

SUMMARY
The discovery of H3K27M mutations in pediatric gliomas marked a new chapter in cancer epigenomics. Numerous studies have investigated the effect of this mutation on H3K27 trimethylation, but only recently have we started to realize its additional effects on the epigenome. Here, we use isogenic glioma H3K27M+/− cell lines to investigate H3K27 methylation and its interaction with H3K36 and H3K9 modifications. We describe a “step down” effect of H3K27M on the distribution of H3K27 methylation: me3 is reduced to me2, me2 is reduced to me1, whereas H3K36me2/3 delineates the boundaries for the spread of H3K27me marks. We also observe a replacement of H3K27me2/3 silencing by H3K9me3. Using a computational simulation, we explain our observations by reduced effectiveness of PRC2 and constraints imposed on the deposition of H3K27me by antagonistic H3K36 modifications. Our work further elucidates the effects of H3K27M in gliomas as well as the general principles of deposition in H3K27 methylation.
Graphical Abstract
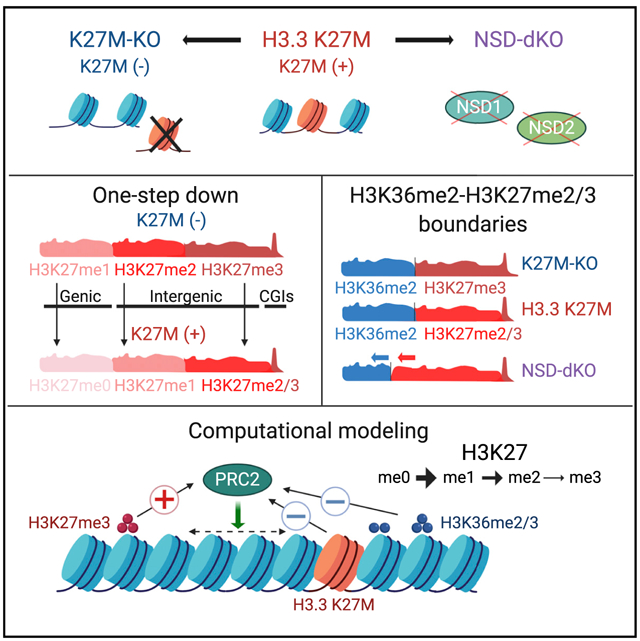
In Brief
Harutyunyan et al. use isogenic glioma H3K27M+/− cell lines to demonstrate the rewiring of the epigenome, specifically H3K27me1/2/3, H3K36me2/3, and H3K9me3. The dynamic deposition of histone marks is simulated by a stochastic model. This work further advances the understanding of the deposition of H3K27 methylation in H3K27M mutant gliomas.
INTRODUCTION
Pediatric high-grade gliomas (HGGs) are aggressive brain tumors with a dismal prognosis and are a major cause of cancer-related mortality in children. Most pediatric HGGs arising in the brain midline carry somatic mutations leading to lysine-27-to-methionine substitution (K27M) in histone 3 (H3) genes (Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Wu et al., 2012). These H3K27M mutations are fully clonal (Nikbakht et al., 2016) and are the founding driver events in HGGs, important for tumor initiation but also for its maintenance (Harutyunyan et al., 2019; Larson et al., 2019; Pathania et al., 2017; Silveira et al., 2019). The primary effect of histone H3 mutations is widespread changes in the epigenomic landscape, which globally deregulate gene expression.
H3K27M can occur in both canonical (H3.1, H3.2) and non-canonical (H3.3) H3 genes. Previous studies have shown that, despite comprising only 3%–17% of the H3 pool, H3K27M mutant histones dominantly inhibit the polycomb repressive complex 2 (PRC2), leading to a global reduction of H3K27me2 and H3K27me3 levels with concomitant increases of the H3K27ac mark (Bechet et al., 2014; Bender et al., 2013; Lewis et al., 2013). Multiple studies have investigated the molecular mechanisms responsible for H3K27M-induced changes in H3K27me3 (Bender et al., 2013; Chan et al., 2013; Fang et al., 2018; Harutyunyan et al., 2019; Lewis et al., 2013; Mohammad et al., 2017; Piunti et al., 2017; Stafford et al., 2018). The general consensus is that H3K27me3 is retained at certain unmethylated CpG islands (CGIs), whereas it is mostly lost elsewhere in the genome. There is less agreement upon the mechanisms responsible for these changes, and several theories have been suggested: preferential recruitment of PRC2 to H3K27M containing nucleosomes with subsequent retention and inhibition by sequestration (Chan et al., 2013; Diehl et al., 2019; Fang et al., 2018; Justin et al., 2016; Lewis et al., 2013); PRC2 and H3K27me3 retention at strong PRC2 targets (Mohammad et al., 2017); exclusion of PRC2 from regions containing H3K27M nucleosomes and increase of H3K27ac on such heterotypic nucleosomes (Piunti et al., 2017); H3K27M allowing the deposition of H3K27me3 at unmethylated CGIs but affecting the spread of the mark (Harutyunyan et al., 2019); and H3K27M having partly irreversible inhibition on PRC2 (Stafford et al., 2018).
Notably, most of the studies so far have focused on the H3K27me3 mark as the primary target of H3K27M, although the role of increased levels of H3K27ac in tumorigenesis has also been considered (Krug et al., 2019; Lewis et al., 2013; Mohammad et al., 2017; Nagaraja et al., 2017; Piunti et al., 2017). Other histone marks, possibly equally important in mediating H3K27M pathogenesis, have received much less attention. We have previously shown that, in addition to the decrease and restriction of H3K27me3 to unmethylated CGIs, the genome-wide distribution of H3K27me2 is also markedly changed (Harutyunyan et al., 2019). Another study suggested that, as a consequence of H3K27me3 loss in H3K27M mutant cells, the levels of H3K36me2 increase (Stafford et al., 2018). Other than these reports, there has been limited investigation of potential genome-wide changes in other histone modifications, even though they could prove integral to the pathogenesis of H3K27M-mutant HGGs.
In this study, we have used our model system of CRISPR-edited H3.3K27M-mutant HGG cell lines to comprehensively characterize genome-wide changes in a number of histone marks (H3K27me1, H3K27me2, H3K27me3, H3K36me2, H3K36me3, and H3K9me3) and correlate those modifications with each other, aiming to understand the basic rules of co-occurrence and mutual exclusivity of these histone marks in the presence and in the absence of H3.3K27M.
RESULTS
We have previously used CRIPSR/Cas9-editing to remove the H3.3K27M mutation in two HGG cell lines (BT245 K27M-KO and SU-DIPGXIII K27M-KO), for comparison with their parental cell lines in an isogenic tumor context (Harutyunyan et al., 2019). We have also described a wild-type GBM cell line, G477, overexpressing H3.3K27M or H3.3K27R (control). Here, we extended this analysis with another isogenic H3.3K27M cell line (HSJ019 K27M-KO; Figures S1A and S1B) and a wider range of histone marks. For simplicity, we refer to H3.3K27M-mutant lines as K27M+ and their isogenic H3.3K27M-KO, as well as K27R overexpression as K27M− (Table S1).To better understand the changes in the epigenomic landscape induced by H3.3K27M mutation, it is important to consider not only H3K27 modifications in isolation but also in conjunction with other histone marks. We focus on H3K36me2/3 and H3K9me3 because they also define large genomic domains and have been reported to interact with H3K27me (Chen and Dent, 2014; Streubel et al., 2018). We illustrate most of the results in the main body and figures of the manuscript using the BT245 cell line, but all of our observations—with the exception of the NSD1/2 knockout (KO)—are corroborated in two additional H3.3K27M cell lines and one wild-type cell line engineered to overexpress H3.3K27M, thus, generalizing across genetic backgrounds, and the complete results are provided in Supplemental Information.
“One-Step” Reduction of H3K27 Methylation in H3.3K27M
As we previously showed (Harutyunyan et al., 2019), the normally broad deposition of H3K27me2 and H3K27me3 is greatly reduced and restricted in K27M+ cells (Figure 1A). Mass spectrometry analysis showed that the presence of H3.3K27M results in a drastic reduction of H3K27me3 levels, significant reduction of H3K27me2, but no change in H3K27me1 (Figure 1B). Unexpectedly, chromatin immunoprecipitation sequencing (ChIP-seq) profiling of H3K27me1 revealed profound genome-wide reorganization of this mark (Figures 1A and S1C). The distribution of H3K27me1 in K27M+ cells appears inverted compared with those in K27M−, i.e., regions highly enriched for this mark in K27M+ are depleted of it in K27M− and vice versa (Figures 1A and 1C). Furthermore, there is substantial similarity between deposition of H3K27me1 in K27M+ and H3K27me2 in K27M−. At the next methylation level, we observed similarity between H3K27me2 in K27M+ and H3K27me3 in K27M−. At the highest methylation level, in K27M+, H3K27me3 is restricted to PRC2 nucleation sites at unmethylated CGIs (Figures 1A, 1C, S1D, and S1E). Although we do not have ChIP-seq data for H3K27me0, we can make reasonable inference of its behavior based on the levels of H3K27me1/2/3 observed from the ChIP-seq data: the level of H3K27me0 should equal the proportion of histones that are not in any of the other methylation states. Because, in K27M+, the levels of all three H3K27 methylation states decrease within genes, we can expect that the unmethylated H3K27 is particularly enriched in the genic regions in K27M+, similar to that of H3K27me1 in K27M−. Summarizing these observations, a striking pattern for the distribution of H3K27 methylation marks emerges: in K27M+, one methyl group seems uniformly removed genome-wide, thereby, downgrading H3K27me3 to H3K27me2, H3K27me2 to H3K27me1, and H3K27me1 to H3K27me0.
Figure 1. One-Step Reduction of H3K27 Methylation in H3.3 K27M.
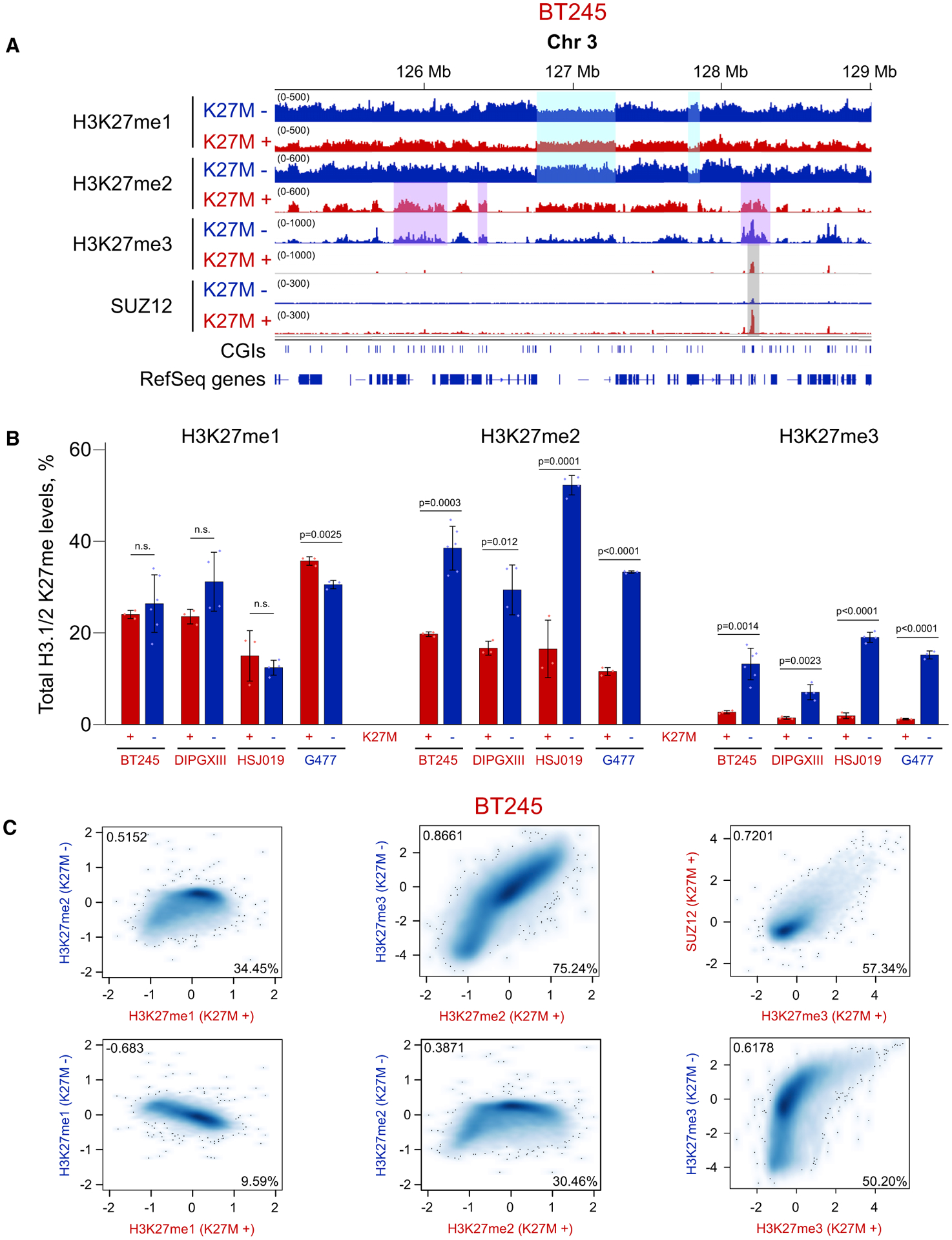
(A) Visualization of ChIP-seq results for H3K27me1/2/3 in BT245 K27M+/− cells. Highlighted regions illustrate inversion of H3K27me1 distribution, 1-step reduction of H3K27me2 → me1, H3K27me3 → me2, and retention of H3K27me3 at PRC2 (SUZ12) nucleation sites in K27M+ as compared with K27M−.
(B) Quantitative mass spectrometry of H3K27me in K27M+/− lines. We include the normally H3 wild-type (WT) cell line G477 that has been transfected to stably express either H3.3K27M or H3.3K27R (negative control) mutant histones to illustrate the bidirectionality of the changes. Data from four cell lines; means ± SD, n ≥ 3 replicates, Student’s t test. Dots show individual replicate values.
(C) Genome-wide relationships among different levels of H3K27 methylation. Correlation of H3K27me1/2/3 and SUZ12 levels in 100-kb bins in BT245 isogenic cell line system. H3K27me1 levels in K27M+ are anticorrelated with those in K27M− and more similar to H3K27me2 in K27M−; H3K27me2 in K27M+ correlates well with H3K27me3 in K27M−; and high levels of H3K27me3 in K27M+ correspond to increased levels of SUZ12. Axis scales are log2 (ChIP versus input); darker color indicates a higher density of bins. Total numbers of bins in the six panels (left to right, top to bottom) are 28,307, 28,297, 28,292, 28,307, 28,304, and 28,292.
Pearson correlation coefficients are shown in the upper left corners of the plots, whereas the percentage of the domain overlap of the two marks are shown in the lower right corners.
H3K36me2 Forms the Boundaries of H3K27me3 Spread in Wild-Type and H3K27me2 in K27M
H3K36 methylation has been shown to inhibit the deposition of H3K27 methylation, and H3K27me3 rarely coexists with H3K36me2/3 on the same nucleosomes (Schmitges et al., 2011; Yuan et al., 2011). Considerable insight into the differences in spreading of H3K27 methylation between H3K27M mutant and wild-type cells can be obtained from its interaction with the intermediate K36 methylation level, H3K36me2, which has previously been shown to oppose intergenic propagation of H3K27me2/3 (Alabert et al., 2020; Lu et al., 2016; Stafford et al., 2018).
Mass spectrometry shows that the overall levels of H3K36me2 remain nearly unchanged in the presence or absence of H3.3K27M across all four assayed cell lines (Figure 2A). This suggests that the spread of H3K27me2/3 after the removal of K27M in glioma cell lines does not displace pre-existing H3K36me2. The spatial relationship between the spreading of H3K27me2/3 and the presence of H3K36me2 is better illustrated by the ChIP-seq data. In K27M−, we observed a strong anticorrelation of H3K27me3 and H3K36me2, with alternating domains of each mark (Figures 2B and S2A), whereas, in K27M+, we see practically total exclusion of H3K27me3 from H3K36me2 domains, largely because of the restriction of this mark to unmethylated CGIs. An interesting pattern was observed for the H3K27me2/H3K36me2 interaction: in K27M−, those marks co-occur in many regions, but in K27M+, they appear antagonistic (anticorrelated), similar to H3K27me3/H3K36me2 in K27M− (Figures 2B, S2A, and S2B). These data suggest that H3K36me2 forms boundaries restricting the spread of H3K27me3 in K27M− and H3K27me2 in K27M+ cells.
Figure 2. H3K36me2 Forms Boundaries for H3K27me3 in WT and for H3K27me2 in K27M.
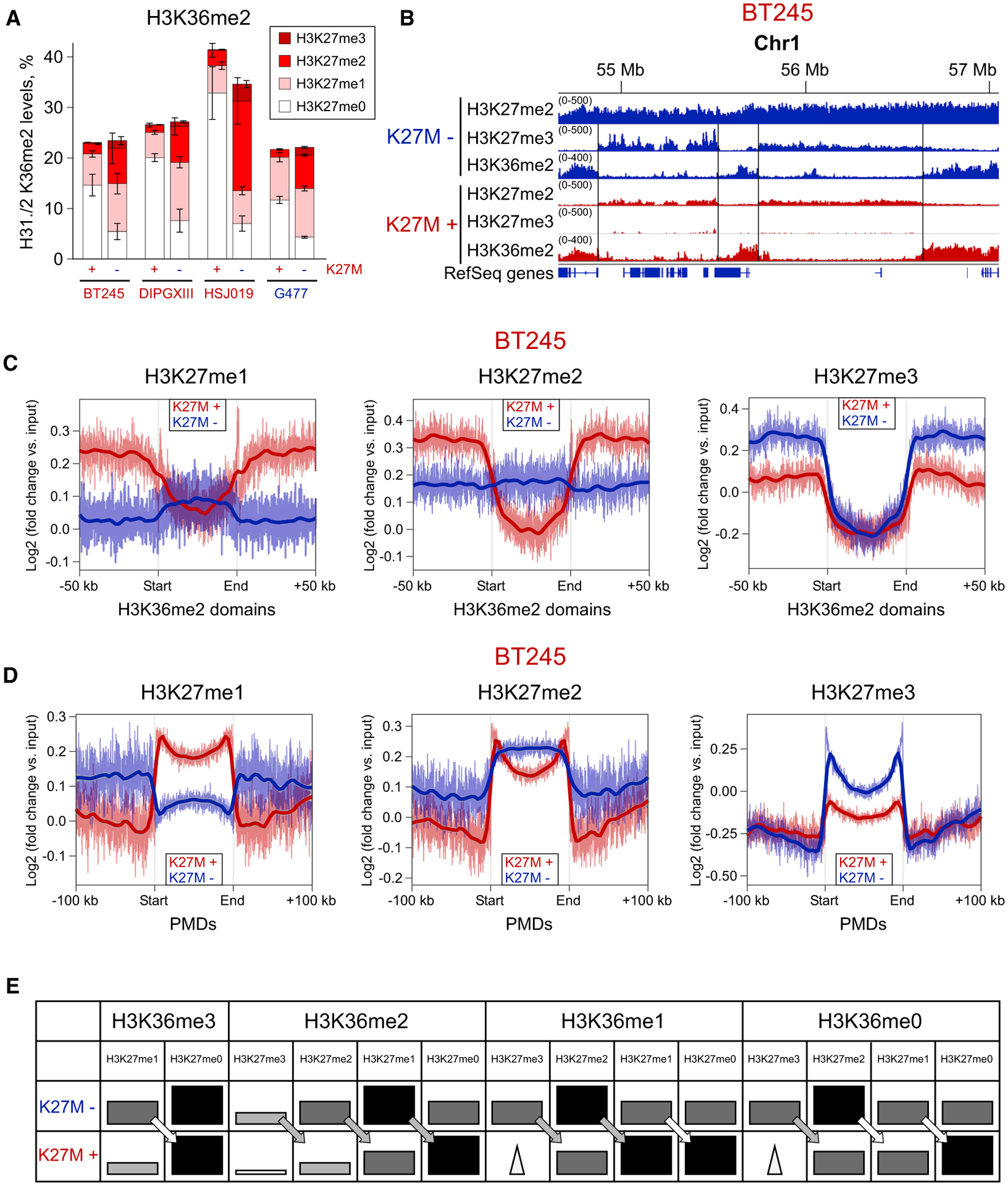
(A) Combinatorial mass spectrometry showing a large increase in the co-occurrence of H3K27me2/H3K36me2 after removal of H3.3K27M. Bars heights indicate the levels of the H3K36me2 mark, and differently colored sections indicate the proportion of H3K36me2 combination with H3K27me marks. Data from four isogenic cell lines; means ± SD for each mark combination, n ≥ 3 replicates.
(B) ChIP-seq signal showing alternating H3K36me2/H3K27me3 domains in K27M−, corresponding to alternating H3K36me2/K27me2 domains in K27M+ in BT245 cell line. We do not observe large-scale changes in the H3K36me2 distribution, suggesting that removal of the H3.3K27M mutation and subsequent spread of H3K27me3 does not displace H3K36me2.
(C) Aggregated signal of H3K27me1/2/3 over the intergenic H3K36me2 domains in the BT245 cell line. Domains containing active genes have been removed to exclude the effect of H3K36me3. In the K27M+, all H3K27 methylation levels are excluded from the H3K36me2 domains, whereas, in K27M−, only H3K27me3 is excluded, and H3K27me1 is slightly elevated because of its lack of conversion into the higher levels. Domains overlapping with H3K36me3 have been removed to exclude its effect.
(D) Aggregated signal of H3K27me1/2/3 over partially methylated domains (PMDs) in the BT245 cell line. PMD domains represent the most permissive regions for the spread of H3K27 methylation in both conditions. H3K27me1 levels are higher in PMDs in K27M+ condition, whereas H3K27me3 is depleted, compared with K27M−. H3K27me2 is depleted at the central part of PMDs, being retained at the edges.
(E) Schematic illustration of the patterns of H3K27 methylation marks in H3K36me0/1/2/3-enriched regions, with K27M+ or without K27M−. The schema is derived from mass spectrometry data. Darker and bigger rectangles indicate higher levels of the respective histone mark in broad domains. Triangles denote narrow peaks of H3K27me3 across the regions (at unmethylated CGIs). The arrows show the one-step reduction of H3K27me marks in K27M+.
This is further illustrated at a genome-wide scale by aggregate plots of H3K27me marks in H3K36me2 domains (Figures 2C and S2C). A sharp drop of the mark in H3K36me2 domains indicates a boundary effect. Although, in K27M−, the H3K27me2 remains stable, a sharp drop is observed in K27M+. H3K27me3, drops in both K27M− and K27M+ cells, although the magnitude of the drop is less pronounced in K27M+, probably because of the overall genome-wide depletion of that mark (Figures 2C and S2C). We also estimated the co-occurrence of these H3 marks by calculating the proportion of H3K27me ChIP-seq signal in H3K36me2 domains (Figure S2B). Consistent with Figure 2C, in K27M, we observed an increase of H3K27me1 and H3K27me2, but a decrease of H3K27me3. This again shows that H3K27me2 can spread into H3K36me2 domains in contrast to H3K27me3, which cannot go into these domains upon removal of H3.3K27M.
Next, we considered changes in H3K27me within partially methylated domains (PMDs) (Figures 2D and S2D), a compartment of the genome characterized by low levels of DNA methylation and the absence of H3K36me2. In both PMD and H3K36me2 domains, we observed a clear inversion of the distribution of H3K27me1 between K27M+ and K27M− cells (Figures 2C, 2D, and S2D). H3K27me1 levels are reduced in H3K36me2 domains and elevated in PMDs in K27M+ cells, whereas the opposite trends are observed in K27M− cells. H3K27me2 is enriched at the edges of PMDs in K27M+ and central regions in K27M−. The relative depletion of H3K27me2 in the interiors of PMDs in K27M+ may reflect the inhibition of PRC2 and reduced ability to reach regions that are distant from nucleation sites. Conversely, the enrichment of H3K27me2 at the edges of PMDs corresponds to the higher density of PRC2 nucleation sites. The presence of H3K9me3 may also prevent H3K27me2 from spreading into PMDs. H3K27me3 is depleted in PMDs in K27M+, as previously shown (Harutyunyan et al., 2019), whereas, in K27M−, it gains—particularly at the edges, where it replaces H3K27me2 (Figures 2C and S2D).
Finally, combinatorial mass spectrometry, which allows joint detection of the modifications at the K36 and K27 positions located on the same histone peptide, corroborated the interactions observed in the ChIP-seq data (Figure 2A). In K27M+, the H3K36me2 mark predominantly co-occurs with unmodified H3K27 or with H3K27me1. This reflects the inability of H3K27me2 to spread past the H3K36me2 domain boundaries and the restriction of H3K27me3 to PRC2 nucleation sites. In K27M−, there is a significant increase of the co-occurrence of H3K36me2 and H3K27me2 peptide combination (Figure 2A). This corresponds to the more extensive spread of H3K27me2, which is now produced at higher levels and able to spread over the H3K36me2 domains. There is also a slight increase of the H3K27me3/H3K36me2 combination, reflecting a slight encroachment of H3K27me3 into regions occupied by H3K36me2, although this mark is mostly unable to spread into pre-existing H3K36me2 domains, despite a large increase in its overall levels. To show the global patterns of H3K27me marks in combination with H3K36me0/1/2/3, we derived a table from mass spectrometry data (Figure 2E). Overall, the lower marks of H3K27me tend to be more abundant within regions marked by H3K36 methylation. The “step-down” from K27M− to K27M+ always holds for “me3 → me2,” shown by gray arrows, whereas the “me2 → me1” and “me1 → me0” steps are less consistent in certain regions, shown by white arrows.
Depletion of H3K36me2 Allows the Spread of H3K27me2/3 in H3.3K27M
To further assess the interaction of H3K27me2/3 and H3K36me2 marks, we used CRISPR/Cas9 to knock out NSD1 and NSD2, the two main H3K36me2 methyltransferases (Wagner and Carpenter, 2012), in a H3.3K27M cell line (BT245) (Figures S3A and S3B). Mass spectrometry showed a significant loss of H3K36me2 and a gain of H3K27me2, but no detectable change in H3K27me3 (Figure 3A, left panel). Importantly, the gain of H3K27me2 co-occurred with unmodified H3K36 or H3K36me1, but not with H3K36me2 (Figure 3A, right panel), suggesting that H3K36me2 may form boundaries for H3K27me2; thus, it can only spread into the regions vacated by H3K36me2 (i.e., regions with H3K36me0/1). Consequently, ChIP-seq data showed a general depletion of H3K36me2 domains, observable mostly in intergenic regions (Figures 3B and S3C). Furthermore, we observed a concomitant increase in H3K27me2 and, to a lesser extent, in H3K27me3 in those depleted domains (Figures S3D and S3E). Aggregating the signal of H3K27me2 and H3K27me3 across all H3K36me2 domains demonstrated that, at a genome-wide scale, regions of H3K36me2 depletion exhibit a general gain of H3K27me2 and H3K27me3 (Figures 3C and S3F). The ability of PRC2 to deposit H3K27me2 and low levels of H3K27me3 in H3K36me2-depleted regions is also reflected in a genome-wide anticorrelation of their changes (Figure 3D). Finally, when identifying regions that undergo the largest changes in H3K27me, we observed that the gain of H3K27me2 and, to a lesser extent, H3K27me3 is most enriched in regions that had high H3K36me2 levels in parental cells and lose H3K36me2 upon NSD1/2 KO (Figures 3E and S3G).
Figure 3. Depletion of H3K36me2 Domains Allows Increase of H3K27me2/3.
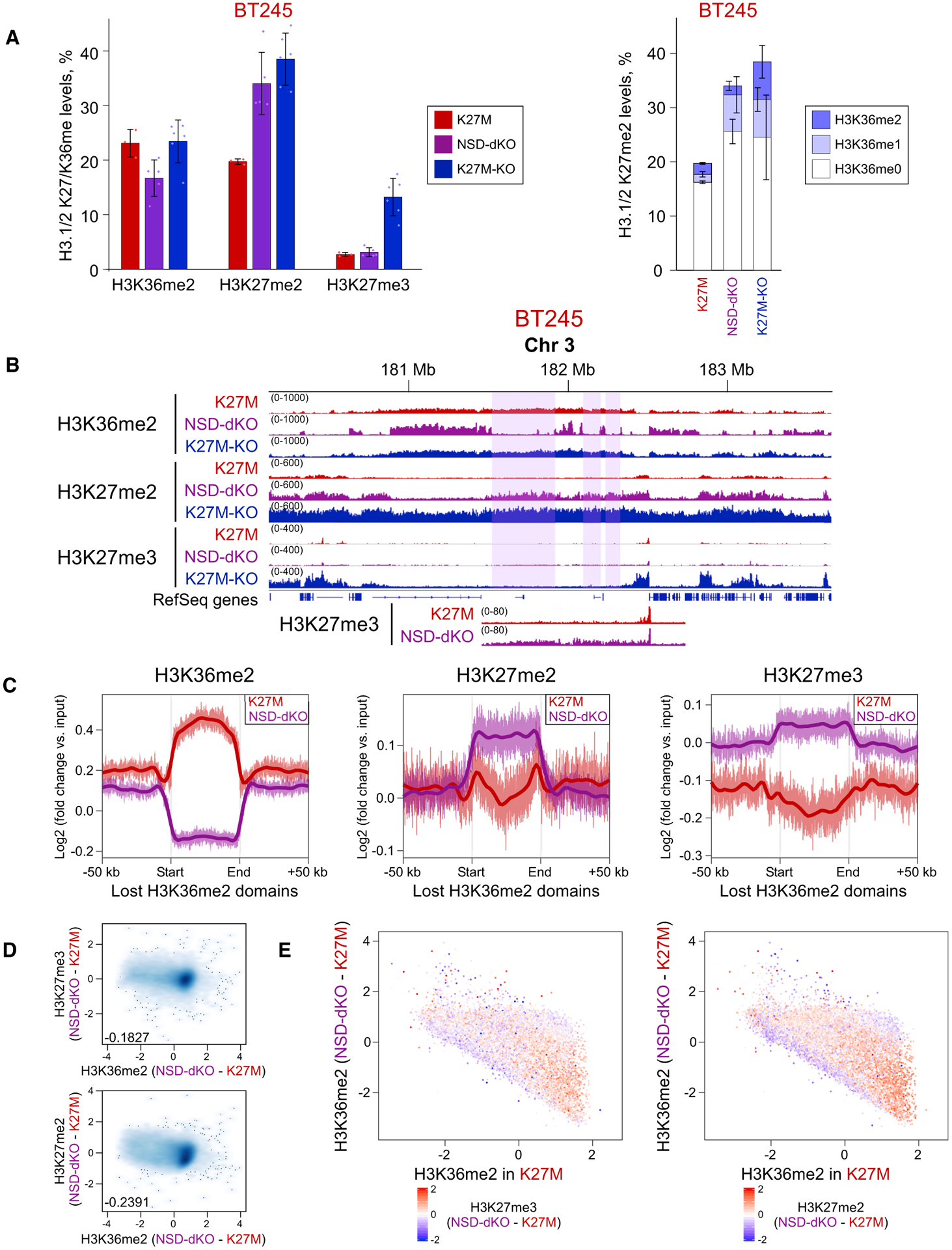
(A) Mass spectrometry shows increase of H3K27me2 (left panel) and its increased co-occurrence with H3K36me0/1 (right panel), reflecting its spread into regions that lose H3K36me2. In the right panel, H3K27me2 is shown in combination with H3K36me0/1/2 (increasingly dark color). Means ± SD (for each bar section on the right), n ≥ 3 replicates. Dots show individual replicate values.
(B) ChIP-seq data of BT245 cell line showing H3K27me2/3 spreading into the regions of H3K36me2 loss. Highlighted regions illustrate domains of H3K36me2 loss. The region is shown at a different scale at the bottom, to visualize the low-level increase in H3K27me3.
(C) Aggregate signal of H3K36me2, H3K27me2/3 in the regions of H3K36me2 loss shows an increase of H3K27me2 and a subtler increase in H3K27me3.
(D) Correlation of H3K27me2/3 with H3K36me2 levels in 100-kb bins in BT245. Genome-wide loss of H3K36me2 in NSD1/2-DKO correlates with gains of H3K27me2/3.
(E) H3K36me2 signal change in genome-wide 100-kb bins of BT245 and NSD1/2-dKO (first clone); color is coded by H3K27me3 (left panel) and H3K27me2 (right panel) levels. The y axis shows the difference in H3K36me2 levels in K27M versus NSD1/2-dKO (log2), whereas x axis shows H3K36me2 levels in K27M. Regions with largest loss of H3K36me2 experience the highest gain of H3K27me2/3 (bottom right).
Loss of Association between H3K36me3 and H3K27me1 and Lack of Effect on Gene Expression in H3.3K27M
Although H3K36me3 has been previously shown to be antagonistic to H3K27me2/3 (Schmitges et al., 2011; Yuan et al., 2011), it has been reported to be tightly associated with H3K27me1 (Ferrari et al., 2014). In addition, the presence of H3K27me1 in active gene bodies has been suggested to be important for promoting transcription (Ferrari et al., 2014). Although overall levels of H3K27me1 are not affected by the H3K27M mutation (Figure 1B), in K27M+, there is a marked redistribution of H3K27me1 away from gene bodies (Figures 1A, 4A, 4B, S4A, and S4B). Thus, we investigated the relationship between the distribution of H3K36me3, H3K27me1, and gene expression changes in the context of H3.3K27M. In the absence of H3.3K27M, as expected, H3K27me1 coexists with H3K36me3 and follows the enrichment pattern across gene bodies of highly expressed genes (Figures 4A, 4B, S4A, and S4B). However, that co-occurrence is absent in K27M+ because H3K27me1 becomes depleted in gene bodies (Figures 4B and S4B). Accordingly, in K27M+, we observed significantly less of the H3K27me1-H3K36me3 combination by mass spectrometry (Figure 4C). When correlating the distribution of those marks to the transcriptome, we observed a consistent, positive relationship of gene expression with H3K36me3 in both K27M− and K27M+ cells (Figures 4D, 4E, and S4C). However, for H3K27me1, we detect an anticorrelation with gene expression in the presence of H3.3K27M (Figures 4E and S4C), suggesting that H3K27me1 is not a major factor in determining gene expression.
Figure 4. H3K27me1 Association with H3K36me3 and Gene Expression Is Absent in H3.3 K27M Samples.
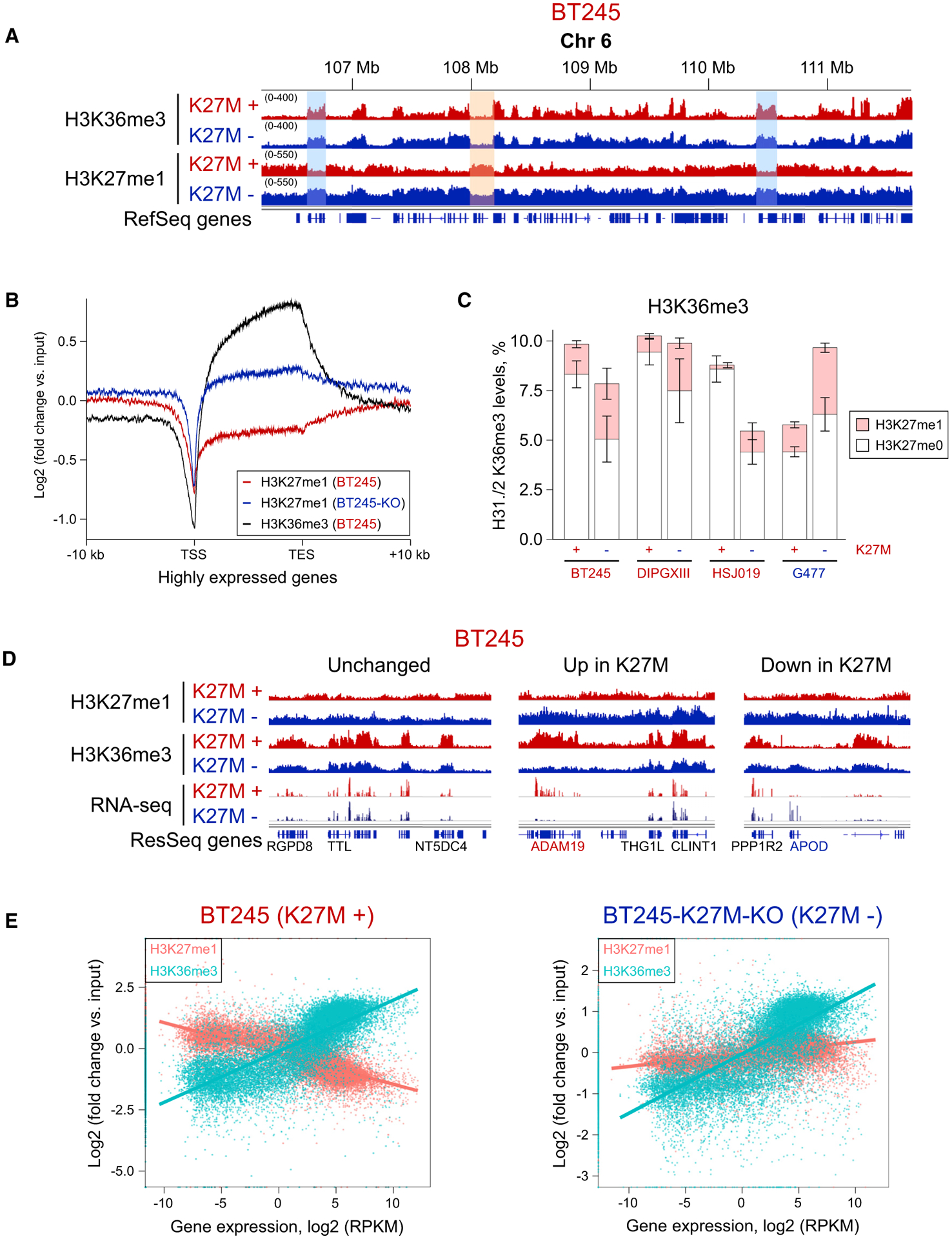
(A) ChIP-seq tracks of BT245 isogenic cell line showing H3K27me1 domains co-occurring with H3K36me3 in K27M−, whereas, in K27M+, H3K27me1 is mainly depleted in H3K36me3-rich regions. No major changes for H3K36me3 are observed.
(B) Aggregate plots of H3K27me1 overexpressed genic regions, showing higher enrichment in K27M− cells.
(C) Histone mass spectrometry showing changes in H3K36me3-H3K27me1 combinatorial mark in K27M− and K27M+ conditions. Means ± SD (for each bar section), n ≥ 3 replicates.
(D) Comparison of H3K27me1, H3K36me3, and gene expression (RNA-seq) for representative examples of unchanged, upregulated, and downregulated genes.
(E) Correlation plots of H3K27me1 and H3K36me3 signal with gene expression of respective genes in K27M− and K27M+ conditions. Although H3K36me3 levels positively correlate with gene expression in both conditions, H3K27me1 levels show mild positive correlation in K27M− condition and is negatively correlated with gene expression in K27M+ cells.
H3K9me3 Is Deposited in the Regions of H3K27me2 Loss in H3.3K27M Cells
Finally, we profiled our cell lines for H3K9me3, a repressive mark mostly present in constitutive heterochromatin (Figures 5A and S5A). Mass spectrometry showed a consistent decrease of this mark in K27M− (Figure 5B). H3K9me3 loss was observed in both PMDs and genes (Figures 5C, 5D, and S5B). We hypothesized that the change in H3K9me3 levels may be a secondary consequence of changes and redistribution of H3K27me marks. Hence, we investigated genome-wide correlations between variation in H3K9me3 and H3K27me1/2/3. The difference in H3K9me3 shows the strongest relationship (anticorrelation) with the difference in H3K27me2 and, to a lower extent, with H3K27me1, whereas there seems to be a slight positive correlation with H3K27me3 (Figures 5E and S5C). These correlations are stronger in PMD regions (Figures 5F and S5D), where ChIP-seq tracks indicate the replacement of H3K27me2 by H3K9me3 in a number of PMDs in K27M+ cells (Figure 5A). This is further illustrated by the aggregated plots (Figure 5C), where H3K9me3 is enriched in PMDs in K27M+ but is greatly reduced—especially in the center of PMDs—in K27M−, whereas H3K27me2 shows the opposite trends (Figures 2D, 5C, and S5B). Consistent with the repressive role of H3K9me3, it is found negatively correlated with gene expression in PMDs (Figure S5E), where genes with low H3K9me3 in promoters have higher expression levels than genes with high H3K9me3 have.
Figure 5. Moderate H3K9me3 Increase in Domains Losing H3K27me2/3 in H3.3 K27M.
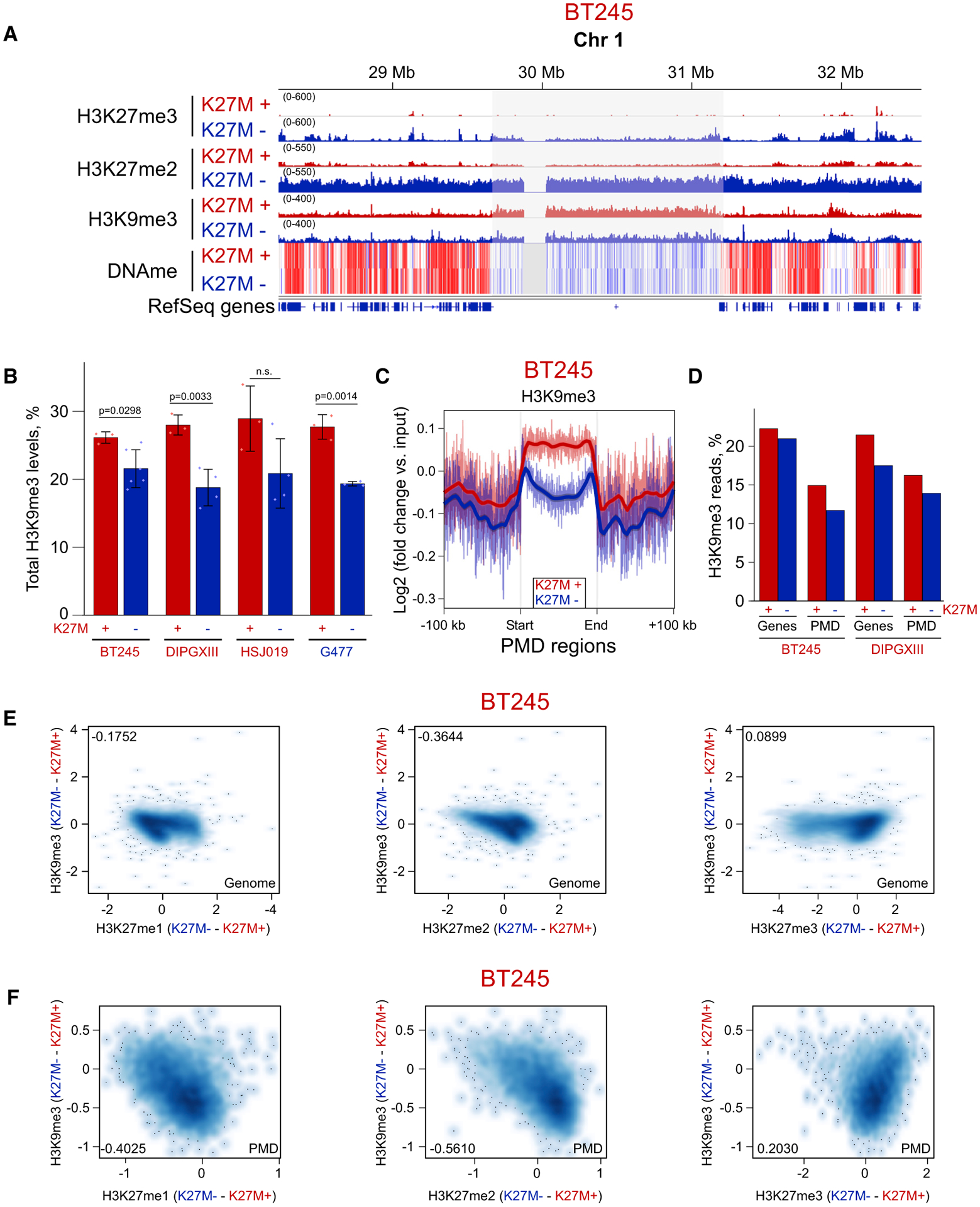
(A) ChIP-seq and whole-genome bisulphite sequencing tracks of BT245 cell line showing H3K9me3 enrichment in some H3K27me2-depleted PMDs.
(B) Histone mass spectrometry showing higher levels of H3K9me3 in K27M+ cell lines and its reduction in K27M−. Data from four cell lines; means ± SD, n ≥ 3 replicates, Student’s t test. Dots show individual replicate values.
(C) Aggregate plots of H3K9me3 over PMD domains, summarizing the observed changes on ChIP-seq tracks.
(D) Proportion of H3K9me3 reads is decreased both in PMD regions and in genes in K27M−.
(E and F) Correlation plots of H3K9me3 change with changes in H3K27me1/2/3 marks genome wide (E) and in PMD regions (F). Axis scales are log2 (ChIP versus input), darker color indicates a higher density of bins. Pearson correlation coefficients are shown in the corners of the plots.
Computational Modeling of H3K27me Deposition by PRC2
To aid interpretation of our results, we conceived a stochastic simulation model for the dynamic deposition of methylation marks on the H3K27 residue by the PRC2 complex (Figure 6A), STOPHIM, a program for simulating the stochastic process of histone modification. The full details of the model are described in the Method Details section. We represent a chromatin segment as a 1-dimensional nucleosome array and incorporate the key properties of PRC2 that are supported by current literature: (1) PRC2 molecules are first recruited at specific genomic loci, where they have a high binding affinity (Li et al., 2017) and (2) PRC2 then propagates outward and catalyzes conversion to H3K27me (1, 2, and 3) at each encountered nucleosome. To our knowledge, no clear mode of PRC2 propagation has been described, and we propose a simple diffusion, random-walk model. We further propose a stepwise catalytic process (me0 → me1, me1 → me2, or me2 → me3), with increasing difficulty for adding additional methyl groups. The H3K27me levels are maintained at equilibrium by cell division; during which, half of the nucleosomes are removed at random and replaced by naked nucleosomes. This basic version of the model is sufficient to simulate the H3K27me3 dynamics after EZH2 induction in EZH2-null mouse embryonic stem cells (ESCs) (Video S1) described by Højfeldt et al. (2018).
Figure 6. Computational Modeling of PRC2 Function in WT Condition and Various Modes of PRC2 Inhibition.
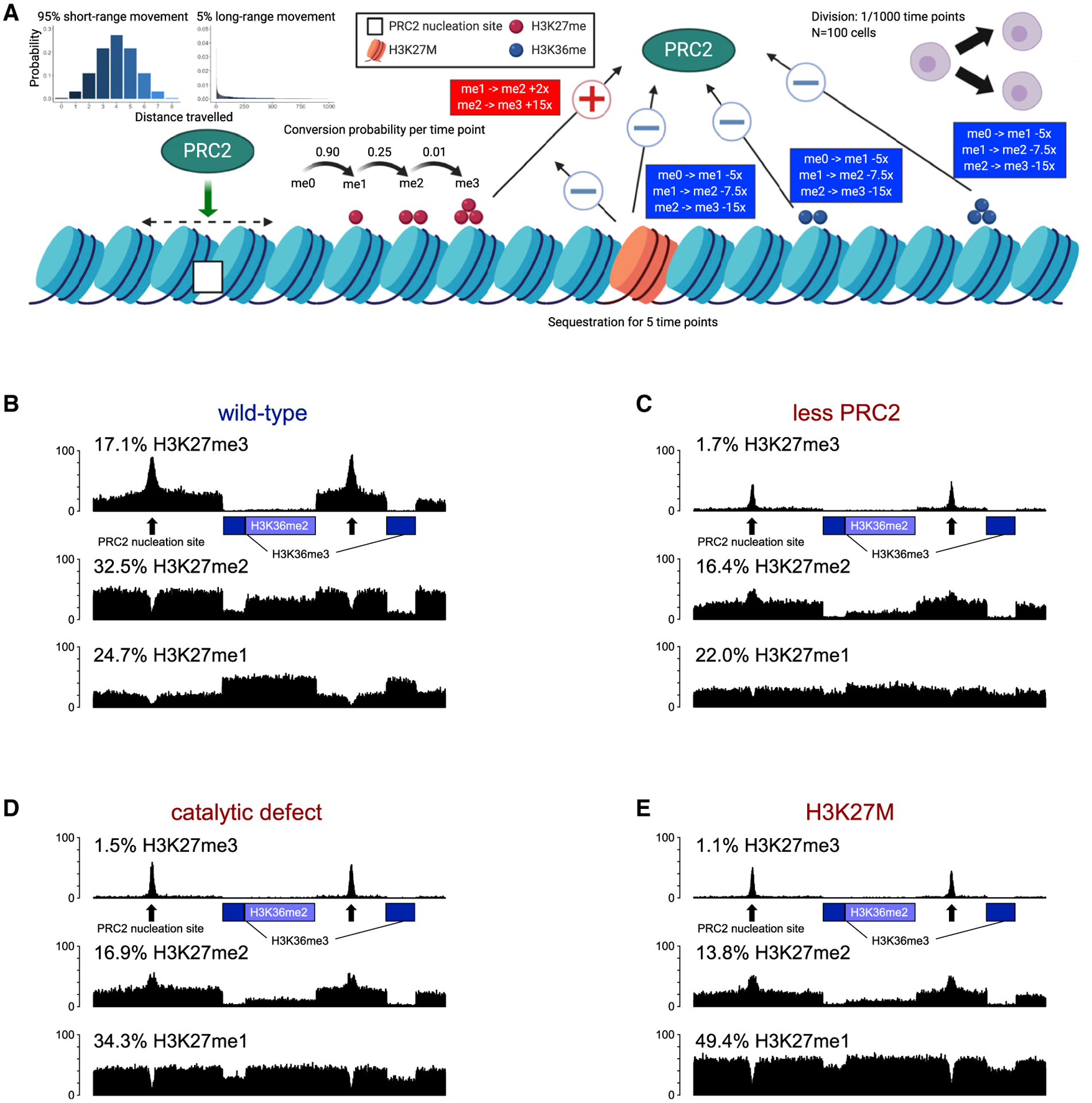
(A) Illustration of the computational model, STOPHIM. All modifiable parameters and the effects of histone marks, as well as H3K27M mutation on PRC2 are shown. Created with BioRender.com.
(B) Simulated pattern of H3K27me1/2/3 deposition in the WT condition. The two regions of marked by H3K36me3 are denoted with green boxes, whereas the region with H3K36me2 is denoted with an orange box. Two PRC2 nucleation sites are shown with black arrows.
(C–E) H3K27me1/2/3 landscape simulated under different modes of PRC2 inhibition: 4-fold less PRC2 molecules available (C), 3-fold reduced catalytic activity for all steps of H3K27me deposition (D), and the presence of 5% H3K27M mutant histones, which retain PRC2 for a short period and permanently damage it by reducing its catalytic activity by 2-, 5-, and 10-fold for each subsequent step of H3K27 methylation (E). The equilibrium levels (percentages) of H3K27me1/2/3 achieved in the simulation model are shown above the simulated “tracks.”
We incorporate additional concepts to the model for simulating the observed patterns in this study. First, we introduce allosteric activation (Margueron et al., 2009), where the rate of conversion to the me2 and me3 states are enhanced after PRC2 encounters H3K27me3. Second, we allow two modes of PRC2 propagation: (1) in proximity to the nucleation sites, PRC2 remains associated with the chromatin and is able to translocate only short distances at each time interval, or (2) after dissociating from the chromatin (with low probability), PRC2 can diffuse freely and travel much further at each step. Third, we introduce the antagonistic marks H3K36me2/3 to reduce the deposition rates of H3K27me with variable effects (Schmitges et al., 2011; Zheng et al., 2012). Fourth, we model the effect of the K27M mutation on PRC2. We explored three modes of inhibition: (1) a general inhibition model, in which the catalytic activity of PRC2 is reduced; (2) a sequestration model, in which PRC2 is retained or trapped locally at the mutant histones, slowing down its further movement and effectively reducing the number of available PRC2 complexes; and (3) a “poison” model (Stafford et al., 2018), in which PRC2 partially loses catalytic activity when encountering K27M, and the loss persists after leaving K27M.
To illustrate the process, we simulated a genomic region containing two PRC2-binding loci, two active chromatin regions marked by H3K36me3 (genic) and H3K36me2 (intergenic), respectively. We extensively explored the parameter space of the model to simulate the levels and distribution of H3K27me observed in glioma cell lines (Figure 6). Several key parameters need to be optimized to simulate the observed data, such as the traveling distance of PRC2 from nucleation sites or the antagonistic effects of H3K36me2/3 on H3K27me1/2/3. The detailed setup of parameters is provided in the Method Details section.
We tested our model in a wild-type state (Figure 6B) and in three possible modes of PRC2 inhibition by H3K27M: it reduced the number of available PRC2 molecules (Figure 6C); it reduced the catalytic activity (Figure 6D) and the presence of H3K27M nucleosomes, which retain PRC2; and it permanently reduced its catalytic activity, even after release (Figure 6E; Video S2). All three modes of inhibition are generally able to recapitulate the patterns of histone mark distribution observed in our experimental data: (1) retention of H3K27me3 within peaks around nucleation sites, accompanied by greatly reduced H3K27me3 levels genome-wide; (2) reduced levels of H3K27me2 genome-wide, and significant exclusion from H3K36me2/3 regions (similarly to the distribution of H3K27me3 in wild-type samples); and (3) maintenance of the overall genome-wide levels of H3K27me1, but characterized by redistribution from genic to intergenic regions.
DISCUSSION
Interactions between post-translational histone modifications are complex and dynamic and have key regulatory roles in cellular state and functioning. Understanding those interactions is important, both at the normal physiological level and in pathological states, such as cancers. The H3K27M mutation is a key oncogenic event in pediatric HGGs and has profound epigenetic effects (histone modifications and DNA methylation). In this work, we profile a wide range of histone modifications in isogenic H3.3K27M+/− primary HGG cell lines to gain new insights into the epigenomic consequences of H3K27M.
The first key outcome of this study is an in-depth characterization of changes at all levels of H3K27 methylation in the presence of H3.3K27M mutation (Figure 7). In addition to the previously described H3K27me3 restriction to unmethylated CGIs, we find that H3K27me2 replaces H3K27me3 in facultative heterochromatin domains and is depleted in other genomic regions that it normally occupies. Surprisingly, the H3K27me1 mark, which, in contrast to H3K2me2/3, is not globally decreased in K27M+, is also drastically reorganized genome wide, displaying an “inverted” distribution compared with the K27M− condition. H3K27me1 in K27M+ replaces H3K27me2 in intergenic regions and is depleted from its usual localization in gene bodies of expressed genes. Using mass spectrometry, we observed a significant overall increase in H3K27me0 in K27M+, and because all methylated H3K27 marks are depleted in genic regions, we predict that those regions are enriched in unmethylated H3K27. We can generalize the changes in the distributions of all three K27 methylation marks by a simplified rule: the effect of the K27M mutation in glioma cell lines is the removal—one-step reduction—of one K27 methyl group throughout the genome.
Figure 7. A Model of Global Histone Modification Changes in H3.3 K27M Cells.
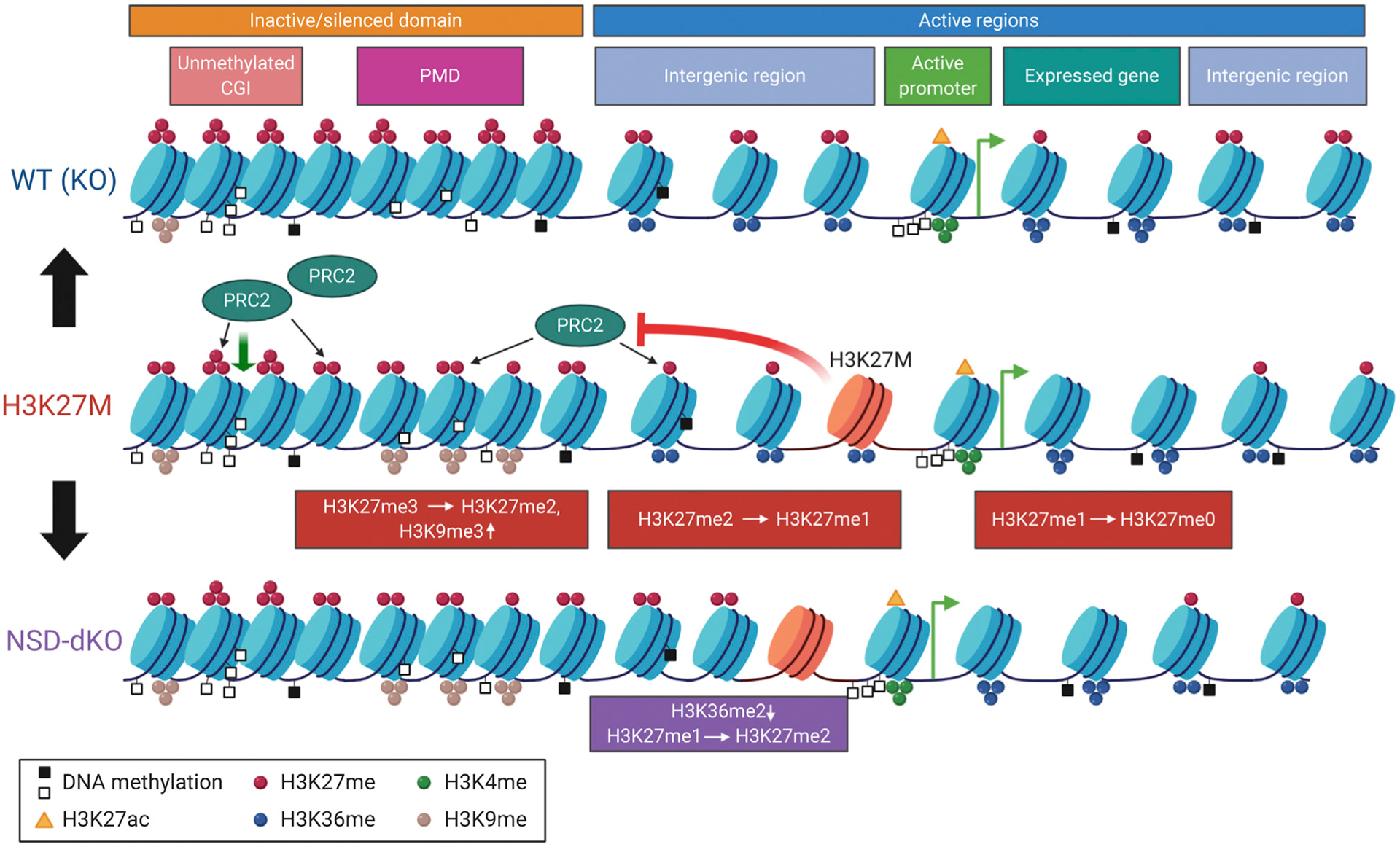
The effect of H3K36me2-mark depletion by knocking out NSD1/2 on H3K27me2/3 marks is depicted. Colored circles illustrate different histone methylation marks (red, H3K27me; blue, H3K36me; green, H3K4me; and brown, H3K9me). Orange triangles are H3K27ac, whereas empty and filled black squares are unmethylated and methylated CGIs, respectively. Created with BioRender.com.
The antagonistic distribution and behavior of H3K27me and H3K36me marks has previously been investigated under normal conditions and in cancers (Lu et al., 2016; Oksuz et al., 2018; Stafford et al., 2018; Streubel et al., 2018). Increasing number of reports show that, upon depletion of one of the marks, other modifications move into the vacated genomic regions, such as in the case of a H3K36M mutation (Lu et al., 2016). Although, in our isogenic cell lines, we have not observed drastic changes in the bulk levels of the H3K36me2/3 marks in the presence or absence of the H3.3K27M mutation, there are profound changes in the co-occurrence of the K27–K36 methylation. H3K27me2-H3K36me2 domains represent one of the most abundant combinatorial states in the K27M− condition and disappear almost completely in K27M+. That is not only due to a global reduction of the H3K27me2 mark (which is still present in about 10% of the nucleosomes) but also to its predominant deposition in the regions previously occupied by H3K27me3. Thus, although, in the K27M− condition, H3K36me2 forms the boundaries for the spread of H3K27me3, in K27M+, the same boundary restricts the spread of H3K27me2.
We provide further experimental evidence for this boundary effect by knocking out NSD1/2 H3K36 methyltransferases and depleting H3K36me2 from many intergenic domains. In the absence of the H3K36me2 mark, despite the presence of H3.3K27M, PRC2 is able to deposit H3K27me2 and low levels of H3K27me3 in those regions. This result also supports our previous finding that PRC2 is not fully sequestered by H3K27M, but rather, it is still able to operate within facultative heterochromatin (Harutyunyan et al., 2019). Interestingly, although most of the residual H3K27me3 in K27M+ is deposited in the proximity of unmethylated CGIs (PRC2 nucleation sites), there is a very low level of the mark still present in heterochromatic domains (compared with euchromatic domains), similar to these newly acquired domains upon H3K36me2 removal. This could be explained by a reduced functional state of PRC2, being able to deposit H3K27me2 and very low levels of H3K27me3 in the regions normally abundant in H3K27me3.
Our characterization of changes in H3K9me3 showed that this mark is also actively involved in epigenomic changes induced by H3.3K27M. H3K9me3 is increased in the regions losing H3K27me2/3 marks, possibly compensating for the absence of their repressive effect. It is likely that cells are able to compensate for the loss of epigenomic regulatory effects of one mark by relying on other repressive mechanisms. Although the repressive effects of H3K27me2/3, H3K9me3, and DNA methylation are not identical, in emergency situations (e.g., the catastrophic loss of H3K27me3 caused by the H3K27M mutation), cells may be forced to use other available mechanisms to survive and function. It is also likely that cellular context has an important role in determining which compensatory mechanism will be used in each case. We have not observed an increase of the H3K36me2 mark in our isogenic HGG CRISPR/Cas9 KO system; however, in other systems, such as for overexpression of the H3K27M mutation in HEK293T cells, H3K36me2 does seem to increase in domains losing H3K27me3 (Stafford et al., 2018).
To better understand the H3K27me deposition patterns described above, we created a computational simulation describing the movement and activity of the PRC2 complex. Under wild-type conditions, the deposition patterns can result from a few key assumptions: (1) although PRC2 has a preferential affinity for specific nucleation sites around which it exhibits increased occupancy (as illustrated by ChIP-seq data), it must travel sufficiently freely between nucleation sites to deposit approximately uniform levels of all H3K27me marks; (2) deposition of each subsequent methylation level is increasingly less efficient; (3) the presence of antagonistic histone modifications, such as H3K36me2/3, reduces the efficiency of PRC2, and the reduction is stronger for higher methylation levels; and (4) the system is maintained in a steady state by cell division and passive dilution. Although PRC2 is allowed to diffuse and sample the entire genome, it has reduced efficiency in the presence of antagonistic marks: H3K36me3 (active genes) and H3K36me2 (active intergenic domains), which restrict deposition of H3K27me3 and H3K27me2 in those regions. We propose that the increase of H3K27me1 within active genes is not a result of preferential deposition within those regions, but rather, the outcome of decreased deposition of H3K27me2/3 and the lack of conversion of H3K27me1 to higher methylation states, which occurs in other regions of the genome.
A similar principle can be used to explain the one-step reduction in K27 methylation observed in the presence of the H3.3K27M mutation. The inhibition by K27M makes deposition of a new histone mark less likely during any time point of the simulation. As a result, H3K27me3 is predominantly present in the regions of highest occupancy or PRC2 (nucleation sites) because passive dilution removes it from all other regions of the genome, and there is not enough “critical mass” for allosteric activation to be effective. H3K27me2 can spread to regions previously occupied by H3K27me3, partly because it is not converted to the higher state; however, the antagonism presented by H3K36me2 in active domains is now sufficient to reduce the propagation of H3K27me2. As for H3K27me1, because it is now not efficiently converted to H3K27me2/3, it is retained throughout the genome and depleted in active genes, where it was not efficiently deposited in the first place.
We found that we can simulate the distribution of H3K27 methylation in the presence of the K27M mutation under many of the proposed models: sequestration of PRC2 at the chromatin or cytosolic capture by unincorporated mutant histones (Fang et al., 2018; Justin et al., 2016; Lewis et al., 2013) and stable poisoning of PRC2 after encountering K27M (Bender et al., 2013; Stafford et al., 2018). The only proposed model for which we could not identify suitable parameters to fit the data was exclusion of PRC2 from the chromatin by mutant histones (Piunti et al., 2017). Although strong opinions exist in the field regarding the underlying mechanism of PRC2 inhibition by H3K27M, we feel that more experimental evidence is necessary to fully elucidate the process. As it stands, our simulation model provides insight into how changes of relevant parameters lead to downstream changes in histone modification levels and patterns. Moreover, this is a model in development and further adjustments and improvements are certain to yield more insightful results. Possible future developments include various non-canonical subunits of PRC2, which may change the complex’s properties and interactions with chromatin, and spatial chromatin conformation. We provide the code openly and encourage other researchers to explore and improve its functioning.
In a broader context, identifying the interactions and balance of the epigenetic marks is important not only for cancer but also for understanding basic biological functions of those histone modifications. In that sense, histone mutations identified in cancers (H3K27M, H3K36M, etc.) have been invaluable tools for advancing our understanding of the functions of histone marks. This is particularly true for less-studied histone modifications of lower methylation levels. Recently, important insight has been obtained regarding the function of the H3K36me2 mark, as being able to recruit DNMT3A and guiding de novo DNA methylation (Weinberg et al., 2019). There is currently much less understanding of the effects of H3K27me1 and H3K27me2 marks. A report characterizing those marks (Ferrari et al., 2014) has implicated H3K27me2 in repressive functions, whereas it suggests H3K27me1 likely contributes to transcription, being enriched in highly expressed genes. Our findings show that, at least in the presence of the H3.3K27M mutation, H3K27me1 is not linked to gene expression, and thus a reassessment of its functional significance is required. Even though we may currently lack knowledge of the direct functional roles of most histone modifications, particularly the intermediate states like mono and dimethylation, an understanding of their individual properties is an important component in our understanding of the global epigenome dynamics.
The epigenomic landscape constitutes a finely tuned and delicately balanced distribution of histone modifications with different functions, regulating cell fate and function. This balanced state is disturbed in many diseases, particularly in cancer. Histone mutations take epigenomic disorganization to a new level. It is, therefore, imperative to understand all secondary, indirect changes that occur as a result of the primary effect of the histone mutation. Armed with that knowledge, we will be more likely to find actionable targets in those deadly cancers and make improvements in treatment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jacek Majewski (jacek.majewski@mcgill.ca).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the raw human data are Gene Expression Omnibus (GEO): GSE147783. RNA-seq data for BT245 and SU-DIPGXIII cell lines (parental and K27M-KO) were published previously (Krug et al., 2019) and deposited in GEO under the accession number GSE128745.
An open-sourced R program for conducting simulations is available at https://github.com/tianyuan-lu/STOPHIM
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Samples and Clinical Information
This study was approved by the Institutional Review Board of the respective institutions from which the samples were collected. We thank Keith Ligon and Michelle Monje for generously sharing primary tumor cell lines established from patients with high-grade glioma.
Cell culture
Three H3.3 K27M mutant patient-derived cell lines - BT245 (8 years old, male), SU-DIPGXIII (6 years old, female), HSJ019 (13 years old, female), and one H3 wild-type cell line - G477 (15 years old, female), were used in this study (Key Resources Table; Table S1). Tumor-derived cell lines were maintained in NeuroCult NS-A proliferation media (StemCell Technologies) supplemented with bFGF (10ng/mL) (StemCell Technologies), rhEGF (20 ng/mL) (StemCell Technologies) and heparin (0.0002%) (StemCell Technologies) on plates coated in poly-L-ornithine (0.01%) (Sigma) and laminin (0.01 mg/mL) (Sigma). All lines tested negative for mycoplasma contamination, checked monthly using the MycoAlert Mycoplasma Detection Kit (Lonza). Tumor-derived cell lines were confirmed to match original samples by STR fingerprinting.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-H3K27me3 | Cell Signaling Technology | Cat # 9733; RRID:AB_2616029 |
| Rabbit monoclonal anti-H3K27me2 | Cell Signaling Technology | Cat # 9728; RRID:AB_1281338 |
| Mouse monoclonal anti-H3K27me1 | Active Motif | Cat #61015; RRID:AB_2715573 |
| Rabbit monoclonal anti-H3K36me2 | Cell Signaling Technology | Cat # 2901; RRID:AB_1030983 |
| Mouse monoclonal anti-H3K36me3 | Active Motif | Cat #61021; RRID:AB_2614986 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat # ab8898; RRID:AB_306848 |
| Rabbit monoclonal anti-SUZ12 | Cell Signaling Technology | Cat # 3737; RRID:AB_2196850 |
| anti-H3K27M | Millipore | Cat # ABE419; RRID:AB_2728728 |
| anti-H3K27me3 | Millipore | Cat # ABE44; RRID:AB_10563660 |
| anti-total H3 | Abcam | Cat#ab1791; RRID:AB_302613 |
| anti-beta-actin | Cell Signaling Technology | Cat # 4970; RRID:AB_2223172 |
| anti-NSD1 | NeuroMab | Cat# 75–280; RRID:AB_11001827 |
| anti-NSD2 | Millipore | Cat# MABE191 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NeuroCult Proliferation Kit | STEMCELL Technologies | Cat # 05751 |
| EGF | STEMCELL Technologies | Cat# 78006.1 |
| FGF | STEMCELL Technologies | Cat # 78003.2 |
| Poly-L-Ornithine | Sigma Aldrich | Cat # A-004-M |
| Laminin | Sigma Aldrich | Cat # L2020 |
| Heparin | STEMCELL Technologies | Cat # 07980 |
| Propionic anhydride | Sigma Aldrich | Cat # 240311–50G |
| Critical Commercial Assays | ||
| iDeal ChIP-seq Kit for Histones | Diagenode | Cat # C01010051 |
| Alt-R® CRISPR-Cas9 tracrRNA, ATTO 550 | Integrated DNA Technologies | Cat# 1075927 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081058 |
| Kapa HTP Library Preparation Kit Illumina Platforms | Roche | Cat #07961901001 |
| Kapa Hyper Prep Kit | Roche | Cat # 07962363001 |
| Empore Extraction Disk, 3M C18 | Fisher Scientific | Cat # 143863–2 |
| Epitect Fast DNA Bisulfite Kit | QIAGEN | Cat # 59826 |
| Kapa HiFi Uracil+ Kit | Roche | Cat # 07959079001 |
| AllPrep DNA/RNA/miRNA Universal Kit | QIAGEN | Cat # 80224 |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Human/Mouse/Rat | Illumina | Cat# RS-122–2201 |
| Deposited Data | ||
| Raw human data | Krug et al., 2019 | GEO: GSE128745 |
| Raw human data | This paper | GEO: GSE147783 |
| Experimental Models: Cell Lines | ||
| BT245 | Keith Ligon | N/A |
| SU-DIPGXIII | Michelle Monje | N/A |
| HSJ019 | Nada Jabado | N/A |
| G477 | Peter Dirks | N/A |
| Oligonucleotides | ||
| GAGGGCGCACTCATGCGAG | Integrated DNA Technologies | N/A |
| GCCCTATCGGCAGTACTACG | Integrated DNA Technologies | N/A |
| GTGAATGGAGATACCCGTGT | Integrated DNA Technologies | N/A |
| GCAGCTCGGAGTCTTCCCGT | Integrated DNA Technologies | N/A |
| CGGGTGTTTAATGGAGAACC | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Feng Zhang | Addgene plasmid # 48138 |
| Software and Algorithms | ||
| BWAv0.7.17 | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Bedtools v2.22.1 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Integrative Genomic Viewer | Thorvaldsdóttir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| deepTools v3.1.0 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| STAR v2.5.3a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.3 | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| Samtools v0.1.18 | Li et al., 2009 | https://github.com/samtools/samtools |
| UCSC tools | UCSC | https://genome.ucsc.edu/util.html |
| R 3.6.1 | The R Project for Statistical Computing | https://www.r-project.org/ |
METHOD DETAILS
CRISPR/Cas9 genome editing
BT245 and SU-DIPGXIII CRISPR edited cell lines were already used previously (Harutyunyan et al., 2019; Krug et al., 2019). HSJ019 cell line was CRIPSR edited for this study. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138). In primary HGG lines heterozygous for H3F3A-K27M, clones were derived with the mutant allele edited using PX458 and the guide sequence GAGGGCGCACTCATGCGAG. CRISPR-Cas9 editing was carried out as described in Ran et al. (2013). Eight hundred thousand cells were harvested and electroporated with Amaxa Human Embryonic Stem Cell Nucleofector kit (Lonza) according to the manufacturer’s protocol. Flow cytometry sorted single GFP+ cells in 96 well plates, 72 hours post-transfection. Clones were expanded and the target locus sequenced by Sanger sequencing. Select clones were screened by Illumina MiSeq system for the target exon to confirm complete mutation of the K27M allele. Mass spectrometry confirmed the absence of K27M mutant peptide in these clones.
CRISPR-Cas9 editing of NSD1/2 in BT245 cell line was performed using the Alt-R CRISPR-Cas9 System (IDT) and designing synthetic crRNA guides targeting the NSD1 gene at two positions simultaneously (guide 1 in PWWP domain: GCCCTATCGGCAGTACT ACG; guide 2 in SET domain: GTGAATGGAGATACCCGTGT). Clones were obtained and CRISPR-Cas9 editing was performed again targeting the NSD2 gene using two crRNA guides simultaneously in the same conserved exon (GCAGCTCGGAGTCTTCCCGT and CGGGTGTTTAATGGAGAACC). For all CRISPR editing, a duplex with Alt-Râ CRISPR-Cas9 tracrRNA, ATTO 550 was formed and coupled to the Cas9 Nuclease V3 and transfection was performed using Lipofectamine CRISPRMAX reagent and Cas9 PLUS Reagent (Thermo Fisher Scientific) following IDT instructions for Cationic lipid delivery of CRISPR ribonucleoprotein complexes into mammalian cells and cell culture methods for GBM cells. After 48h transfection, single cells were sorted using fluorescence-activated cell sorting (FACS) detecting the red dye ATTO550 in 4 coated 96-well plate at 1 cell per well. Clones were expanded and screened for editing events at the target locus by targeted PCR, detection of large deletions on gel followed by Sanger sequencing and targeted deep sequencing using Illumina MiSeq. To address the possibility of off-target effects of CRISPR editing, first we checked the predicted hits by guide RNA design tool and all of the off-target hits had at least 2 mismatches, which makes cutting in off-target sites unlikely. Additionally, we looked at all the predicted off-target hits which were mapping within genes and checked RNA-seq, ChIP-seq and whole genome bisulphite sequencing (WGBS) data from those cell lines in order to find any possible cuts and there were none.
Lentiviral transduction
Lentiviruses were gifts from Dr. Peter Lewis. Lentiviruses expressing H3.3-K27R and H3.3-K27M were applied for 24 hours and G418 (Wisent) selection (500 ng/mL) was maintained for the duration of growth.
Western blotting
Cells were lysed using RIPA buffer with added protease inhibitors (Roche). Whole lysate protein concentration was determined with the Bradford assay reagent (Bio-Rad). Ten micrograms of protein was separated on NuPAGE 3%–8% Tris Acetate Protein gels (ThermoFisher Scientific) and wet-transferred to a nitrocellulose membrane (Bio-Rad). Membrane blocking was performed with 5% skim milk in tris buffered saline (50 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.4) (TBST) for 1 hour. Membranes were incubated overnight with primary antibody solutions in 1% skim milk in TBST: anti-H3K27M (1:200, Millipore ABE419), anti-H3K27me3 (1:1000, Millipore ABE44), anti-total H3 (1:2000, Abcam 1791). Membranes were washed 3 times in TBST, and the ECL anti-rabbit IgG Horseradish Peroxidase linked whole antibody (GE Healthcare) was applied for 1 hour, at 1:1000 dilution in 1% skim milk in TBST. Membranes were washed 3 times and the signal was resolved with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare) and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
For NSD1 and NSD2 blots, the following procedure was used. Cell pellets were resuspended in 100ul RIPA buffer (CST) containing 1:100 Protease inhibitor cocktail (Sigma) and 0.1mM PMSF and incubated one hour on ice. After 10 minutes centrifugation at 4°C, the supernatant was used for quantification using BCA-Pierce Protein assay (ThermoFisher Scientific) in a 96 well microplate, read on Infinite 200 Pro (Tecan). 50ul of sample in 1x Laemmli buffer (6x: 0.35M Tris HCl pH 6.8, 30% Glycerol, 10% SDS, 20% Beta-mercaptoethanol, 0.04% Bromophenol blue, in water) was loaded on 4%–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) and transferred using Trans-Blot Turbo RTA Mini 0.45 μm LF PVDF Transfer Kit (Bio-Rad) according to manufacturer instructions and transfer at the High MW program 10min for NSD1 and normal setting 7min for NSD2.
Gel was cross-linked on Bio-Rad imager system and whole protein images were captured on both gel and membranes. Membrane blocking was performed with 5% skim milk in tris buffered saline (50 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.4) (TBST) for 1 hour. Membranes were incubated overnight at 4°C with primary antibody solutions in 2% skim milk in TBST: anti-NSD1 (1:1000, Antibodies Inc./NeuroMab N312/10), anti-NSD2 (1:1000, Millipore MABE191). Membranes were washed 3 times in TBST, and the Peroxidase-AffiniPure Goat Anti-Mouse IgG (H+L) antibody (Jackson IR) was applied for 1 hour, at 1:10,000 dilution in 2% skim milk in TBST. Membranes were washed again 3 times and the signal was resolved with Clarity Western ECL Substrate (Bio-Rad) and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
Histone modification quantification with nLC-MS
The complete workflow for histone extraction, LC/MS, and data analysis was recently described in detail (Karch et al., 2016). Briefly, cell pellets (approx. 1×106 cells) were lysed on ice in nuclear isolation buffer supplemented with 0.3% NP-40 alternative. Isolated nuclei were incubated with 0.4 N H2SO4 for 3 hours at 4°C with agitation. 100% trichloroacetic acid (w/v) was added to the acid extract to a final concentration of 20% and samples were incubated on ice overnight to precipitate histones. The resulting histone pellets were rinsed with ice cold acetone + 0.1% HCl and then with ice cold acetone before resuspension in water and protein estimation by Bradford assay. Approximately 20 μg of histone extract was then resuspended in 100 mM ammonium bicarbonate and derivatized with propionic anhydride. 1 μg of trypsin was added and samples were incubated overnight at 37°C. After tryptic digestion, a cocktail of isotopically-labeled synthetic histone peptides was spiked in at a final concentration of 250 fmol/μg and propionic anhydride derivatization was performed a second time. The resulting histone peptides were desalted using C18 Stage Tips, dried using a centrifugal evaporator, and reconstituted using 0.1% formic acid in preparation for nanoLC-MS analysis.
nanoLC was performed using a Thermo ScientificTM Easy nLCTM 1000 equipped with a 75 μm × 20 cm in-house packed column using Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany). Buffer A was 0.1% formic acid and Buffer B was 0.1% formic acid in 80% acetonitrile. Peptides were resolved using a two-step linear gradient from 5% to 33% B over 45 min, then from 33% B to 90% B over 10 min at a flow rate of 300 nL/min. The HPLC was coupled online to an Orbitrap Elite mass spectrometer operating in the positive mode using a Nanospray Flex Ion Source (Thermo Scientific) at 2.3 kV. Two full MS scans (m/z 300–1100) were acquired in the orbitrap mass analyzer with a resolution of 120,000 (at 200 m/z) every 8 DIA MS/MS events using isolation windows of 50 m/z each (e.g., 300–350, 350–400…650–700). MS/MS spectra were acquired in the ion trap operating in normal mode. Fragmentation was performed using collision-induced dissociation (CID) in the ion trap mass analyzer with a normalized collision energy of 35. AGC target and maximum injection time were 10e6 and 50 ms for the full MS scan, and 10e4 and 150 ms for the MS/MS can, respectively. Raw files were analyzed using EpiProfile.
ChIP-sequencing
Experimental procedures for chromatin immunoprecipitation and sequencing (ChIP-seq) are similar to those described in Harutyunyan et al. (2019) and Krug et al. (2019).
Cells were fixed with 1% formaldehyde (Sigma). Fixed cell preparations were washed, pelleted and stored at −80°C. Sonication of lysed nuclei (lysed in a buffer containing 1% SDS) was performed on a BioRuptor UCD-300 for 60 cycles, 10 s on 20 s off, centrifuged every 15 cycles, chilled by 4°C water cooler. Samples were checked for sonication efficiency using the criteria of 150–500bp by gel electrophoresis. After the sonication, the chromatin was diluted to reduce SDS level to 0.1% and before ChIP reaction 2% of sonicated Drosophila S2 cell chromatin was spiked-in the samples for quantification of total levels of histone mark after the sequencing (see below).
ChIP reaction for histone modifications was performed on a Diagenode SX-8G IP-Star Compact using Diagenode automated Ideal ChIP-seq Kit. 25ul Protein A beads (Invitrogen) or 70ul of sheep anti-mouse IgG beads (Invitrogen) were washed and then incubated with antibodies (protein A beads with: anti-H3K27me3 (1:40, CST 9733), anti-H3K27me2 (1:50, CST 9728), anti-H3K36me2 (1:50, CST 2901), anti-H3K9me3 (1:66, Abcam ab8898); anti-mouse IgG beads with: anti-H3K27me1 (1:50, Active Motif 61015), anti-H3K36me3 (1:100, Active Motif 61021)), and 2 million cells of sonicated cell lysate combined with protease inhibitors for 10 hr, followed by 20 min wash cycle with provided wash buffers.
ChIP reaction for SUZ12 was performed as follows: antibodies (anti-SUZ12 (1:150, CST 3737)) were conjugated by incubating with 40ul protein A beads at 4°C for 6 hours, then chromatin from ~4 million cells was added in RIPA buffer, incubated at 4°C o/n, washed using buffers from Ideal ChIP-seq Kit (1 wash with each buffer, corresponding to RIPA, RIPA+500mM NaCl, LiCl, TE), eluted from beads by incubating with Elution buffer for 30 minutes at room temperature.
Reverse cross linking took place on a heat block at 65°C for 4 hr. ChIP samples were then treated with 2ul RNase Cocktail at 65°C for 30 min followed by 2ul Proteinase K at 65°C for 30 min. Samples were then purified with QIAGEN MiniElute PCR purification kit as per manufacturers’ protocol. In parallel, input samples (chromatin from about 50,000 cells) were reverse crosslinked and DNA was isolated following the same protocol.
Library preparation was carried out using Kapa HTP or HyperPrep Illumina library preparation reagents. Briefly, for HTP kit, 25ul of ChIP sample was incubated with 45ul end repair mix at 20°C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50ul buffer enzyme mix for 30°C 30 min, followed by PEG/NaCl purification. Adaptor ligation: bead bound sample was incubated with 45ul buffer enzyme mix and 5ul of different TruSeq DNA adapters (Illumina) for each sample, at 20°C for 15 min, followed by PEG/NaCl purification (twice). Library enrichment: 12 cycles of PCR amplification. Size selection was performed after PCR using a 0.6×/0.8× ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments.
For HyperPrep kit, end-repair and A tailing were performed in one reaction: 15ul of ChIP sample was incubated with 45ul end repair+A-tailing mix at 20°C for 30 min, then 65°C for 30 min. Directly proceeded to adaptor ligation by adding 5ul of IDT for Illumina Unique Dual Indexes (IDT) adapters and 45ul ligation buffer enzyme mix, incubating at 20°C for 15 min, followed by Ampure XP bead purification. Library enrichment: 10 cycles of PCR amplification. Size selection was performed after PCR using a 0.6×/0.8× ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments.
ChIP libraries were sequenced using Illumina HiSeq 2000, 2500 or 4000 at 50bp single reads or Illumina NovaSeq 6000 at 50bp paired-end reads (one read used in the analysis for compatibility).
RNA-sequencing
Total RNA was extracted from cell pellets and mouse tumors using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN) according to instructions from the manufacturer. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicenter) to achieve greater coverage of mRNA and other long non-coding transcripts.
Paired-end sequencing (100 bp) was performed on the Illumina HiSeq 4000 platform.
Whole genome bisulphite sequencing
DNA was extracted from cell lines using the AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN) following manufacturer instructions. Whole-genome sequencing libraries were generated from 700 to 1000 ng of genomic DNA spiked with 0.1% (w/w) unmethylated λ DNA (Promega) previously fragmented to 300–400 bp peak sizes using the Covaris focused-ultrasonicator E210. Fragment size was controlled on a Bioanalyzer DNA 1000 Chip (Agilent) and the KAPA High Throughput Library Preparation Kit (KAPA Biosystems) was applied. End repair of the generated dsDNA with 3′ or 5′ overhangs, adenylation of 3′ ends, adaptor ligation, and clean-up steps were carried out as per KAPA Biosystems’ recommendations. The cleaned-up ligation product was then analyzed on a Bioanalyzer High Sensitivity DNA Chip (Agilent) and quantified by PicoGreen (Life Technologies). Samples were then bisulfite converted using the Epitect Fast DNA Bisulfite Kit (QIAGEN) according to the manufacturer’s protocol. Bisulfite-converted DNA was quantified using Oli-Green (Life Technologies) and, based on quantity, amplified by 9–12 cycles of PCR using the Kapa Hifi Uracil + DNA polymerase (KAPA Biosystems) according to the manufacturer’s protocol. The amplified libraries were purified using Ampure XP Beads (Beckman Coulter), validated on Bioanalyzer High Sensitivity DNA Chips, and quantified by PicoGreen.
Sequencing of the WGBS libraries was performed on the Illumina HiSeq2500/HiSeqX system using 125 or 150-bp paired-end sequencing.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of ChIP-seq data
Raw reads from ChIP-seq were aligned to hg19 genome and Drosophila dm6 genome using BWA (Li and Durbin, 2009) version 0.7.17 with default parameters. After alignment, we counted reads in different genomic compartments, e.g., bins (1kb, 10kb and 100kb), CpG islands (CGIs), promoters and genic regions. All the read counting was done using bedtools (Quinlan and Hall, 2010) version 2.22.1. The annotations of CGIs, promoters and RefSeq transcripts for hg19 genome were downloaded from UCSC Table Browser. Promoters were defined as 5kb regions centered on RefSeq TSS. For genic regions, we took the region with the longest length of (TES-TSS) if multiple TSS and/or TES exist.
We normalized the ChIP-seq enrichments of samples by their input data, Rx ratios, or read depth (total read counts). Unless specified, all ChIP data in the scatterplots of this paper are input-normalized. The procedure of input normalization is as follows. Let Si and Ni be the read counts in the i-th genomic compartment of the sample and input respectively, and TS and TN be the total read counts of the sample and input, the normalization on Si was done as follows:
ChIP-Rx (ChIP with reference exogenous genome) is a technique which applies spike-in Drosophila chromatin as internal control (Orlando et al., 2014). For each ChIP-seq profile, we calculated the ChIP-Rx ratio (denoted as Rx) as follows:
where s is the percentage of reads mapped to human genome in the target sample, s_dmel is the percentage of spike-in Drosophila genome in the sample, and similarly i and i_dmel are defined for the input sample. We normalized the ChIP-seq tracks using ChIP-Rx ratios as follows: for each sample, the read counts in the genomic compartments (e.g., bins) were first divide by the total number of reads, then multiplied by ChIP-Rx ratio of the sample, and then multiplied by a normalization factor (which is the same for all samples, set as 10°10 here) to avoid very small values.
ChIP-sequencing coverage tracks were visualized using IGV 2.3 software (Robinson et al., 2011; Thorvaldsdóttir et al., 2013). The correlation plots of H3K27me between K27M and KO (Figure S1E) were generated as described in Baubec et al. (2015), where they aggregated 1kb bins into 1000 windows, while here we aggregated 10kb bins into 100 windows.
All the scatterplots (Figures 1C, 3D, 5E, 5F, S1D, and S5D) were done in 100kb bins (excluded those overlapping the ENCODE DAC blacklisted regions), unless specified. The color scale in the colored scatterplots (e.g., Figures 3E and S3G) was restricted within [−2, 2] to enhance the color effect. The restriction was done in this way: all values lower than −2 were assigned as −2, and all values larger than 2 were assigned as 2.
The aggregate plots of H3K27me and H3K9me3 were generated using deepTools v3.1.0 (Ramírez et al., 2016). The normalization was done using deepTools’ bamCompare. After input normalization, the signals were aggregated around the regions (flanking regions of H3K36me2 domains, PMDs and genes are ± 50 kb, +/− 100 kb and ± 10 kb respectively) using deepTools’ computeMatrix and visualized by averaging the score in bins equidistant from the mid-point (with NaN values discarded). For the plots around PMDs, the aggregated signals were noisy because of the relatively small number of PMDs. To reduce noise, we set “-binSize 100” in “computeMatrix” to average the signals over the bins of 100bp.
The broad domains of H3K36me2, H3K36me3 and H3K27me were called using the same method as described in Weinberg et al. (2019). ChIP signals (1kb bins, after input normalization) were segmented to identify changes in signal average and variance using PELT (Killick et al., 2012; Wambui et al., 2015). Segments were merged within a distance of 10 bp, and the segments greater than 500 kb were outputted as the large domains. For Figure 2 in main text, the regions of H3K36me2 domains were obtained in this way: subtracting the H3K36me2 domain by H3K36me3 domains using “bedtools subtract”; excluding the remaining regions with size smaller then 10kb; removing also the regions whose flanking regions (+/−50kb) overlap H3K36me3 domain. In Figure 3, the regions “lost H3K36me2 domains” in BT245-NSDdko were obtained as follows: take all the 10kb bins (excluded those overlapping the ENCODE DAC blacklisted regions) with the difference of H3K36me2 (i.e., BT245-NSDdko subtracting parental) lower than −0.5, subtract genes from those regions, exclude the remaining regions with size smaller than 50kb, and merge the neighboring regions with distance less than 50kb.
The tornado plots in Figure S3E were generated using deepTools plotHeatmap, sorted by the first sample, setting “-colorMap coolwarm,-heatmapHeight 20.” Regions “Lost H3K36me2 domains” were obtained by subtracting the H3K36me2 broad domains in NSDdko from those in parental cells, i.e., BT245_parental subtracting BT245_NSDdko. Regions “Retained H3K36me2 domains” were obtained by taking intersect of the H3K36me2 broad domains in parental BT245 and NSDdko. Both “Lost H3K36me2 domains” and “Retained H3K36me2 domains” exclude the regions with size smaller than 50kb.
The boxplots of H3K9me3 and gene expression in Figure S5E were generated as follows: first mapped H3K9me3 to the promoters of the genes overlapping with PMDs, then extracted the genes in two groups: H3K9me3 high and low, representing top and bottom 20% of promoter-H3K9me3 levels (normalized by read-depth) respectively; and then plotted expression levels (RPKM) of genes in these two groups.
To calculate the percentage of reads in particular regions, we first counted the reads using bedtools (Quinlan and Hall, 2010) version 2.22.1, then we aggregated the number of reads in those regions and calculated the percentage by dividing the total number of reads.
Analysis of RNA-seq data
STAR (Dobin et al., 2013) (version 2.5.3a) was used to align raw reads from RNA-seq to hg19 genome. After alignment, we counted the mapped reads for each gene from annotation files in GTF format (from UCSC Table Browser) using featureCounts program (version 1.5.3).
The expression levels (RPKM) of genes were calculated as: RPKM = #Reads / ((transcripLen/1000) * (totalReads /1e6)), where #Reads is the number of reads mapped to this gene, transcripLen is the transcript length, totalReads is the total number of reads for this sample.
We classify genes into four groups based on their expression levels (RPKM). The lowest group includes only genes with 0 read. The rest three groups were divided with equal numbers of genes. The “Highly expressed genes” and “Active genes” are referred to the group of genes with the highest expression levels.
Analysis of WGBS data
We aligned the raw reads from WGBS to hg19 genome using BWA (Li and Durbin, 2009) (version 0.6.1) after converting the reference genome to bisulfite mode. Low-quality sequence at the 3′ ends were trimmed. And for the overlapping paired-end reads, we clipped the 3′ end of one of them to avoid double counting. This was done on both forward and reverse strand. After alignment, we filtered reads that were duplicate, or were poorly mapped (with more than 2% mismatches), or not mapped at the expected distance based on the library insert size.
To call methylation level of individual CpGs, Samtools (Li et al., 2009) (version 0.1.18) in mpileup mode was applied. CpGs covered by less than five reads were removed. We also discarded the CpGs that overlapped with SNPs from dbSNPs (build 137) or were located within the ENCODE DAC blacklisted regions or Duke excluded regions (ENCODE Project Consortium, 2012).
To call partially methylated domains (PMDs), we used a window size of 10 kb and slid the window on the measurements of methylation level at each CpG. For each 10 kb window where the average methylation level of mCGs was less than 70% (with each CpG covered by at least 5 MethylC-Seq reads), we extended the region with increments of 10 kb until the average methylation level of the region was greater than 70%. After that, we reported the regions with size larger than 1 Mb as PMDs.
Computational modeling of PRC2
We conceived a stochastic propagation model to simulate the dynamic process of deposition of H3K27 methylation marks by PRC2, STOPHIM: program for simulating STOchastic Process of HIstone Modification. In this study, we simulated a genomic region (length = 5,000 nucleosomes, corresponding to approximately 1 Mb of DNA) containing two PRC2 binding regions (1,490–1,510 and 3,190–3,210 nucleosomes), in 100 cells. We introduced two actively transcribed genic regions marked with H3K36me3 (2,100–2,300 and 3,500–3,750 nucleosomes) and one in intergenic region marked with H3K36me2 (2,301–2,900 nucleosomes).
Bi-modal random walk of PRC2
In this conceptual model, PRC2 complexes were first recruited at specific genomic loci (nucleation sites) where they had a high binding affinity, and then randomly diffused along the chromatin. We simulated the diffusion by a random walk process:
We created a one-dimensional vector with continuous integers to represent coordinates of histones wrapped around by DNA sequence; We next assigned two narrow regions to harbor nucleation sites.
We initiated eight PRC2 complexes at each of the two nucleation sites and recorded their coordinates, at time (t) = 0.
- For a PRC2 complex, we drew a random value:
-
(3.1)with 95% probability, from a binomial distribution Binom(8, 0.5) to represent short-range diffusion due to spontaneous thermal motion; or
-
(3.2)with 5% probability, from a gamma distribution Gamma(0.5, 0.001) to represent possible long-range traveling of the complex after dissociating from the chromatin or across interacting genomic regions.
-
(3.1)
We approximated this random value with the nearest (non-negative) integer and regarded this integer as the distance the PRC2 complex traveled within a time unit.
We changed the coordinate of the PRC2 complex at t = 1 if the traveling distance was non-zero, upstream or downstream with equal probability, based on the distance obtained in (4).
For each PRC2 complex, we repeated (3) – (5) independently, so that we obtained the coordinates for all complexes at t = 1.
We repeated (3) – (6) for a given period of time.
We repeated (2) – (7) for a population of 100 cells, each cell independently yet having identical representation of the simulated chromatin and nucleation sites; We recorded the cumulative distribution of methylation marks until the relative abundance of different methylation marks reached an equilibrium.
When a PRC2 complex traveled beyond the border of our simulated region, we introduced a new complex initiating at a nucleation site to replace the one we lost track of, and independently simulated its trajectory together with other complexes.
Catalytic features of PRC2
During the process described above, whenever a PRC2 complex encountered and resided on a histone, it catalyzed methylation of the unmethylated or lowly-methylated H3K27 to a higher methylated status with differential catalytic activity. We simulated this dynamic process by conducting independent Bernoulli trials with pre-defined conversion rates at each time point at loci occupied by PRC2 complexes. We then iteratively recorded methylation status at each simulated locus in each cell. Generally, PRC2 more easily converted unmethylated H3K27 (i.e., H3K27me0) to mono-methylated H3K27 (i.e., H3K27me1; 0.90 per time unit), than converting H3K27me1 to H3K27me2 (0.25 per time unit), and H3K27me2 to H3K27me3 (0.01 per time unit). However, when an adjacent histone was marked by H3K27me3, PRC2 would have largely enhanced catalytic capacity, especially for tri-methylating H3K27me2 (by 15 times), known as the allosteric activation process (Margueron et al., 2009). The boosted capacity of an allosterically activated PRC2 would be maintained for a given short period of time (t = 2) after the complex dissociated from the histone. Meanwhile, antagonistic histone modifications, in particular H3K36me3 in actively transcribed regions and H3K36me2 in intergenic regions, decreased the catalytic activity of PRC2 complexes such that the conversion rates from lowly-methylated status to heavily-methylated status were reduced proportionally.
Cell division actively removes methylation
For each cell independently, we simulated events of cell division occurring periodically (t = 1,000). During cell division, half of the methylation marks were randomly removed, replaced by unmethylated histones. Meanwhile, half of the PRC2 complexes were removed from current positions and re-initiated at the nucleation sites, while the other half remained where they were.
Key parameters in PRC2 modeling
Several parameters were key to our model and were tuned by numerical in silico experiments in order to recover the observed distributional patterns. First was the balance between traveling distance of PRC2 from nucleation site and the conversion rates. PRC2 must be able to diffuse far away enough from the nucleation sites to produce an approximately uniform distribution - outside of restricted regions - of all three H3K27me modifications. Here we used a combination of short-range and long-range movements to ensure a centered cumulative exposure around the nucleation sites to PRC2 complexes while permitting long-range deposition of H3K27me1 and H3K27me2. The second parameter was related to the antagonistic effect of H3K36me2 on the deposition of H3K27me in intergenic regions. We set the resistance to be the strongest to H3K27me3 and weakest to H3K27me1. Specifically, the H3K27me0 to H3K27me1, H3K27me1 to H3K27me2 and H3K27me2 to H3K27me3 conversion rates were reduced two-fold, three-fold and five-fold, respectively. Third, within active genes, the deposition of H3K27me was hindered by the presence of H3K36me3. Again, the inhibition had the strongest effect on H3K27me3 and the weakest on H3K27me1. Specifically, the H3K27me0 to H3K27me1, H3K27me1 to H3K27me2 and H3K27me2 to H3K27me3 conversion rates were reduced five-fold, 7.5-fold and 15-fold, respectively. These key parameters are summarized in Figure 6A.
Modeling Mechanisms of H3K27M mutation
We tested whether plausible mechanisms for the reorganization of epigenome caused by K27M mutation could be elucidated by this model:
We reduced the number of PRC2 complexes four-fold to examine whether this lack of enzyme could lead to observed phenomena.
We explored whether systematically reducing the conversion rates and weakening allosteric activation, three-fold each, could be the determinants.
In the simulated scenario of harmful K27M mutants, we randomly introduced 5% K27M mutant histones for each cell independently. Upon residing on a mutant histone which cannot be methylated, the catalytic capacity of PRC2 complexes will be permanently impaired (Figure 6A). Specifically, these PRC2 complexes became five-fold less likely to di-methylate H3K27me1, and ten-fold less likely to tri-methylate H3K27me2. Moreover, the allosteric activation effect would be blocked. Moreover, the K27M mutant histones were able to sequester PRC2 complexes for a longer period of time (t = 5) than normal histones. As a consequence, the propagation of heavily-methylated marks was largely hindered.
Supplementary Material
Highlights.
The H3K27M mutation induces a one-level decrease in H3K27 methylation
H3K36me2/3 domains determine the boundaries for the spread of H3K27 methylation
H3K9me3 increases in the regions losing H3K27me2/3 due to H3K27M
A computational model is developed for the dynamic deposition of H3K27 methylation
ACKNOWLEDGMENTS
We would like to thank many past and current members of the Jabado and Majewski laboratories for prior and ongoing work that has made this research possible. Simon Cavanagh-Papillon has participated in many critical discussions that have helped our ideas crystallize. Bo Hu provided analytical avenues that helped guide this study, even when they were not directly used in the analysis. The work in the N.J. and J.M. laboratory is supported by the Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l’Économie, de la Science et de l’Innovation du Québec and by the NIH grant P01-CA196539. The work in the B.A.G. laboratory is supported by NIH grants R01AI118891 and P01CA196539 and by Leukemia and Lymphoma Robert Arceci Scholar Award. T.L. is supported by the Quantitative Life Sciences Graduate Program and the W.R. Lasha Graduate Research Fellowship, Faculty of Medicine, McGill University. Data analyses were enabled by computer and storage resources provided by Compute Canada and Calcul Québec.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108390.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alabert C, Loos C, Voelker-Albert M, Graziano S, Forne I, Reveron-Gomez N, Schuh L, Hasenauer J, Marr C, Imhof A, et al. (2020). Domain model explains propagation dynamics and stability of histone H3K27 and H3K36 methylation landscapes. Cell Rep. 30, 1223–1234.e1228. [DOI] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, and Schübeler D (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. [DOI] [PubMed] [Google Scholar]
- Bechet D, Gielen GG, Korshunov A, Pfister SM, Rousso C, Faury D, Fiset PO, Benlimane N, Lewis PW, Lu C, et al. (2014). Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol. 128, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. [DOI] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, and Dent SY (2014). Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet 15, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl KL, Ge EJ, Weinberg DN, Jani KS, Allis CD, and Muir TW (2019). PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc. Natl. Acad. Sci. USA 116, 22152–22157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Gan H, Cheng L, Lee JH, Zhou H, Sarkaria JN, Daniels DJ, and Zhang Z (2018). H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. eLife 7, e36696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, Fischle W, Bonaldi T, and Pasini D (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62. [DOI] [PubMed] [Google Scholar]
- Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, et al. (2019). H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun 10, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, and Helin K (2018). Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol 25, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. (2016). Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch KR, Sidoli S, and Garcia BA (2016). Identification and quantification of histone PTMs using high-resolution mass spectrometry. Methods Enzymol. 574, 3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killick R, Fearnhead P, and Eckley IA (2012). Optimal detection of change-points with a linear computational cost. J. Am. Stat. Assoc 107, 1590–1598. [Google Scholar]
- Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, Bertrand KC, Mikael LG, McConechy MK, Chen CCL, et al. (2019). Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 35, 782–797.e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, Fan Y, Shaw TI, Silveira AB, Qu C, et al. (2019). Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35, 140–155.e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, et al. (2017). Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al. (2017). Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho CY, Kambhampati M, Hwang EI, Faury D, et al. (2016). Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun 7, 11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee C-H, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PR, Garcia BA, et al. (2018). Capturing the onset of PRC2-mediated repressive domain formation. Mol. Cell 70, 1149–1162.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. (2017). H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32, 684–700.e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. (2011). Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, Shaw TI, Zhu X, Larson JD, Easton J, Shao Y, et al. (2019). H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 137, 637–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, et al. (2018). The H3K36me2 methyltransferase Nsd1 demarcates PRC2-mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol. Cell 70, 371–379.e5. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, and Carpenter PB (2012). Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol 13, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambui GD, Waititu GA, and Wanjoya A (2015). The Power of the pruned exact linear time(PELT) test in multiple changepoint detection. Am. J. Theor. Appl. Stat 4, 581–586. [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. ; St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, and Zhu B (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem 286, 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Sweet SM, Popovic R, Martinez-Garcia E, Tipton JD, Thomas PM, Licht JD, and Kelleher NL (2012). Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. USA 109, 13549–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw human data are Gene Expression Omnibus (GEO): GSE147783. RNA-seq data for BT245 and SU-DIPGXIII cell lines (parental and K27M-KO) were published previously (Krug et al., 2019) and deposited in GEO under the accession number GSE128745.
An open-sourced R program for conducting simulations is available at https://github.com/tianyuan-lu/STOPHIM