
Figure 5.
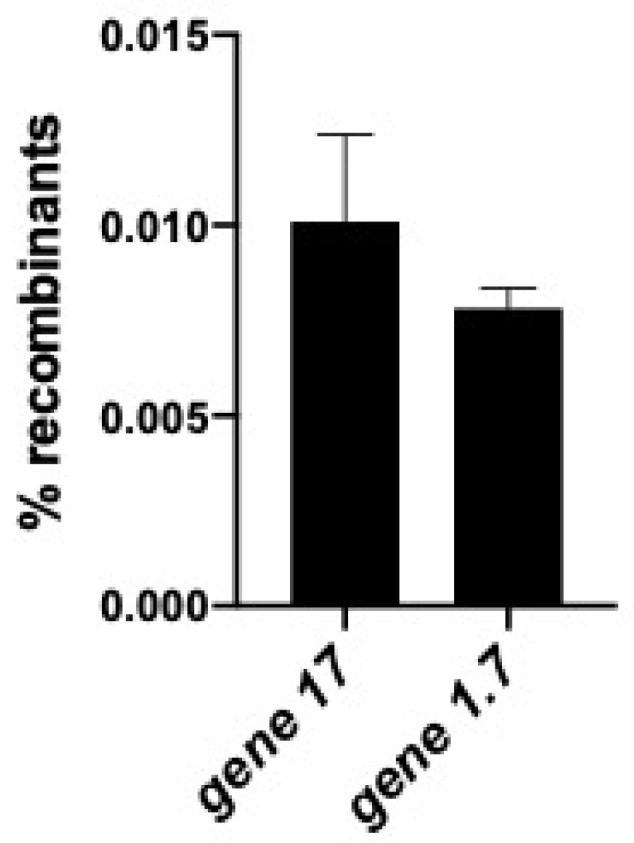
Replacement of T7 genes g1.7 and g17 with the bacterial trxA gene. Recombineering was used to replace two different T7 genes with the bacterial trxA gene. One gene, g1.7, is non-essential; the other gene, g17, is essential and required for plaque formation. The trxA dsDNA PCR fragments, g1.7<>trxA and g17<>trxA were amplified using the bacterial trxA gene as a template to generate the trxA gene flanked by T7 DNA homology that targets recombination to g1.7 or g17, respectively. Oligos ARP301 and ARP302 used for PCR are designed with trxA having T7 flanking homologies to replace g1.7 with trxA. Oligos ARP356 and ARP357 used for PCR are designed with trxA having T7 flanking homologies to replace g17 with trxA (Table 2). LT976 cells were induced for Red-mediated recombination and then co-electroporated with genomic T7 DNA and either the g1.7 or g17 targeted trxA PCR products. The electroporation reactions were out-grown in SOC medium at 37 °C. Gene 1.7 trxA replacements were detected on E. coli B 3783, a host that lacks the trxA gene in its genome. The g17 replacements were detected on ARP3750 at 42 °C. Non-recombinant T7 cannot grow on these two bacteria that lack trxA. T7 recombinant frequencies in the lysates were calculated by the following equations: [(T7 g1.7<>trxA recombinant phage titer on E. coli B3783 ΔtrxA/(total phage titer on E. coli B)] × 100; [(T7 g17<>trxA recombinant phage titer on ARP3750)/(total phage titer on ARP3607)] × 100. Values represent an average of at least three biological replicates with the standard error of the mean calculated. The identity and accuracy of the gene substitutions on the recombinant phages were confirmed by sequence.