
Figure 1.
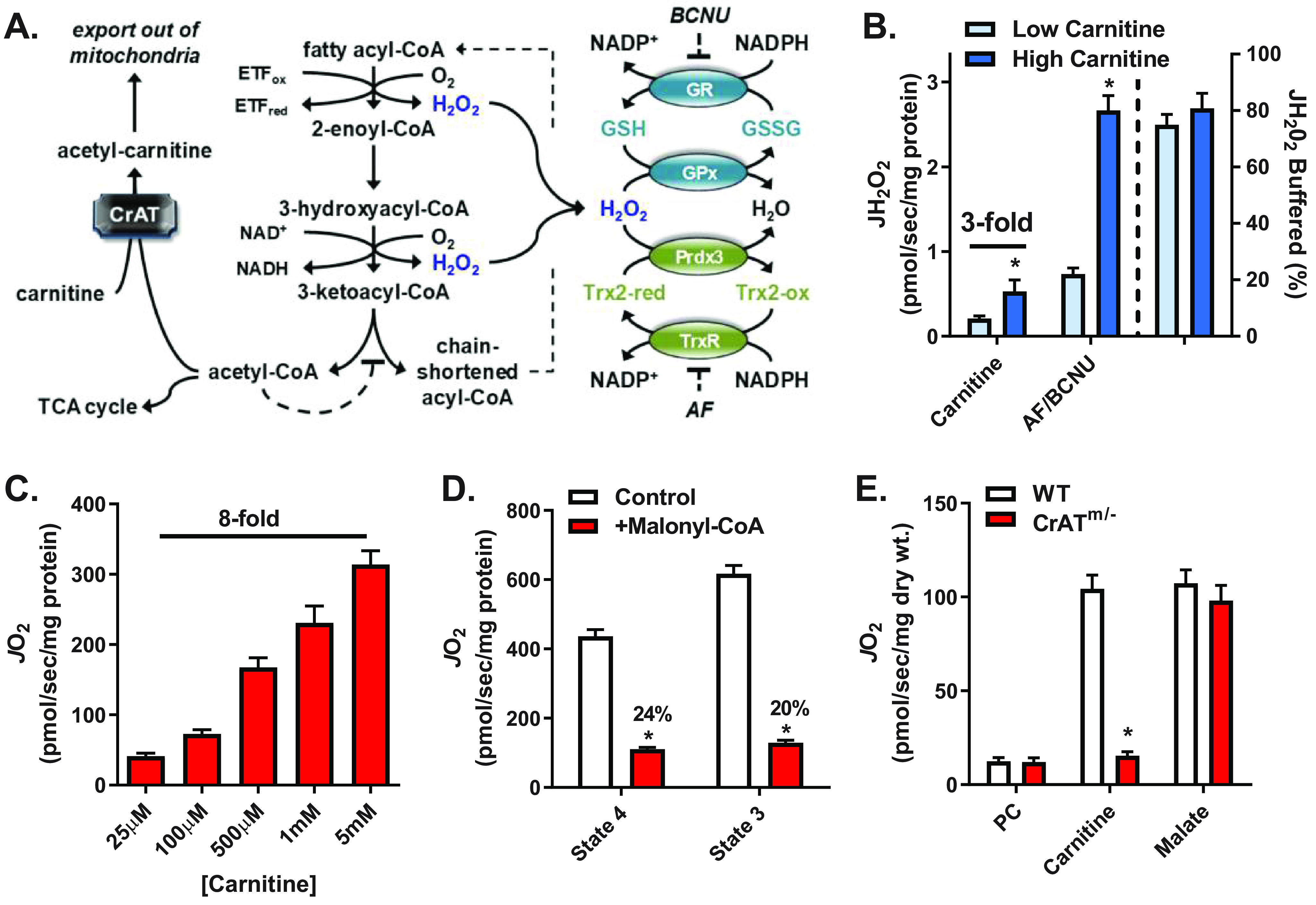
Increased flux through β-oxidation increases JO2, JH2O2 production, and electron flux through redox buffering circuits. A, schematic showing the mechanism by which carnitine via CrAT relieves acetyl-CoA–mediated inhibition of β-oxidation (left), sites of H2O2 generation from β-oxidation dehydrogenases (blue type, center), and the thioredoxin (Trx) and GSH redox buffering circuits (right). B, JH2O2 (left y axis) measured in mitochondria isolated from mouse skeletal muscle supported by PCoA (10 μm) and either low (25 μm) or high (5 mm) carnitine followed by the addition of AF/BCNU (1 μm/100 μm), inhibitors of Trx and GSH reductases, respectively. The percentage of JH2O2 buffered (right of dotted line, right y axis) reflects the percentage of JH2O2 produced but not emitted (i.e. (JH2O2 production (AF/BCNU rate) – JH2O2 emission (PCoA + carnitine rate))/JH2O2 production × 100). C, JO2 measured in mitochondria isolated from mouse skeletal muscle during basal (no ADP) respiration supported by PCoA (10 μm) and increasing concentrations of carnitine. D, PCoA plus carnitine (5 mm) supported JO2 during basal (state 4) or ADP-stimulated (state 3) respiration in the absence or presence of malonyl-CoA (100 μm), a CPT-1 inhibitor. E, JO2 measured in PmFBs from WT and CrATm/− mice during respiration supported by palmitoyl-carnitine (20 μm) followed by the additions of carnitine (5 mm) and malate (0.2 mm). All data are means ± S.E. (error bars); *, p < 0.05 versus corresponding control by unpaired t test; n = 5–13 mice/group.