

Abstract
Motivation
Tuberculosis (TB) remains one of the main causes of death worldwide. The long and cumbersome process of culturing Mycobacterium tuberculosis complex (MTBC) bacteria has encouraged the development of specific molecular tools for detecting the pathogen. Most of these tools aim to become novel TB diagnostics, and big efforts and resources are invested in their development, looking for the endorsement of the main public health agencies. Surprisingly, no study has been conducted where the vast amount of genomic data available is used to identify the best MTBC diagnostic markers.
Results
In this work, we used large-scale comparative genomics to identify 40 MTBC-specific loci. We assessed their genetic diversity and physiological features to select 30 that are good targets for diagnostic purposes. Some of these markers could be used to assess the physiological status of the bacilli. Remarkably, none of the most used MTBC markers is in our catalog. Illustrating the translational potential of our work, we develop a specific qPCR assay for quantification and identification of MTBC DNA. Our rational design of targeted molecular assays for TB could be used in many other fields of clinical and basic research.
Availability and implementation
The database of non-tuberculous mycobacteria assemblies can be accessed at: 10.5281/zenodo.3374377.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Tuberculosis (TB) is the most lethal infectious disease caused by a single agent, namely bacteria belonging to the Mycobacterium tuberculosis complex (MTBC) (World Health Organization, 2017). Whereas isolating the bacteria from clinical specimens is a time-consuming process, rapid molecular tests have the potential to identify the pathogen DNA in a few hours (Eddabra and Benhassou, 2018; Machado et al., 2018). Over the years, many different molecular assays have been developed for the specific detection of MTBC and its differentiation from non-tuberculous mycobacteria (NTM) (Chin et al., 2018). Most of these assays are based on the PCR amplification of genomic targets that are thought to be specific to the MTBC, like the insertion sequence IS6110, or rely on the design of specific primers that amplify conserved bacterial regions such as the rpoB or rrs genes. Most of these markers were identified in the nineties and have not been evaluated in the light of current genomic databases. In addition, several shortcomings are known for the different assays targeting current MTBC markers, being of special concern the lack of specificity and sensitivity (Chin et al., 2018).
The development of new molecular tools for TB diagnosis is an active area of research, with many companies involved, looking for the endorsement of the World Health Organization (WHO) (Pai et al., 2016). The most successful example has been the Xpert MTB/RIF test (Cirillo et al., 2017), which was endorsed by the WHO back in 2010 for TB diagnosis and recommended as the first-line diagnostic in 2017 (WHO, 2017). The Xpert assay amplifies a conserved region within the rpoB gene to detect both MTBC DNA and drug resistance mutations to rifampicin. With the aim of improving its sensitivity, the new Xpert MTB/RIF Ultra assay also amplifies the MTBC insertion sequence IS6110 and IS1018. However, these insertion sequences were described as MTBC-specific decades ago (Collins and Stephens, 1991; Thierry et al., 1990) and several studies have shown them to be present in non-MTBC organisms while some MTBC strains are known to lack any copy (Liébana et al., 1996; Müller et al., 2015; Pérez-Osorio et al., 2012; Roychowdhury et al., 2015). The fact that the novel assays developed are still based on the amplification of loci that are not specific to the MTBC, highlights the need for the discovery of novel and specific MTBC targets.
Analyzing omic data has been proven to be an effective strategy for the identification of species-specific markers in several organisms (Buchanan et al., 2017; Carmona et al., 2012; Carrera et al., 2017; Koul and Kumar, 2015; Wang et al., 2017; Zozaya-Valdés et al., 2017). In the field of TB, large-scale omic studies have been conducted to identify new biomarkers that are present in patient samples as response to TB infection and genetic markers that are associated with drug-resistance (Cui et al., 2017; Drouin et al., 2016; Ezewudo et al., 2018; Groote et al., 2017; Walzl et al., 2018). In contrast, comparative genomics studies identifying MTBC-specific loci have been scarce and based either on limited Mycobacteria genome databases or on selection criteria that does not assure specificity (Kakhki et al., 2019; Zhao et al., 2014). Furthermore, none of the approaches have analyzed the genetic diversity of the markers using a representative global collection of MTBC strains, a key feature for a universal diagnostic target.
Here, we perform a large-scale comparative genomic analysis to provide a catalog of MTBC-specific loci that will be of great utility for the scientific community working on the development of new research and clinical tools for TB. We assess the global diversity of each MTBC-specific gene among a comprehensive dataset of more than 4700 MTBC strains, spanning all known lineages in which MTBC is divided, showing the value of using the genomic data to identify the best targets for diagnostic assays. We found that the main MTBC markers used up to date are also present in other organisms, mainly NTM. As a proof of concept, we develop a qPCR assay capable of quantifying MTBC DNA with 100% specificity.
2 Materials and methods
2.1 In silico identification of MTBC-specific diagnostic gene markers
To identify MTBC-specific loci, we used blastn (Altschul et al., 1990) to look for all the genes of the M.tuberculosis reference strain H37Rv (NC_000962.3) in the NCBI nucleotide non-redundant database (accessed October 2018) and a custom database comprising 4277 NTM assemblies (Supplementary Methods S1). We filtered the results with a set of stringent parameters that allowed us to provide a diverse catalog of MTBC-specific loci. We focused on the identification of loci having large fractions with no homology outside the MTBC or alignments with low identities, thus enabling the development of highly specific molecular assays minimizing the risk of cross-reaction. We analyzed loci instead of genomic fragments as they are functional units that tend to be more conserved across MTBC strains. We kept loci that aligned with query coverages below 60% and identities lower than 80%. In addition, we also kept genes with query coverages below 25% disregarding the identity of the alignment. In this way, we retained loci containing conserved domains across species but with no global match with genes of other species. Finally, we only considered loci that were present in all the species of the MTBC species genomes in the database.
Once we identified the MTBC-specific loci, we assessed their genetic diversity in circulating MTBC strains. To do this, we analyzed the polymorphisms [single nucleotide polymorphisms (SNPs) and indels] observed at each position across a dataset comprising 4766 genomes of MTBC strains (Chiner-Oms et al., 2019). In the case of indels, we only considered positions showing an indel in at least 10 strains (0.2% of the database) to avoid the noise introduced by single-strain indels spanning large genic regions. Finally, we looked for available information of these genes in the bibliography, what allowed us to discard some candidates based on their genomic context and provide extended information about their physiology. We gathered transcriptomic and proteomic data derived from different published studies: transcriptomic data in response to overexpression of 206 transcription factors (Turkarslan et al., 2015), different genotoxic stresses (Namouchi et al., 2016) and response to nitric oxide stress at different time-points (Cortes et al., 2017), as well as proteomic data in response to nutrient starvation (Albrethsen et al., 2013).
2.2 Set-up of a MTBC-specific qPCR assay for DNA detection and quantification
We selected one of the MTBC-specific targets to set up a qPCR assay for the detection and quantification of MTBC DNA. To select the target for the assay, we took into consideration the number of polymorphisms per base, the absence of high-prevalent polymorphisms, the gene length and its genomic context. We designed the primers and probes for the assay using the web tool Primer-BLAST (Ye et al., 2012). The qPCR assay consisted on the amplification of a 65 bp region within the Rv2341 gene using the following primers: Forward-GCCGCTCATGCTCCTTGGAT, Reverse-AGGTCGGTTCGCTGGTCTTG, Probe-TGAGTGCCTGCGGCCGCAGCGC.
To test the specificity of the assay, we performed qPCR experiments with DNA from different MTBC lineages selected from the MTBC reference dataset described in (Borrell et al., 2019), human DNA, a mock sample with mixed DNA from 20 different bacterial species and 15 different species of NTM (Supplementary Methods S2 and S3). All assays were done per triplicate and with an initial DNA concentration of 0.5 ng/ul. In addition, we evaluated the performance of the assay detecting MTBC DNA in a small test set of clinical samples. We used DNA extracted from 12 homogenized sputum samples from confirmed TB patients, two of them with negative smear microscopy (see ethics statement in Supplementary Material). The reaction efficiency was calculated with decreasing concentrations of H37Ra DNA.
3 Results
3.1 A catalog of MTBC-specific markers
We identified 40 genes to be uniquely present in members of the MTBC according to our filtering parameters (Fig. 1). The median number of SNPs per base (across 4766 MTBC strains) was 0.07, with some of these genes showing either higher or lower diversities (up to 0.10 and 0.04 SNPs/base, respectively). Importantly, although most of the polymorphisms analyzed were strain-specific, we observed high prevalent polymorphisms as well (Fig. 1, Supplementary File S1). For instance, Rv0610c showed a SNP present in 4182 strains and Rv2823c showed an insertion in 4345 strains. Analysis of the phylogenetic distribution of these polymorphisms confirmed that they mapped to deep branches in the phylogeny. For example, the SNP in Rv0610c affected all modern lineages (L2, L3, L4).
Fig. 1.

The 40 MTBC-specific loci. Gene names in red indicate the 10 loci that were discarded as diagnostic markers for being within RD (Rv2274c and Rv2816c-2820c), associated to CRISPR (Rv2816c-2823c) or duplicated in the genome (Rv3424c). Concentric circles represent different genetic diversity metrics. Outer heatmap: number of SNPs per base. Blue circle: prevalence of each SNP of each gene across the database of MTBC strains. Inner, red circle: prevalence of each indel of each gene across the database of MTBC strains. Note that both inner circles have two scales, one from 0 to 300 strains and other from 300 to 4800 strains. The region of the Rv2341 gene amplified in our qPCR assay is indicated in light yellow. RD 182 and 207 are clearly detected in our analysis, indicated as contiguous deleted regions in a high number of strains.
Among the initial 40 MTBC-specific genes, 9 were discarded as potential diagnostic markers since they were included in large genomic deletions known as regions of difference (RD) 182 (Rv2274c) and RD 207 (Rv2816c-Rv2820c) (Gagneux et al., 2006) or were in variable genomic regions associated with CRISPR elements (Rv2816c-2823c) (Freidlin et al., 2017). Another gene, Rv3424c was also discarded as we found it to be duplicated in a labile genomic region, between the transposase of the insertion sequence IS1532 and PPE59. Therefore, the curated list of MTBC-specific diagnostic markers finally consisted of 30 genes (Fig. 1).
When looking at published transcriptomic and proteomic data, we found that Rv2003c, Rv2142c and Rv3472 proteins are produced in greater levels (6.19, 3.6 and 100-fold, respectively) when the bacteria is subjected to starvation. Interestingly, Rv2003c is also observed to be overexpressed upon treatment with nitric oxide (Supplementary File S2).
3.2 A specific qPCR assay for MTBC DNA quantification
Based on our genomic analysis, we set up a qPCR assay targeting the Rv2341 gene. This gene, described as ‘probable conserved lipoprotein lppQ’ in the Mycobrowser database (Kapopoulou et al., 2011), is situated in a stable genomic region, between the asparagine tRNA and the gene of the DNA primase. As shown in Figure 1, we were able to design an optimized set of primers avoiding prevalent polymorphisms.
The specificity of the assay was 100% since no cross-reaction was observed with non-MTBC samples. Fluorescence was occasionally detected for some non-MTBC samples in cycles beyond Cq 35 (Fig. 2a). This only happened for 1/9 replicates of the mock bacterial DNA and 16/135 replicates of NTM. Importantly, no NTM sample amplified consistently between replicates, indicating that fluorescence in late cycles are not due to non-specific amplifications but likely to cross-contamination or qPCR artifacts. As shown, the sensitivity of the assay in our small test set was of 100%, since we were able to detect MTBC DNA in all TB patient sputa, including two confirmed TB cases with a negative smear microscopy (Supplementary File S3). The standard curve using purified H37Ra DNA showed an efficiency of the reaction of 100% (2.01) with a limit of detection of 10fg (at Cq 34.43), hypothetically corresponding to two genome equivalents (Fig. 2b). Based on these observations, any result beyond Cq 35 should be considered negative and, therefore, the final setup of our qPCR assay consists in 35 amplification cycles.
Fig. 2.
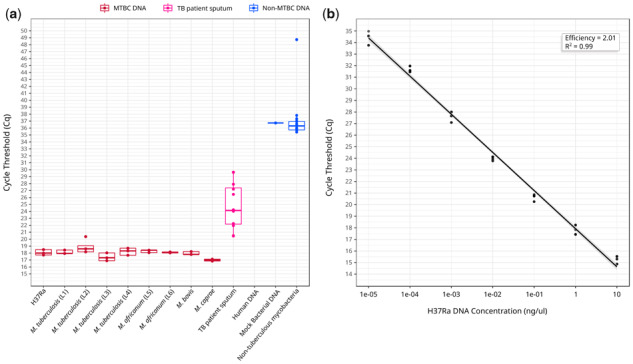
(a) Cq values for MTBC samples of different lineages, sputum samples from confirmed TB patients, and non-MTBC samples. (b) Standard curve for the assay with decreasing concentrations of pure H37Ra DNA (from 10 ng/ul to to 1e–5 ng/ul). The reaction efficiency was of 100% (2.01). In both panels (a) and (b) points represent the mean Cq value of the three sample replicates in each of the three replicated experiments
4 Discussion
Identification of MTBC markers has been an active area of research over the last decades. It is striking that, for such a relevant disease, for which tons of genomic data are already available, the identification of MTBC-specific genes had been relegated to the background. So far, efforts have been focused on the design of MTBC-specific primers and the optimization of assay conditions based on targets identified even decades ago. Recently, Lei Zhou et al. analyzed one loci widely distributed across mycobacteria, the ku gene, against a database of more than 7000 genomes and assessed its suitability to identify several mycobacteria species including MTBC (Zhou et al., 2019). Other works have developed molecular assays based on targets that are claimed to be MTBC-specific, with some of them also trying to identify MTBC-specific loci by using different strategies, including comparative genomics (Kakhki et al., 2019; Zhao et al., 2014). However, the targets described until now are far from specific (Supplementary File S6), most likely due to the limited datasets analyzed and permissive selection criteria.
Our analysis addresses these limitations by analyzing large genomic datasets and using stringent selection criteria to provide a diverse catalog of highly specific MTBC targets to be used by developers of novel molecular assays for TB. Importantly, compared with previous efforts, we also analyzed the genomic diversity of these targets across thousands of strains from all known MTBC lineages, since the conservation of the targets is a key feature to ensure the reproducibility of the diagnostic tests across clinical settings. Additionally, we found that some of the markers that we identify could be targeted to determine the physiological status of MTBC bacteria under certain conditions. For example, Rv2003c, overexpressed during starvation and upon treatment with nitric oxide (Albrethsen et al., 2013; Cortes et al., 2017), is also up-regulated during dormancy (Hegde et al., 2012). Similarly, Rv1374c contains a small RNA that is highly expressed during exponential growth (Arnvig et al., 2011), and hence might be used to evaluate the replicative state of the bacilli.
Strikingly, none of the markers considered to be MTBC-specific up to date and included in most diagnostic tests are in our list of unique MTBC genes. For instance, when examining in which species the IS6110 can be found, we observed several non-MTBC organisms, including 14 NTMs, carrying at least 1 copy. The same is true for IS1081 and mpt64, present in 38 and 6 NTM, respectively (Supplementary Files S4 and S6).
To illustrate the translational potential of our work, we developed a proof-of-concept qPCR assay capable of quantifying MTBC DNA with 100% specificity and a sensitivity up to 2 genome copies. However, its use as a diagnostic tool would require additional evaluations and optimizations with larger sets of samples to ensure the robustness of the assay and to maximize its sensitivity. It is important to note that the aim of our work was not to compare our assay with current diagnostic tests but to provide novel targets for future ones.
Here, we use the genomic data available for a rational, evolutionary-guided, design of a targeted assay for TB. However, this strategy could be used for many other organisms and applications. When possible, the development of next-generation molecular assays should be guided by comprehensive analyses that integrate the data available for each organism.
Funding
This work was supported by projects of the European Research Council (ERC) (638553-TB-ACCELERATE), Ministerio de Economía y Competitividad and Ministerio de Ciencia, Innovación y Universidades (Spanish Government), SAF2013-43521-R, SAF2016-77346-R and SAF2017–92345–EXP (to I.C.), BES-2014-071066 (to G.A.G.), FPU 13/00913 (to A.C.O.).
Conflict of Interest: none declared.
Supplementary Material
References
- Albrethsen J. et al. (2013) Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteomics, 12, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Arnvig K.B. et al. (2011) Sequence-based analysis uncovers an abundance of Non-Coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog., 7, e1002342.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell S. et al. (2019) Reference set of Mycobacterium tuberculosis clinical strains: a tool for research and product development. PLoS One, 14, e0214088.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan C.J. et al. (2017) A genome-wide association study to identify diagnostic markers for human pathogenic campylobacter jejuni strains. Front. Microbiol., 8, 1224 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona S.J. et al. (2012) Diagnostic peptide discovery: prioritization of pathogen diagnostic markers using multiple features. PLoS One, 7, e50748.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera M. et al. (2017) Characterization of foodborne strains of Staphylococcus aureus by shotgun proteomics: functional networks, virulence factors and species-specific peptide biomarkers. Front. Microbiol., 8, 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K.L. et al. (2018) DNA markers for tuberculosis diagnosis. Tuberculosis, 113, 139–152. [DOI] [PubMed] [Google Scholar]
- Chiner-Oms Á. et al. (2019) Genomic determinants of speciation and spread of the complex. Sci. Adv., 5, eaaw3307.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D.M. et al. (2017) Evolution of phenotypic and molecular drug susceptibility testing. Adv. Exp. Med. Biol., 1019, 221–246. [DOI] [PubMed] [Google Scholar]
- Collins D.M., Stephens D.M. (1991) Identification of an insertion sequence, IS1081, in Mycobacterium bovis. FEMS Microbiol. Lett., 67, 11–15. [DOI] [PubMed] [Google Scholar]
- Cortes T. et al. (2017) Delayed effects of transcriptional responses in Mycobacterium tuberculosis exposed to nitric oxide suggest other mechanisms involved in survival. Sci. Rep., 7, 8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J.-Y. et al. (2017) Characterization of a novel panel of plasma microRNAs that discriminates between Mycobacterium tuberculosis infection and healthy individuals. PLoS One, 12, e0184113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A. et al. (2016) Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genomics, 17, 754.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddabra R., Benhassou H.A. (2018) Rapid molecular assays for detection of tuberculosis. Pneumonia, 10, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezewudo M. et al. (2018) Integrating standardized whole genome sequence analysis with a global Mycobacterium tuberculosis antibiotic resistance knowledgebase. Sci. Rep., 8, 15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidlin P.J. et al. (2017) Structure and variation of CRISPR and CRISPR-flanking regions in deleted-direct repeat region Mycobacterium tuberculosis complex strains. BMC Genomics, 18, 168.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S. et al. (2006) Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA, 103, 2869–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groote M.A.D. et al. (2017) Discovery and validation of a six-marker serum protein signature for the diagnosis of active pulmonary tuberculosis. J. Clin. Microbiol, 55, 3057–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S.R. et al. (2012) Understanding communication signals during mycobacterial latency through predicted genome-wide protein interactions and Boolean modeling. PLoS One, 7, e33893.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhki R.K. et al. (2019) The short-chain dehydrogenases/reductases (SDR) gene: a new specific target for rapid detection of Mycobacterium tuberculosis complex by modified comparative genomic analysis. Infect. Genet. Evol., 70, 158–164. [DOI] [PubMed] [Google Scholar]
- Kapopoulou A. et al. (2011) The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Kekkaku, 91, 8–13. [DOI] [PubMed] [Google Scholar]
- Koul S., Kumar P. (2015) A unique genome wide approach to search novel markers for rapid identification of bacterial pathogens. J. Mol. Genet. Med., 9, 1–2. [Google Scholar]
- Liébana E. et al. (1996) Assessment of genetic markers for species differentiation within the Mycobacterium tuberculosis complex. J. Clin. Microbiol., 34, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado D. et al. (2018) Advances in the molecular diagnosis of tuberculosis: from probes to genomes. Infect. Genet. Evol., 72, 93–112. [DOI] [PubMed] [Google Scholar]
- Müller R. et al. (2015) Complications in the study of ancient tuberculosis: non-specificity of IS6110 PCRs. Sci. Technol. Archaeol. Res, 1, 1–8. [Google Scholar]
- Namouchi A. et al. (2016) The Mycobacterium tuberculosis transcriptional landscape under genotoxic stress. BMC Genomics, 17, 791.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai M. et al. (2016) Tuberculosis diagnostics: state of the art and future directions. Microbiol. Spectr., 4, 369–375. [DOI] [PubMed] [Google Scholar]
- Pérez-Osorio A.C. et al. (2012) Rapid identification of mycobacteria and drug-resistant Mycobacterium tuberculosis by use of a single multiplex PCR and DNA sequencing. J. Clin. Microbiol., 50, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury T. et al. (2015) Analysis of IS6110 insertion sites provide a glimpse into genome evolution of Mycobacterium tuberculosis. Sci. Rep., 5, 12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry D. et al. (1990) IS6110, an IS-like element of Mycobacterium tuberculosis complex. Nucleic Acids Res., 18, 188.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkarslan S. et al. (2015) A comprehensive map of genome-wide gene regulation in Mycobacterium tuberculosis. Sci. Data, 2, 150010.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzl G. et al. (2018) Tuberculosis: advances and challenges in development of new diagnostics and biomarkers. Lancet Infect. Dis., 18, e199–e210. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2017) A genoproteomic approach to detect peptide markers of bacterial respiratory pathogens. Clin. Chem., 63, 1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2017) WHO—WHO meeting report of a technical expert consultation. WHO.
- World Health Organization. (2017) Global tuberculosis report 2017. WHO.
- Ye J. et al. (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics, 13, 134.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. et al. (2014) An efficient alternative marker for specific identification of Mycobacterium tuberculosis. World J. Microbiol. Biotechnol., 30, 2189–2197. [DOI] [PubMed] [Google Scholar]
- Zhou L. et al. (2019) A new single gene differential biomarker for complex and non-tuberculosis mycobacteria. Front. Microbiol., 10, 1887.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zozaya-Valdés E. et al. (2017) Target-Specific assay for rapid and quantitative detection of mycobacterium chimaera DNA. J. Clin. Microbiol., 55, 1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.