

Abstract
The molecular and clinical characteristics of non‐ampullary duodenal adenomas and intramucosal adenocarcinomas are not fully understood because they are rare. To clarify these characteristics, we performed genetic and epigenetic analysis of cancer‐related genes in these lesions. One hundred and seven non‐ampullary duodenal adenomas and intramucosal adenocarcinomas, including 100 small intestinal‐type tumors (90 adenomas and 10 intramucosal adenocarcinomas) and 7 gastric‐type tumors (2 pyloric gland adenomas and 5 intramucosal adenocarcinomas), were investigated. Using bisulfite pyrosequencing, we assessed the methylation status of CpG island methylator phenotype (CIMP) markers and MLH1. Then using next‐generation sequencing, we performed targeted exome sequence analysis within 75 cancer‐related genes in 102 lesions. There were significant differences in the clinicopathological and molecular variables between small intestinal‐ and gastric‐type tumors, which suggests the presence of at least two separate carcinogenic pathways in non‐ampullary duodenal adenocarcinomas. The prevalence of CIMP‐positive lesions was higher in intramucosal adenocarcinomas than in adenomas. Thus, concurrent hypermethylation of multiple CpG islands is likely associated with development of non‐ampullary duodenal intramucosal adenocarcinomas. Mutation analysis showed that APC was the most frequently mutated gene in these lesions (56/102; 55%), followed by KRAS (13/102; 13%), LRP1B (10/102; 10%), GNAS (8/102; 8%), ERBB3 (7/102; 7%), and RNF43 (6/102; 6%). Additionally, the high prevalence of diffuse or focal nuclear β‐catenin accumulation (87/102; 85%) as well as mutations of WNT pathway components (60/102; 59%) indicates the importance of WNT signaling to the initiation of duodenal adenomas. The higher than previously reported frequency of APC gene mutations in small bowel adenocarcinomas as well as the difference in the APC mutation distributions between small intestinal‐type adenomas and intramucosal adenocarcinomas may indicate that the adenoma–carcinoma sequence has only limited involvement in duodenal carcinogenesis. © 2020 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: non‐ampullary duodenal adenoma, small bowel adenocarcinoma, methylation, CpG island methylator phenotype, mutation
Introduction
Small intestinal cancers are rare. They comprise only 2–3% of the total annual cancer incidence in the digestive system [1, 2], and a recent epidemiological study in the Unites States of America showed that the age‐adjusted incidence rate (IR) for small intestinal cancers is only 2.10/100 000 person‐years [3]. These small intestinal cancers include neuroendocrine cancers (IR = 0.83), carcinomas (IR = 0.66), sarcomas (IR = 0.20), and lymphomas (IR = 0.38), with small bowel adenocarcinomas (SBAs) accounting for approximately 69% of the carcinomas. The incidence of carcinomas is most prominent in the duodenum, and duodenal carcinomas have increased more markedly than other small intestinal cancers []. Duodenal adenomas are also uncommon lesions, with a reported prevalence of less than 0.1–0.3% in patients undergoing upper gastrointestinal endoscopy [4, 5]. Nonetheless, with the advent of surveillance endoscopy and improvements in endoscopic imaging, these lesions are now being detected incidentally [6]. Moreover, these lesions are thought to progress into duodenal adenocarcinomas (DAs), via the adenoma–carcinoma sequence [7, 8, 9], a common pathway for colorectal cancer (CRC) development [10]. However, their clinicopathological characteristics and natural course have not been investigated in detail, due to their rarity. Because SBAs have a significantly poorer prognosis than CRCs [11], early detection and treatment are crucial. In particular, preoperative diagnosis to distinguish lesions that should be followed up from those that require treatment is an important problem [6], and molecular characterization of premalignant duodenal lesions is essential to address this issue.
Concurrent methylation of multiple CpG islands (CGIs) was first characterized as CpG island methylator phenotype (CIMP) in CRC by Toyota et al [12]. Since then, CIMP has been reported in various types of tumors [13]. Multiple studies also reported the presence of CIMP in SBAs, including DAs [14, 15], and the methylation profiles of DAs reportedly differ from biliary and ampullary carcinomas [14]. Moreover, CIMP positivity, albeit without MLH1 methylation, is reportedly associated with a poor prognosis in DA patients [15]. An earlier study analyzed DNA methylation in ampullary and non‐ampullary duodenal adenomas, but that study did not analyze these tumor types separately [16]. Thus, epigenetic alterations in early non‐ampullary duodenal lesions remain to be elucidated.
By contrast, genetic analyses of SBAs, including non‐ampullary DAs, have been performed by multiple groups [17, 18, 19, 20, 21, 22, 23]. Schrock et al [20] analyzed mutations of cancer‐related genes in 317 SBA samples and showed that the genetic signatures of SBAs were distinct from those in CRCs or gastric cancers. In addition, exome sequencing revealed potential driver genes, dysregulated oncogenic pathways, and targetable mutations in SBAs [18, 19, 21, 22]. Still, the genetic alterations in early duodenal lesions are not fully understood, with only one earlier study analyzing mutations of 50 cancer‐related genes in a limited number of samples [24].
To gain further insight into duodenal carcinogenesis, we analyzed the methylation and mutation status of a large number of non‐ampullary duodenal adenomas and intramucosal adenocarcinoma samples, and assessed their clinicopathological significance. We also evaluated the involvement of the WNT signaling pathway, which is reportedly dysregulated in non‐ampullary duodenal adenomas and adenocarcinomas [25, 26, 27].
Materials and methods
Patients and tissue samples
Specimens of non‐ampullary duodenal adenomas and intramucosal adenocarcinomas (n = 107) were obtained from 107 Japanese patients who underwent endoscopic mucosal resection or endoscopic submucosal dissection at Ishikawa Prefectural Central Hospital or Fukui Prefectural Hospital between 2008 and 2019. Normal samples were also obtained from normal‐appearing adjacent mucosa in 15 patients with small intestinal‐type duodenal adenomas included in the present study. Patients with hereditary cancer syndrome such as familial adenomatous polyposis (FAP) or Peutz–Jeghers syndrome, as well as those with inflammatory bowel diseases, such as Crohn's disease, were excluded. Information on their BMI and smoking status at the time of treatment was also obtained. Approval of this study was obtained from the Institutional Review Board of Ishikawa Prefectural Central Hospital and Fukui Prefectural Hospital, Kanazawa University, Juntendo University, and Sapporo Medical University.
Endoscopic analysis
High‐resolution magnifying endoscopes (GIF‐H290Z; Olympus, Tokyo, Japan) were used for all upper gastrointestinal endoscopic analyses. The morphology of duodenal lesions was determined according to the Paris classification [28]. All lesions detected during esophagogastroduodenoscopy were observed at high magnification using narrow band imaging, after which samples were treated by endoscopic resection for histological analysis. Locations within the duodenum were subcategorized into bulb, descending part, and transverse part. Ninety‐five small intestinal‐type tumors, of which detailed information about location was available, were also subcategorized into tumors within the proximal part (from bulb to periampulla) and those within the distal part (from periampulla to transverse part), as described previously [26].
Histological analysis
Histological studies were first carried out at Ishikawa Prefectural Central Hospital and Fukui Prefectural Hospital. The histological findings for all specimens were then re‐reviewed by two independent pathologists (ST and TY) blinded to the clinical and molecular information. The presence of small intestinal phenotypes was determined based on the presence of a brush border, goblet cells, and Paneth cells in H&E‐stained specimens. Immunohistochemical staining for CD10, MUC2, MUC5AC, and MUC6 was performed as described previously [29]. Small intestinal‐type tumors were defined by CD10 and MUC2 staining. The presence of gastric‐type differentiation was defined by MUC5AC and MUC6 staining. Tumors with the small intestinal phenotype were classified as small intestinal‐type low‐grade adenomas (SLAs), small intestinal‐type high‐grade adenomas (SHAs), or small intestinal‐type intramucosal adenocarcinomas (SCAs) according to the World Health Organization (WHO) tumor classification system (supplementary material, Figure S1) [8]. Tumors with the gastric phenotype were classified as pyloric gland adenomas (PGAs) or gastric‐type intramucosal adenocarcinomas (GCAs). SLAs and PGAs were classified as category 3 tumors, while SHAs, SCAs, and GCAs were classified as category 4 tumors according to the revised Vienna classification of gastrointestinal epithelial neoplasia [30]. SCAs correspond to non‐invasive carcinomas (category 4.2), suspicious for invasive carcinomas (category 4.3), or intramucosal carcinomas (category 4.4). The clinicopathological features of the lesions are summarized in Table 1.
Table 1.
Clinicopathological features of non‐ampullary duodenal lesions.
| Histology | Total | SLA | SHA | SCA | PGA | GCA | P value |
|---|---|---|---|---|---|---|---|
| Vienna classification | Category 3 | Category 4 | Category 3 | Category 4 | |||
| Small intestinal type | Gastric type | (Small intestinal type versus gastric type) | |||||
| Patients' characteristics (n) | 107 | 32 | 58 | 10 | 2 | 5 | |
| Age (years, mean ± SD) | 64 ± 10 | 64 ± 9 | 63 ± 10 | 66 ± 12 | 69 ± 3 | 78 ± 4 | 0.0022 |
| Gender | |||||||
| Male, n (%) | 80 (75%) | 24 (75%) | 43 (74%) | 8 (80%) | 1 (50%) | 4 (80%) | > 0.9999 |
| Female, n (%) | 27 (25%) | 8 (25%) | 15 (26%) | 2 (20%) | 1 (50%) | 1 (20%) | |
| BMI (kg/m2, mean ± SD) | 22.9 ± 3.4 | 23.2 ± 3.5 | 22.4 ± 3.5 | 24.4 ± 2.4 | 22.2 ± 1.1 | 23.7 ± 3.2 | 0.79 |
| Smoking status | |||||||
| Current smoker, n (%) | 29 (27%) | 7 (22%) | 20 (34%) | 0 (0%) | 0 (0%) | 2 (40%) | > 0.9999 |
| Former/non‐smoker, n (%) | 78 (73%) | 25 (78%) | 38 (66%) | 10 (100%) | 2 (100%) | 3 (60%) | |
| Lesion characteristics | |||||||
| Location in duodenum | |||||||
| Bulb, n (%) | 17 (16%) | 4 (13%) | 6 (10%) | 1 (10%) | 2 (100%) | 4 (80%) | < 0.0001 |
| Descending part, n (%) | 86 (80%) | 27 (84%) | 49 (85%) | 9 (90%) | 0 (0%) | 1 (20%) | (Bulb versus non‐bulb) |
| Transverse part, n (%) | 3 (3%) | 1 (3%) | 2 (3%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| Others, n (%) | 1 (1%) | 0 (0%) | 1 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| Lesion size (mm, mean ± SD) | 13 ± 8 | 11 ± 6 | 14 ± 8 | 19 ± 10 | 10 ± 0 | 18 ± 5 | 0.41 |
| Morphology | |||||||
| Protruding (0‐Ip, 0‐Is, 0‐Isp), n (%) | 16 (15%) | 4 (13%) | 4 (7%) | 3 (30%) | 1 (50%) | 4 (80%) | 0.0002 |
| Mixed (0‐I + IIa), n (%) | 5 (5%) | 0 (0%) | 3 (5%) | 1 (10%) | 1 (50%) | 0 (0%) | (Protruding or mixed versus nonprotruding) |
| Nonprotruding and nonexcavated (0‐IIa, 0‐IIc, 0‐IIa + IIc), n (%) | 85 (79%) | 28 (87%) | 50 (86%) | 6 (60%) | 0 (10%) | 1 (20%) | |
| N/A, n (%) | 1 (1%) | 0 (0%) | 1 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | |
GCA, gastric‐type intramucosal adenocarcinoma; N/A, not available; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
DNA isolation
DNA was isolated from formalin‐fixed, paraffin‐embedded (FFPE) sections using a QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany). A TaqMan RNase P Detection Reagents kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to quantify the purified DNA.
DNA methylation analysis
DNA methylation was analyzed using bisulfite pyrosequencing as described previously [31]. A cut‐off value of 15% was used to define genes as methylation‐positive. We used five CIMP markers (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1) proposed by Weisenberger et al [32]. Tumors were defined as CIMP‐positive if two or more loci showed methylation and as CIMP‐high (CIMP‐H) if three or more loci showed methylation, as previously reported [16]. In addition, methylation of the MLH1 gene was investigated. Primer sequences are shown in supplementary material, Table S1.
Targeted amplicon sequencing analysis
A customized panel, encompassing all exons for 75 cancer‐related genes including those frequently mutated in SBAs [17, 18, 19, 20, 21, 33, 34], was created using the Ion AmpliSeq Designer (Thermo Fisher Scientific) (supplementary material, Table S2). Genes whose mutations had been reported in duodenal adenomas were also included [24]. The assay design consisted of 3663 amplicons with an average length of 112 bp, covering 95.5% of the 366 kb target sequence. Library preparation and sequencing with an Ion Proton sequencer were performed as described previously [35, 36]. The templates were sequenced after emulsion PCR with 12–16 samples per Ion PI chip using an Ion PI HI‐Q Chef kit (Thermo Fisher Scientific).
Identification of somatic mutations and copy number variations
Somatic mutations and copy number variations (CNVs) were detected as described previously [35]. Human genome build 19 (hg19) was used as a reference. Signal processing, base‐calling, mapping to the hg19 reference, alignment, and further quality filtering were performed using Torrent Suite version 5.0 (Thermo Fisher Scientific). Somatic mutations, including point mutations, insertions, and deletions, were detected using Ion Reporter Software 5.0 (Thermo Fisher Scientific). Because matched normal controls were not available, we utilized Ion Reporter software tumor–normal workflow using Demo AmpliSeq Exome control as the normal control for excluding common single nucleotide polymorphisms (SNPs). A sequencing coverage of 20x and a minimum variant frequency of 5% of the total number of distinct tags were used as cut‐offs. The pathogenic status of the variant was stated if it was a missense variant with less than 0.1% global minor allele frequency in the dbSNP and/or the variant was suggested as being pathogenic in the ClinVar, COSMIC, SIFT, or PolyPhen‐2 databases. Variants with allele frequencies between 0.4 and 0.6 or greater than 0.9 were considered germline variants unless listed as a pathogenic variant. Integrative Genomics Viewer (IGV) software (http://software.broadinstitute.org/software/igv/) was used to filter out possible strand‐specific errors, such as a mutation that was identified in the forward or reverse DNA strand but not in both strands. CNVs were also detected using Ion Reporter Software with an algorithm based on the Hidden Markov Model. Recurrent genomic regions with CNVs were detected using copy numbers greater than 3 and less than 1 for gains and losses, respectively.
Immunohistochemistry
Immunohistochemical studies of β‐catenin expression were performed in 102 samples, as described previously [37]. A mouse anti‐β‐catenin monoclonal antibody (1:1000 dilution, Clone 14; BD Biosciences, San Jose, CA, USA) was used. Expression of β‐catenin was evaluated semi‐quantitatively in tumor cells with β‐catenin‐positive nuclei, as reported previously [38]: negative, 0–9%; focal, 10–49%; and diffuse, > 50%. All slides were evaluated by two pathologists (ST and TY) blinded to the clinical and molecular data. In addition, immunohistochemical studies of two mismatch repair (MMR) proteins, MLH1 (1:1000 dilution, Ab92312; Abcam, Cambridge, UK) and MSH2 (1:1000 dilution, Ab 227 841; Abcam), was performed in 107 samples to assess microsatellite instability (MSI) status. To evaluate expression, lymphocytes in adjacent normal tissue were used as an internal positive control. When nuclear staining was identified in epithelial cells, the lesion was defined as positive for MMR proteins. All slides were evaluated by a pathologist (SK) blinded to the clinical and molecular data.
Statistical analysis
Continuous data were analyzed using t‐tests (for two groups) or ANOVA with a post hoc Tukey's HSD test (for more than two groups). Comparison of categorical data between two or more groups was performed using the Fisher's exact test or chi‐squared test. Values of p < 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA).
Results
Clinicopathological characteristics of non‐ampullary duodenal lesions
The clinicopathological and molecular characteristics of 107 non‐ampullary duodenal lesions analyzed in this study are summarized in Table 1. The average age at endoscopic treatment was 64 years. The male‐to‐female ratio was approximately 3:1, which is consistent with previous studies from Japan [9, 26, 39]. When compared between cases with small intestinal‐type (SLA, SHA, and SCA) and gastric‐type (PGA and GCA) tumors, age at treatment was significantly higher in those with gastric‐type tumors (t‐test, p = 0.0022). In addition, tumors with the gastric phenotype were more prevalent in the duodenal bulb (6/7, 86%) than were those with the small intestinal phenotype (11/100, 11%) (Fisher's exact test, p < 0.0001). On endoscopic observation, most tumors with gastric differentiation were protruding lesions or mixed lesions that included a protruding portion (6/7, 86%), and differed significantly from tumors with small intestinal differentiation, most of which were non‐protruding and non‐excavated lesions (84/100, 84%) (Fisher's exact test, p = 0.0002, Table 1). We also compared the clinicopathological characteristics of small intestinal‐type tumors based on the proximal or distal locations (supplementary material, Table S3). Significant differences were found in the tumor histological types (SLA, SHA or SCA, chi‐squared test, p = 0.017) and categories (3 or 4, Fisher's exact test, p = 0.0075) between tumors in the proximal and distal duodenum. In addition, the prevalence of nonprotruding and nonexcavated type tumors was significantly higher in the distal part (Fisher's exact test, p = 0.037).
Methylation analysis of CIMP markers and MLH1
Among non‐ampullary duodenal lesions, we assessed the methylation status of cancer‐associated genes in 107 lesions and assessed the CIMP and CIMP‐H status in 106 lesions. Twenty‐five lesions (25/106, 24%) were defined as CIMP‐positive, and seven lesions (7/106, 7%) were defined as CIMP‐H (Figure 1 and Table 2). The prevalence of CIMP‐positive lesions was significantly associated with male gender (Fisher's exact test, p = 0.033), older age (cut‐off value 75 years, Fisher's exact test, p = 0.043), and larger tumor size (cut‐off value 15 mm, Fisher's exact test, p = 0.032) (Table 3). By contrast, BMI, smoking status, and the location and endoscopic morphology of the lesions were not associated with CIMP positivity. When samples were divided into adenomas (SLA, SHA, and PGA) and intramucosal adenocarcinomas (SCA and GCA), the prevalence of CIMP‐positive lesions was higher in intramucosal adenocarcinomas (6/15, 40%) than in adenomas (19/91, 21%), although there were no statistical significant differences (Fisher's exact test, p = 0.18, Table 3). When only small intestinal‐type tumors were analyzed, CIMP positivity was not associated with tumor histological type (SLA, SHA, and SCA). Moreover, the prevalence of CIMP‐positive lesions was not associated with either histological classification (small intestinal type or gastric type) or Vienna classification (category 3 or 4). Although two MLH1 methylation‐positive lesions were detected among the SHA samples, methylation levels were below 20% (15.8% and 16.5%) in these lesions. In addition, there was no association between MLH1 methylation status and CIMP or CIMP‐H positivity (Figure 1). Although there was no relationship between CIMP‐H status and age, gender, smoking status, or disease location, CIMP‐H positivity was associated with BMI (t‐test, p = 0.022) and lesion size (t‐test, p = 0.012) (supplementary material, Table S4). In addition, the prevalence of CIMP‐H tumors did not significantly differ among histological subtypes or between small intestinal‐type and gastric‐type tumors.
Figure 1.
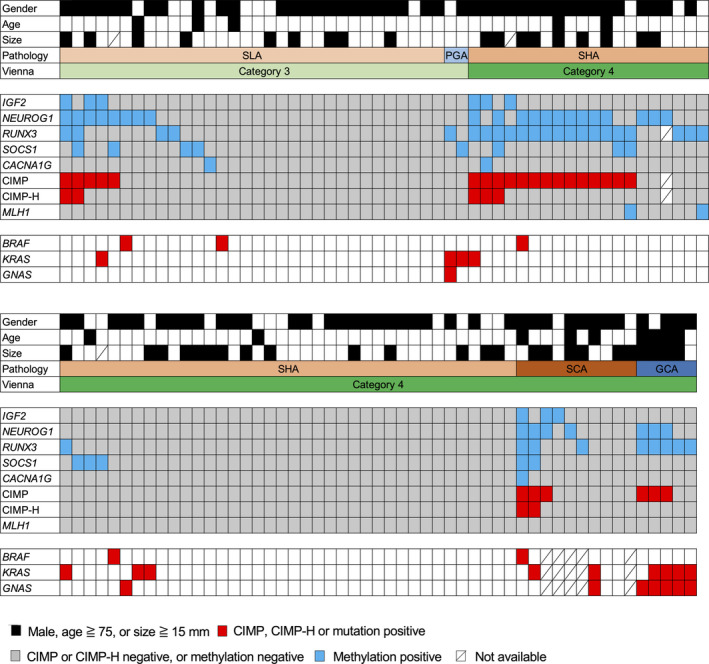
Methylation and mutation profiles in non‐ampullary duodenal adenomas and intramucosal adenocarcinomas. Summarized results for CIMP marker methylation, CIMP status, MLH1 methylation, and BRAF/KRAS/GNAS mutations in tumors with the indicated histological types are shown. CIMP, CpG island methylator phenotype; GCA, gastric‐type intramucosal adenocarcinoma; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
Table 2.
Molecular characteristics and β‐catenin expression of the respective histological types of non‐ampullary duodenal lesions.
| Histology | Total | SLA | SHA | SCA | PGA | GCA | P value |
|---|---|---|---|---|---|---|---|
| Small intestinal type | Gastric type | (small intestinal type versus gastric type) | |||||
| Gene mutation (n) | 102 | 32 | 58 | 5 | 2 | 5 | |
| APC mutation, n (%) | 56 (55%) | 17 (53%) | 34 (59%) | 3 (60%) | 1 (50%) | 1 (20%) | 0.24 |
| BRAF mutation, n (%) | 5 (5%) | 2 (6%) | 2 (3%) | 1 (20%) | 0 (0%) | 0 (0%) | > 0.9999 |
| KRAS mutation, n (%) | 13 (13%) | 1 (3%) | 4 (7%) | 2 (40%) | 2 (100%) | 4 (80%) | < 0.0001 |
| GNAS mutation, n (%) | 8 (8%) | 0 (0%) | 1 (2%) | 1 (20%) | 1 (50%) | 5 (100%) | < 0.0001 |
| Epigenetic alteration (n) | 107 | 32 | 58 | 10 | 2 | 5 | |
| CIMP*, n (%) | 25 (24%) | 5 (16%) | 14 (24%) | 3 (30%) | 0 (0%) | 3 (60%) | 0.36 |
| CIMP‐high*, n (%) | 7 (7%) | 2 (6%) | 3 (5%) | 2 (20%) | 0 (0%) | 0 (0%) | > 0.9999 |
| MLH1 methylation, n (%) | 2 (2%) | 0 (0%) | 2 (3%) | 0 (0%) | 0 (0%) | 0 (0%) | > 0.9999 |
| β‐catenin expression (n) | 102 | 32 | 58 | 5 | 2 | 5 | |
| Negative, n (%) | 15 (15%) | 3 (10%) | 5 (8%) | 1 (20%) | 1 (50%) | 5 (100%) | < 0.0001 |
| Focal, n (%) | 33 (32%) | 11 (34%) | 19 (33%) | 2 (40%) | 1 (50%) | 0 (0%) | (focal or diffuse versus negative) |
| Diffuse, n (%) | 54 (53%) | 18 (56%) | 34 (59%) | 2 (40%) | 0 (0%) | 0 (0%) | |
CIMP, CpG island methylator phenotype; GCA, gastric‐type intramucosal adenocarcinoma; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
β‐catenin expression was categorized as negative (0–9%), focal (10–49%), and diffuse (> 50%).
CIMP status and CIMP‐high status were analyzed in 106 lesions.
Table 3.
Relationship between CIMP status and clinicopathological characteristics of non‐ampullary duodenal lesions.
| Characteristics | Total | CIMP positive | CIMP negative | P value |
|---|---|---|---|---|
| Patients (n) | 106 | 25 | 81 | |
| Age (years, mean ± SD) | 64 ± 10 | 68 ± 9 | 63 ± 10 | 0.026 |
| < 75, n (%) | 91 (86%) | 18 (72%) | 73 (90%) | 0.043 |
| ≥ 75, n (%) | 15 (14%) | 7 (28%) | 8 (10%) | |
| Gender | ||||
| Male, n (%) | 79 (75%) | 23 (92%) | 56 (69%) | 0.033 |
| Female, n (%) | 27 (25%) | 2 (8%) | 25 (31%) | |
| BMI (kg/m2, mean ± SD) | 22.9 ± 3.4 | 23.4 ± 3.4 | 22.7 ± 3.4 | 0.39 |
| Smoking status | ||||
| Current smoker, n (%) | 29 (27%) | 4 (16%) | 25 (31%) | 0.20 |
| Former/non‐smoker, n (%) | 77 (73%) | 21 (84%) | 56 (69%) | |
| Location | ||||
| Bulb, n (%) | 16 (15%) | 5 (20%) | 11 (14%) | 0.52 |
| Descending – transverse part, n (%) | 89 (84%) | 20 (80%) | 69 (85%) | (Bulb versus non‐bulb) |
| Other, n (%) | 1 (1%) | 0 (0%) | 1 (1%) | |
| Size (mm, mean ± SD) | 13 ± 8 | 17 ± 10 | 12 ± 7 | 0.032 |
| < 15, n (%) | 61 (57%) | 9 (36%) | 52 (64%) | 0.032 |
| ≥ 15, n (%) | 42 (40%) | 14 (56%) | 28 (35%) | |
| N/A, n (%) | 3 (3%) | 2 (8%) | 1 (1%) | |
| Morphology | ||||
| Protruding, n (%) | 16 (15%) | 4 (16%) | 12 (15%) | 0.78 |
| Mixed, n (%) | 5 (5%) | 0 (0%) | 5 (6%) | (Protruding or mixed versus nonprotruding) |
| Nonprotruding and nonexcavated, n (%) | 84 (79%) | 21 (84%) | 63 (78%) | |
| N/A, n (%) | 1 (1%) | 0 (0%) | 1 (1%) | |
| Histology | ||||
| SLA, n (%) | 32 (30%) | 5 (20%) | 27 (34%) | 0.51* |
| SHA, n (%) | 57 (54%) | 14 (56%) | 43 (53%) | |
| SCA, n (%) | 10 (9%) | 3 (12%) | 7 (9%) | |
| PGA, n (%) | 2 (2%) | 0 (0%) | 2 (2%) | |
| GCA, n (%) | 5 (5%) | 3 (12%) | 2 (2%) | |
| Small intestinal type, n (%) | 99 (93%) | 22 (88%) | 77 (95%) | 0.35 |
| Gastric type, n (%) | 7 (7%) | 3 (12%) | 4 (5%) | |
| Adenoma, n (%) | 91 (86%) | 19 (76%) | 72 (89%) | 0.18 |
| Adenocarcinoma, n (%) | 15 (14%) | 6 (24%) | 9 (11%) | |
| Vienna classification | ||||
| Category 3, n (%) | 34 (32%) | 5 (20%) | 29 (36%) | 0.14 |
| Category 4, n (%) | 72 (68%) | 20 (80%) | 52 (64%) |
CIMP, CpG island methylator phenotype; GCA, gastric‐type intramucosal adenocarcinoma; N/A, not available; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
Analyzed with SLA, SHA, and SCA.
Targeted amplicon sequencing of non‐ampullary duodenal lesions
We performed targeted sequencing of all exons in 75 cancer‐related genes frequently mutated in SBAs, including DAs, as well as duodenal adenomas. A sequencing overview, including reads, coverage, and uniformity of the read coverage distribution, is shown in supplementary material, Table S5. Each FFPE sample underwent an average of 8.5 million sequencing reads after quality filtering. A mean coverage depth of 2461.0 reads (737.0–10 950.0) per base was observed. All single nucleotide variations and insertions/deletions detected through bioinformatics analysis underwent visual inspection using the Integrative Genomics Viewer for confirmation. We identified a mean of 1.9 somatic nonsynonymous mutations (range 0–8) per sample (supplementary material, Table S6). At least one somatic nonsynonymous mutation was observed in 45 of the 75 genes. The ten most commonly mutated genes in non‐ampullary duodenal lesions are illustrated in Figure 2. APC was the most frequently mutated gene in these lesions (56 of 102 samples; 55%), followed by KRAS (13/102; 13%), LRP1B (10/102; 10%), GNAS (8/102; 8%), ERBB3 (7/102; 7%), and RNF43 (6/102; 6%).
Figure 2.
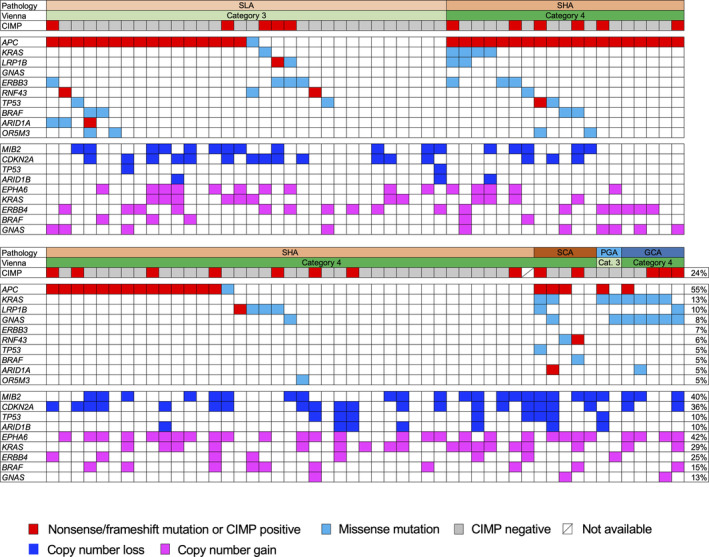
Mutation and CNV profiles in non‐ampullary duodenal adenomas and intramucosal adenocarcinomas. Upper panels show histological types, Vienna classification, and CIMP status. Middle panels show summarized results for targeted sequencing of cancer‐related genes in tumors with the indicated histological types. Lower panels show frequently detected CNVs. Frequencies of CIMP status as well as mutations and CNVs in respective genes are shown on the right. CIMP, CpG island methylator phenotype; GCA, gastric‐type intramucosal adenocarcinoma; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
For APC, a single mutation per sample was detected in 48 subjects, and two different mutations per sample were detected in eight subjects, resulting in a total of 64 mutations in this study. Most APC mutations were nonsense (43 mutations) or frameshift (16 mutations), though a number of missense mutations (five mutations) were also detected. The mutation distribution within APC was visualized using MutationMapper in cBioPortal (https://www.cbioportal.org) [40, 41] with several modifications (Figure 3A). Most of the deleterious mutations were distributed between codons 700 and 1200 or between codons 1400 and 1600. Although a prior study suggested that T1556fs is a mutation hotspot within APC in duodenal adenomas [24], we most frequently found a mutation at R1450X (in ten samples), which is similar to CRC [42] but different from ampullary carcinoma [34] (Figure 3B). Although the frequencies of mutations within mutation cluster regions (codons 700–1200 and 1400–1600) among the total mutations detected in each histological type were similar between SLAs and SHAs, the mutation frequency in small intestinal‐type adenomas (SLAs + SHAs) (86%; 51/59 mutations) within these regions was higher than in SCAs (33%; 1/3 mutations) (Fisher's exact test, p = 0.065). Overall, mutations within WNT signaling pathway components were detected in 59% (60/102) of the samples (Figure 4).
Figure 3.
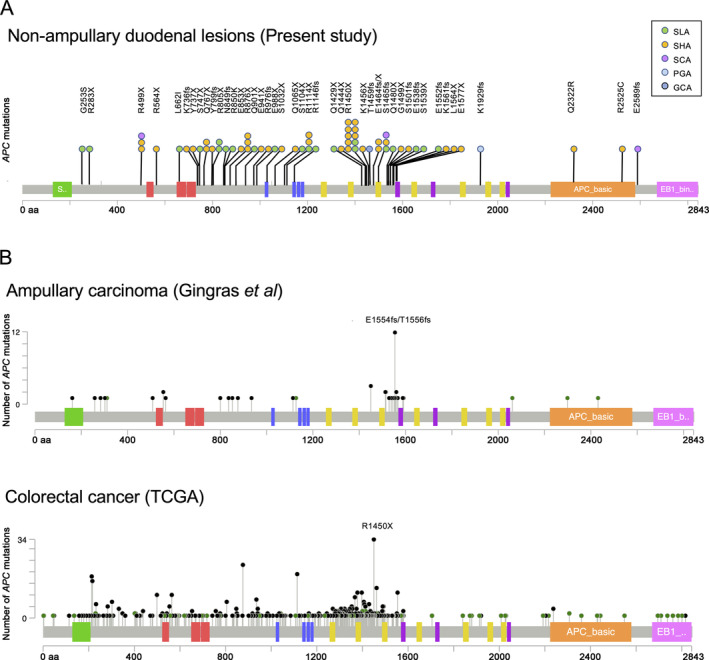
APC mutations detected in non‐ampullary duodenal lesions. (A) Schematic representation of 64 APC mutations detected in the present study. Most of the somatic mutations detected in SLAs and SHAs were clustered within codons 700–1200 and 1400–1600, where hotspot mutation (R1450X) is located. Each circle represents an individual mutation in each patient. (B) Mutations previously reported in ampullary carcinomas [34] and colorectal cancers [42] are also indicated for comparison. GCA, gastric‐type intramucosal adenocarcinoma; PGA, pyloric gland adenoma; SCA, small intestinal‐type intramucosal adenocarcinoma; SHA, small intestinal‐type high‐grade adenoma; SLA, small intestinal‐type low‐grade adenoma.
Figure 4.
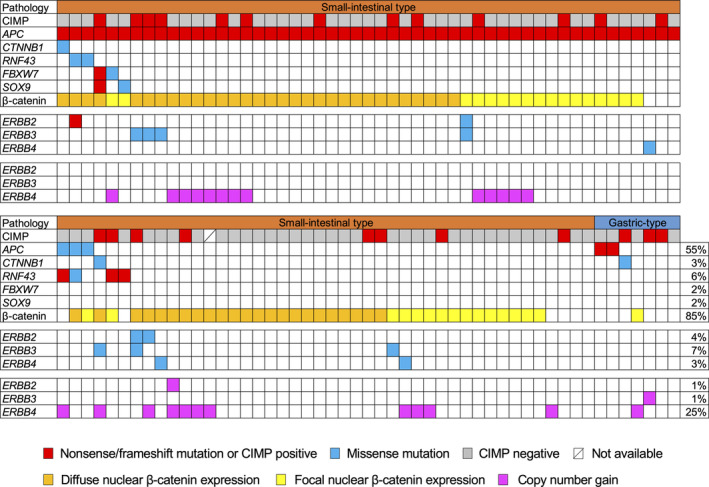
Mutation and copy number gains of genes related to the WNT signaling pathway and ERBB receptor family members in non‐ampullary duodenal lesions. Pathological types (small intestinal or gastric type) and CIMP status, mutations in WNT signaling‐associated genes, and β‐catenin expression status are shown at the top. Mutations and copy number gains in ERBB receptor family members are shown in the middle panels and lower panels. Frequencies of mutations and copy number gains in respective genes as well as β‐catenin expression positivity are shown on the right. CIMP, CpG island methylator phenotype.
There were significant differences in the prevalence of KRAS mutations among the histological subtypes of small intestinal‐type tumors: SLA (1/32; 3%), SHA (4/58; 7%), and SCA (2/5; 40%) (chi‐squared test, p = 0.013), though there was no significant difference in the prevalence of APC mutations among histological subtypes (Table 2). TP53 mutations were detected in five samples (5%), and samples harboring a mutation within at least one gene among APC, KRAS, and TP53 were observed in 62 patients (61%) (Figure 2). All eight detected GNAS mutations were at codon 201, and six of those are associated with KRAS mutations. When compared between small intestinal‐type tumors and gastric‐type tumors, GNAS and KRAS mutations were significantly associated with gastric‐type tumors (Table 2).
ERBB2 mutations, which have been frequently reported in SBAs, were also detected in four of the 102 samples (4%). The frequency of lesions harboring at least one mutation in an ERBB receptor family member (ERBB2, ERBB3, or ERBB4) was 12% (12/102) (Figure 4). BRAF mutation was detected in five samples (5%), which is comparable to earlier studies reporting frequencies of 6–11% in SBA samples [17, 20, 21]. BRAF V600E mutation was detected in only one SCA sample with CIMP and CIMP‐H (Figure 1), which is consistent with an earlier report that only about 10% of BRAF‐mutated SBAs harbor V600E mutations [20].
Targeted amplicon sequencing detects CNVs
We also detected CNVs in segments of the genome that could be duplicated or deleted from the sequencing data (Figure 2 and supplementary material, Table S7). Based on copy number gains in all samples, the most frequently affected genes that were considered oncogenic were EPHA6 (43/102; 42%), followed by KRAS (30/102; 29%), ERBB4 (25/102; 25%), BRAF (12/102; 15%), and GNAS (13/102; 13%). On the other hand, the most frequently affected tumor suppressive genes by copy number losses were MIB2 (41/102; 40%), CDKN2A (37/102; 36%), TP53 (10/102; 10%), and ARID1B (10/102; 10%). The distributions of CNVs were not different among tumor histological subtypes. As for ERBB family genes, copy number gains were observed in one case each for ERBB2 and ERBB3, while frequent copy number gains (25/102, 25%) were detected in ERBB4 (Figure 4). Lastly, the frequency of lesions harboring at least one mutation or copy number gain in an ERBB family member was 34% (35/102).
Immunohistochemistry
Of the 102 non‐ampullary duodenal lesions analyzed, 33 (32%) showed focal nuclear expression and 54 (53%) showed diffuse nuclear expression, whereas 15 (15%) showed no nuclear expression of β‐catenin (Table 2, Figure 4, and supplementary material, Figure S2). The prevalence of diffuse and focal nuclear β‐catenin accumulation in duodenal adenomas (SLA, SHA, and PGA) (90%; 83/92) was consistent with earlier reports of nuclear β‐catenin accumulation in 64–84% of non‐ampullary adenomas [25, 26, 27]. Lesions showing nuclear β‐catenin accumulation (diffuse or focal) were not associated with APC mutation‐positive tumors or tumors with mutations of WNT signaling components. The prevalence of nuclear β‐catenin accumulation was significantly higher in small intestinal‐type tumors (86/95, 91%) than in gastric‐type tumors (1/7, 14%) (Fisher's exact test, p < 0.0001, Table 2). However, when we focused on tumors with the small intestinal phenotype, the prevalence of nuclear β‐catenin accumulation did not significantly differ among SLAs, SHAs, and SCAs. We found no significant differences between β‐catenin expression or molecular variables and the locations (proximal and distal) of the 90 small intestinal‐type tumors (supplementary material, Table S3).
Among MMR proteins, expression of MLH1 and MSH2 was evaluated immunohistochemically across all non‐ampullary duodenal lesions. All samples stained positively for MLH1, which was largely consistent with the methylation data. By contrast, loss of MSH2 expression was detected in one SCA sample (KT23) (supplementary material, Figure S3). That sample harbored the largest number of somatic mutations (eight) among the 75 genes investigated (supplementary material, Table S6). Six of the eight mutations were insertions of one base resulting in frameshift mutations, which supports the possibility of MSI.
Discussion
In the present study, we performed integrated genetic and epigenetic analyses of non‐ampullary duodenal lesions. In addition to clinicopathological variables, there were significant differences in molecular variables between small intestinal‐type and gastric‐type tumors. High prevalences of KRAS and GNAS mutations in PGAs and GCAs were consistent with earlier reports [43, 44]. The low prevalences of nuclear β‐catenin expression in PGAs and GCAs, which is also consistent with an earlier study [26], may suggest that the WNT signaling pathway is less involved in development of non‐ampullary duodenal tumors with the gastric phenotype. These results indicate that small intestinal‐type and gastric‐type tumors arise via separate carcinogenic pathways. When small intestinal‐type tumors were divided based on whether they were located within the proximal or distal duodenum, SLAs and nonprotruding and nonexcavated type tumors were more prevalent in the distal part. However, there were no significant differences in the molecular characteristics including β‐catenin expression between tumors in these two locations. Previous reports suggested that development of gastric‐type tumors in the proximal duodenum is potentially associated with gastric acid and H. pylori infection, whereas small intestinal‐type tumors in the distal duodenum may be associated with bile acids [26]. Further study will be necessary to clarify the pathological and molecular differences between tumors in the proximal and distal duodenum.
We also found that CIMP and CIMP‐H frequencies are higher in intramucosal adenocarcinomas than in adenomas, indicating that concurrent methylation of CGIs is likely associated with malignant transformation of non‐ampullary duodenal adenomas. In the clinical settings, tumors classified as category 3 according to the revised Vienna classification (SLAs and PGAs) can be monitored and followed up [30], while it is recommended that category 4 tumors (SHAs, SCAs, and GCAs) be treated, especially those that are 20 mm in diameter or larger, as these lesions have a high risk of progression to adenocarcinoma [9]. The fact that CIMP or CIMP‐H positivity was associated with larger lesions in the present study may support this recommendation. At the same time, from the viewpoint of aberrant DNA methylation, SHAs could be followed up at a later time, especially if they are small in size. Despite recent advances in endoscopic technology, including magnifying endoscopy and image enhanced endoscopy, it remains difficult to distinguish between lesions that should be followed up or treated [6]. Pretreatment diagnosis by using biopsy is also difficult because the biopsy procedure itself may induce unintended fibrosis, possibly causing unsuccessful endoscopic resection. If specific endoscopic findings for lesions with concurrent methylation are determined, detailed endoscopic assessment could contribute to the prediction of premalignant lesions, as was previously reported for colorectal serrated lesions [45].
MLH1 methylation has been reported in 12% of non‐ampullary adenomas [16] as well as in 14% [15] or 25% [14] of DAs. MSI has also been reported in 20% [15] or 33% [17] of DAs, where it is reportedly associated with MLH1 methylation [15]. In the present study, only two MLH1 methylation‐positive lesions, with relatively low methylation levels, and no samples with loss of MLH1 immunoreactivity were detected. Additionally, there was only one sample with loss of MSH2 expression in possible association with MSI. These results suggest that acquisition of MSI in association with MMR deficiency, especially with MLH1 methylation, is an infrequent event in early duodenal carcinogenesis.
Recent studies indicated that the genes recurrently mutated in SBAs are TP53 (41–58%), KRAS (27–54%), and APC (11–27%) [17, 18, 19, 20, 21, 22]. An earlier study analyzed 50 hotspot mutations in cancer‐related genes in 19 patients with non‐ampullary duodenal adenomas and adenocarcinomas, and found prevalent mutations in KRAS (63%), APC (47%), and TP53 (37%) [24]. In the present study, we found mutations in APC (55%) and KRAS (13%); TP53 mutations were observed in only 5% of the samples. These differences are likely due to the proportion of the adenocarcinomas or high‐grade adenomas. In the present study, most KRAS mutations appeared to occur at the progression of intramucosal adenocarcinomas. In addition, the lower prevalence of TP53 mutations in early duodenal lesions than in SBAs suggests that most TP53 mutations occur at a later stage of tumorigenesis. These results in SBAs appear to some degree consistent with the adenoma–carcinoma sequence previously seen in CRC [10]. We also detected a higher prevalence of gene mutations associated with the WNT signaling pathway (59%) and nuclear β‐catenin accumulation (85%) in non‐ampullary duodenal lesions. Together with earlier immunohistochemical and gene expression studies of duodenal adenomas [25, 26, 27], these results indicate the importance of the WNT signaling pathway to duodenal adenoma development.
Interestingly, we detected APC mutations (55%) more frequently in early non‐ampullary duodenal lesions than did earlier studies in advanced lesions, which reported frequencies of 13–27% in SBAs or 8% in DAs [17, 20, 21, 22]. Kojima et al separately analyzed multiple components in non‐ampullary duodenal lesions that exhibited different histological grades, and observed APC mutations more frequently in the adenoma (58%) than in the adenocarcinoma (25%) components [24]. The earlier observation that abnormal nuclear localization of β‐catenin is more frequent in non‐ampullary duodenal adenomas than in adenocarcinomas (84% versus 33%) may support these results [26]. Although a number of studies have reported that gastric intramucosal neoplasias with the small intestinal phenotype also frequently show APC mutations, a few dysplasia/intramucosal neoplasias with APC mutation reportedly progress to gastric cancer [46]. Together with the different mutation distribution patterns between duodenal adenomas (SLAs + SHAs) and SCAs, the higher prevalence of APC mutations in duodenal adenomas than in SBAs or DAs may suggest that most duodenal adenomas, especially those with mutations within mutation cluster regions, also have low malignant potential and do not progress to DA. This suggests that the adenoma–carcinoma sequence has only limited impact on duodenal carcinogenesis. Targeted deep DNA sequencing with tumor variant allele frequency analysis in larger numbers of adenoma, intramucosal adenocarcinoma, and DA samples may confirm these findings and improve our understanding of the mechanisms underlying duodenal carcinogenesis, as has been described for gastric intramucosal neoplasias with the small intestinal phenotype [46].
ERBB2 mutations and amplifications have been detected in 12–23% of SBAs, which suggests that they are potential therapeutic targets [17, 20, 21, 22]. ERBB2 alterations are also significantly associated with a duodenal location when compared to other parts of the small intestine [17, 20]. In the present study, ERBB2 mutations were observed in only 4% of samples. Because a significant number of ERBB2 mutations were observed in SBAs with MMR deficiency [17, 21], most of these mutations may have been acquired after the cancer progression and/or acquisition of MSI. A recent study using SBA patient‐derived cell lines demonstrated that small‐molecule ERBB2 inhibitors have anti‐cancer activity both in vitro and in vivo [22]. Such small‐molecule ERBB2 inhibitors may be useful not only for the treatment of metastatic SBA but also for the chemoprevention of multiple non‐ampullary duodenal adenoma in patients with FAP, if activating ERBB2 mutations are detected.
As for CNVs, previous conventional and array comparative genomic hybridization studies reported that in SBAs, the regions most commonly exhibiting copy number gains were at chromosomes 5p, 7, 8q, 13, 16, and 20, while copy number losses were detected at chromosomes 4, 5q, 8p, 17p, and 18 [23]. In the present study, the frequent occurrence of copy number losses at the TP53 locus (17p) as well as the gains at the BRAF (7q) and GNAS (20q) loci was consistent with those earlier results. Notably, frequent copy number gains at the ERBB4 locus were observed in the present study. In ovarian serous carcinoma, immunohistochemical detection of high ERBB4 expression reportedly correlated with cisplatin resistance and a poorer prognosis [47]. Because recent studies suggest that somatic CNVs at oncogenic loci are not always associated with gene expression [42, 48, 49], further validation of the effect of CNVs through comparison with expression is needed before the utilization of CNVs as biomarkers.
This study has several limitations. One is the lack of MSI analysis other than the immunohistochemical comparison of MMR proteins. In addition, the sample sizes were small when the specimens were divided into intestinal‐ and gastric‐type tumors or CIMP‐H and non‐CIMP‐H tumors. Furthermore, analysis of corresponding normal samples for validation of somatic mutations was not performed. Although rigorous bioinformatics pipelines were used to discriminate somatic from germline mutations, the results of mutation analyses may still include germline mutations. However, we have several novel findings. First, molecular and clinicopathological characteristics differ among small intestinal‐type tumors and gastric‐type tumors, which is suggestive of separate carcinogenic pathways. Second, concurrent methylation of CGIs appears to be associated with the development of non‐ampullary duodenal lesions. Third, prevalent WNT pathway gene mutations and positive staining for nuclear β‐catenin indicate the involvement of WNT signaling in the development of duodenal tumors. Finally, we observed a higher frequency of APC mutations than previously reported in SBA patients as well as differences in the mutation distributions within APC between adenoma and intramucosal adenocarcinoma samples. This suggests that the adenoma–carcinoma sequence has only limited involvement in duodenal carcinogenesis.
At present, it remains difficult to predict the progression from adenoma to adenocarcinoma or to determine endoscopically which lesions can be followed and which must be treated. The differences between the pathogenesis of small intestinal‐type tumors in the proximal duodenum and those in the distal part are also uncharacterized. Accumulation of data from comprehensive genetic and epigenetic analyses and comparison with more detailed endoscopic and pathological findings will likely provide new insight into non‐duodenal ampullary carcinogenesis as well as the endoscopic diagnosis of early non‐ampullary duodenal lesions.
Author contributions statement
RO and TS proposed the concept, contributed to the study design, performed statistical analysis, and wrote the manuscript. RO, TS, ST, YK, HM, KN, SK, TY and TM contributed to the collection and interpretation of pathological and immunohistochemical data. RO, TS, YS, HS, EY, EK, HK and TT contributed to the collection and interpretation of molecular data. RO, KH, HN, SI, ST, NY and HD contributed to the collection of clinical data. ST, YS, HS, TY and TM assisted with draft revision of the manuscript. All the authors approved the final draft submitted for publication.
Supporting information
Figure S1. Representative views of duodenal adenomas and intramucosal adenocarcinomas (H&E staining)
Figure S2. Representative views of H&E staining (left) and β‐catenin nuclear expression (right)
Figure S3. Representative views of immunohistochemistry analysis of mismatch repair (MMR) proteins
Table S1. Primer sequences used in this study
Table S2. List of genes in the custom Ampliseq gene panel used in this study
Table S3. Comparison of clinicopathological and molecular characteristics between small intestinal‐type non‐ampullary duodenal lesions located in the proximal and distal parts of the duodenum
Table S4. Relationship between CIMP‐high status and clinicopathological characteristics of non‐ampullary duodenal lesions
Table S5. Summary of targeted amplicon sequencing data
Table S6. Somatic nonsynonymous mutations found in 102 non‐ampullary duodenal lesions
Table S7. Copy number alterations found in 102 non‐ampullary duodenal lesions
Acknowledgements
We thank Mutsumi Toyota, Saeko Ishioka, Isao Kurahayashi, Noriko Sasahara, Satomi Saito, Atsuko Asaka, Naoko Abe, and Aki Iwata for their excellent technical assistance; Rie Oka for helpful advice for statistical analyses; and Dr William F Goldman for editing the manuscript. This study was supported in part by JSPS KAKENHI Grant Nos JP16K09304 and JP19K08463 (to TS), JP17K09374 (to SI), JP19K08367 (to RO), and 16H06279 (PAGS) (to TT); a Grant‐in‐Aid from the Takeda Science Foundation (to TS); and an Extramural Collaborative Research Grant of the Cancer Research Institute, Kanazawa University (to YS).
No conflicts of interest were declared.
Data availability statement
Access to the data from our study is available upon reasonable request.
References
- 1. Pedersen KS, Raghav K, Overman MJ. Small bowel adenocarcinoma: etiology, presentation, and molecular alterations. J Natl Compr Canc Netw 2019; 17: 1135–1141. [DOI] [PubMed] [Google Scholar]
- 2. Schottenfeld D, Beebe‐Dimmer JL, Vigneau FD. The epidemiology and pathogenesis of neoplasia in the small intestine. Ann Epidemiol 2009; 19: 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qubaiah O, Devesa SS, Platz CE, et al Small intestinal cancer: a population‐based study of incidence and survival patterns in the United States, 1992 to 2006. Cancer Epidemiol Biomarkers Prev 2010; 19: 1908–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jepsen JM, Persson M, Jakobsen NO, et al Prospective study of prevalence and endoscopic and histopathologic characteristics of duodenal polyps in patients submitted to upper endoscopy. Scand J Gastroenterol 1994; 29: 483–487. [DOI] [PubMed] [Google Scholar]
- 5. Culver EL, McIntyre AS. Sporadic duodenal polyps: classification, investigation, and management. Endoscopy 2011; 43: 144–155. [DOI] [PubMed] [Google Scholar]
- 6. Tsuji S, Doyama H, Tsuji K, et al Preoperative endoscopic diagnosis of superficial non‐ampullary duodenal epithelial tumors, including magnifying endoscopy. World J Gastroenterol 2015; 21: 11832–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perzin KH, Bridge MF. Adenomas of the small intestine: a clinicopathologic review of 51 cases and a study of their relationship to carcinoma. Cancer 1981; 48: 799–819. [DOI] [PubMed] [Google Scholar]
- 8. Shepherd NA, Carr NJ, Howe JR, et al Carcinoma of the small intestine In WHO Classification of Tumours of the Digestive System, Bosman FT, Carneiro F, Hruban RH, et al (eds). IARC Press: Lyon, 2010; 95–101. [Google Scholar]
- 9. Okada K, Fujisaki J, Kasuga A, et al Sporadic nonampullary duodenal adenoma in the natural history of duodenal cancer: a study of follow‐up surveillance. Am J Gastroenterol 2011; 106: 357–364. [DOI] [PubMed] [Google Scholar]
- 10. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. [DOI] [PubMed] [Google Scholar]
- 11. Overman MJ, Hu CY, Kopetz S, et al A population‐based comparison of adenocarcinoma of the large and small intestine: insights into a rare disease. Ann Surg Oncol 2012; 19: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toyota M, Ahuja N, Ohe‐Toyota M, et al CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96: 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki H, Yamamoto E, Maruyama R, et al Biological significance of the CpG island methylator phenotype. Biochem Biophys Res Commun 2014; 455: 35–42. [DOI] [PubMed] [Google Scholar]
- 14. Kim SG, Chan AO, Wu TT, et al Epigenetic and genetic alterations in duodenal carcinomas are distinct from biliary and ampullary carcinomas. Gastroenterology 2003; 124: 1300–1310. [DOI] [PubMed] [Google Scholar]
- 15. Fu T, Pappou EP, Guzzetta AA, et al CpG island methylator phenotype‐positive tumors in the absence of MLH1 methylation constitute a distinct subset of duodenal adenocarcinomas and are associated with poor prognosis. Clin Cancer Res 2012; 18: 4743–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun L, Guzzetta AA, Fu T, et al CpG island methylator phenotype and its association with malignancy in sporadic duodenal adenomas. Epigenetics 2014; 9: 738–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laforest A, Aparicio T, Zaanan A, et al ERBB2 gene as a potential therapeutic target in small bowel adenocarcinoma. Eur J Cancer 2014; 50: 1740–1746. [DOI] [PubMed] [Google Scholar]
- 18. Alvi MA, McArt DG, Kelly P, et al Comprehensive molecular pathology analysis of small bowel adenocarcinoma reveals novel targets with potential for clinical utility. Oncotarget 2015; 6: 20863–20874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan W, Zhang Z, Dai B, et al Whole‐exome sequencing of duodenal adenocarcinoma identifies recurrent Wnt/β‐catenin signaling pathway mutations. Cancer 2016; 122: 1689–1696. [DOI] [PubMed] [Google Scholar]
- 20. Schrock AB, Devoe CE, McWilliams R, et al Genomic profiling of small‐bowel adenocarcinoma. JAMA Oncol 2017; 3: 1546–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hänninen UA, Katainen R, Tanskanen T, et al Exome‐wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet 2018; 14: e1007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adam L, San Lucas FA, Fowler R, et al DNA sequencing of small bowel adenocarcinomas identifies targetable recurrent mutations in the ERBB2 signaling pathway. Clin Cancer Res 2019; 25: 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haan JC, Buffart TE, Eijk PP, et al Small bowel adenocarcinoma copy number profiles are more closely related to colorectal than to gastric cancers. Ann Oncol 2012; 23: 367–374. [DOI] [PubMed] [Google Scholar]
- 24. Kojima Y, Ohtsuka K, Ohnishi H, et al APC:T1556fs and STK11 mutations in duodenal adenomas and adenocarcinomas. Surg Today 2018; 48: 765–772. [DOI] [PubMed] [Google Scholar]
- 25. Wagner PL, Chen YT, Yantiss RK. Immunohistochemical and molecular features of sporadic and FAP‐associated duodenal adenomas of the ampullary and nonampullary mucosa. Am J Surg Pathol 2008; 32: 1388–1395. [DOI] [PubMed] [Google Scholar]
- 26. Miwa A, Kuwano S, Tomita H, et al The different pathogeneses of sporadic adenoma and adenocarcinoma in non‐ampullary lesions of the proximal and distal duodenum. Oncotarget 2017; 8: 41078–41090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakaguchi Y, Yamamichi N, Tomida S, et al Identification of marker genes and pathways specific to precancerous duodenal adenomas and early stage adenocarcinomas. J Gastroenterol 2019; 54: 131–140. [DOI] [PubMed] [Google Scholar]
- 28. Endoscopic Classification Review Group . Update on the Paris classification of superficial neoplastic lesions in the digestive tract. Endoscopy 2005; 37: 570–578. [DOI] [PubMed] [Google Scholar]
- 29. Murakami T, Akazawa Y, Yatagai N, et al Molecular characterization of sessile serrated adenoma/polyps with dysplasia/carcinoma based on immunohistochemistry, next‐generation sequencing, and microsatellite instability testing: a case series study. Diagn Pathol 2018; 13: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dixon MF. Gastrointestinal epithelial neoplasia: Vienna revisited. Gut 2002; 51: 130–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toyota M, Suzuki H, Sasaki Y, et al Epigenetic silencing of microRNA‐34b/c and B‐cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68: 4123–4132. [DOI] [PubMed] [Google Scholar]
- 32. Weisenberger DJ, Siegmund KD, Campan M, et al CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–793. [DOI] [PubMed] [Google Scholar]
- 33. Yachida S, Wood LD, Suzuki M, et al Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell 2016; 29: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gingras MC, Covington KR, Chang DK, et al Ampullary cancers harbor ELF3 tumor suppressor gene mutations and exhibit frequent WNT dysregulation. Cell Rep 2016; 14: 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakagaki T, Tamura M, Kobashi K, et al Profiling cancer‐related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget 2017; 8: 59113–59122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh RR, Patel KP, Routbort MJ, et al Clinical validation of a next‐generation sequencing screen for mutational hotspots in 46 cancer‐related genes. J Mol Diagn 2013; 15: 607–622. [DOI] [PubMed] [Google Scholar]
- 37. Ougolkov AV, Yamashita K, Mai M, et al Oncogenic β‐catenin and MMP‐7 (matrilysin) cosegregate in late‐stage clinical colon cancer. Gastroenterology 2002; 122: 60–71. [DOI] [PubMed] [Google Scholar]
- 38. Sekine S, Yamashita S, Tanabe T, et al Frequent PTPRK–RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol 2016; 239: 133–138. [DOI] [PubMed] [Google Scholar]
- 39. Goda K, Kikuchi D, Yamamoto Y, et al Endoscopic diagnosis of superficial non‐ampullary duodenal epithelial tumors in Japan: multicenter case series. Dig Endosc 2014; 26(suppl 2): 23–29. [DOI] [PubMed] [Google Scholar]
- 40. Cerami E, Gao J, Dogrusoz U, et al The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao J, Aksoy BA, Dogrusoz U, et al Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsubara A, Sekine S, Kushima R, et al Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum. J Pathol 2013; 229: 579–587. [DOI] [PubMed] [Google Scholar]
- 44. Matsubara A, Ogawa R, Suzuki H, et al Activating GNAS and KRAS mutations in gastric foveolar metaplasia, gastric heterotopia, and adenocarcinoma of the duodenum. Br J Cancer 2015; 112: 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kimura T, Yamamoto E, Yamano HO, et al A novel pit pattern identifies the precursor of colorectal cancer derived from sessile serrated adenoma. Am J Gastroenterol 2012; 107: 460–469. [DOI] [PubMed] [Google Scholar]
- 46. Rokutan H, Abe H, Nakamura H, et al Initial and crucial genetic events in intestinal‐type gastric intramucosal neoplasia. J Pathol 2019; 247: 494–504. [DOI] [PubMed] [Google Scholar]
- 47. Saglam O, Xiong Y, Marchion DC, et al ERBB4 expression in ovarian serous carcinoma resistant to platinum‐based therapy. Cancer Control 2017; 24: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei R, Zhao M, Zheng CH, et al Concordance between somatic copy number loss and down‐regulated expression: a pan‐cancer study of cancer predisposition genes. Sci Rep 2016; 6: 37358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roszik J, Wu CJ, Siroy AE, et al Somatic copy number alterations at oncogenic loci show diverse correlations with gene expression. Sci Rep 2016; 6: 19649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative views of duodenal adenomas and intramucosal adenocarcinomas (H&E staining)
Figure S2. Representative views of H&E staining (left) and β‐catenin nuclear expression (right)
Figure S3. Representative views of immunohistochemistry analysis of mismatch repair (MMR) proteins
Table S1. Primer sequences used in this study
Table S2. List of genes in the custom Ampliseq gene panel used in this study
Table S3. Comparison of clinicopathological and molecular characteristics between small intestinal‐type non‐ampullary duodenal lesions located in the proximal and distal parts of the duodenum
Table S4. Relationship between CIMP‐high status and clinicopathological characteristics of non‐ampullary duodenal lesions
Table S5. Summary of targeted amplicon sequencing data
Table S6. Somatic nonsynonymous mutations found in 102 non‐ampullary duodenal lesions
Table S7. Copy number alterations found in 102 non‐ampullary duodenal lesions
Data Availability Statement
Access to the data from our study is available upon reasonable request.