wahlicht T, Kramer S, Southgate J. Identification of ELF3 as an early transcriptional regulator of human urothelium. Dev Biol 2014;386:321–30. [DOI] [PubMed] [Google Scholar]
- [30].Choi W, Ochoa A, McConkey DJ, et al. Genetic alterations in the molecular subtypes of bladder cancer: illustration in the Cancer Genome Atlas dataset. Eur Urol 2017;72:354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 2004;59:928–42. [DOI] [PubMed] [Google Scholar]
- [33].Horsman MR, Overgaard J. The impact of hypoxia and its modification of the outcome of radiotherapy. J Radiat Res (Tokyo) 2016;57:i90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim J, Kwiatkowski D, McConkey DJ, et al. The Cancer Genome Atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur Urol 2019;75:961–4. [DOI] [PubMed] [Google Scholar]
- [35].Mooi JK, Wirapati P, Asher R, et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: molecular analysis of the AGITG MAX clinical trial. Ann Oncol 2018;29:2240–6. [DOI] [PubMed] [Google Scholar]
- [36].Stintzing S, Wirapati P, Lenz H-J, et al. Consensus molecular sub-groups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. J Clin Oncol 2017;35:3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Warrick JI, Sjödahl G, Kaag M, et al. Intratumoral heterogeneity of bladder cancer by molecular subtypes and histologic variants. Eur Urol 2019;75:18–22. [DOI] [PubMed] [Google Scholar]
- [38].Schuler M, Nogova L, Heidenreich A, et al. 859PAnti-tumor activity of the pan-FGFR inhibitor rogaratinib in patients with advanced urothelial carcinomas selected based on tumor FGFR mRNA expression levels. Ann Oncol 2017;28(suppl_5), mdx371.013. [Google Scholar]
- [39].Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1–3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov 2018;8:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 2019;381:338–48. [DOI] [PubMed] [Google Scholar]
- [41].Petrylak DP, Wit Rde, Chi KN, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): a randomised, double-blind, phase 3 trial. Lancet 2017;390:2266–77. [DOI] [PubMed] [Google Scholar]
- [42].Aine M, Eriksson P, Liedberg F, Höglund M, Sjödahl G. On molecular classification of bladder cancer: out of one, many. Eur Urol 2015;68:921–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.