
Fig. 1. Overview of assessing the reproducibility of phylogenetic inference.
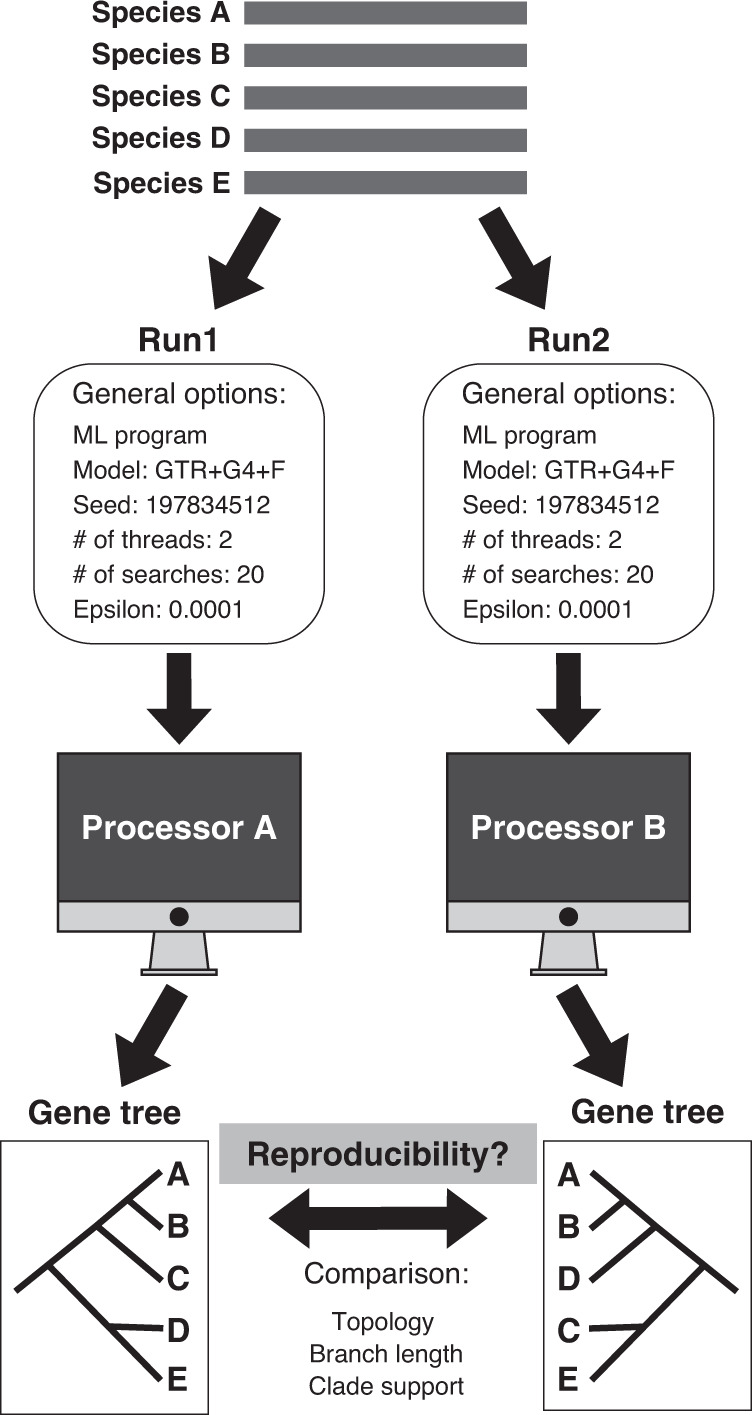
Our assessment begins with a gene sequence alignment. Two replicates (Run1 and Run2) using exactly the same parameters, including substitution model, random starting seed number, number of threads (2), number of tree searches (20), and log-likelihood epsilon for optimization (0.0001) on the same maximum likelihood (ML) program (IQ-TREE or RAxML-NG) were used to evaluate the reproducibility of the phylogenetic tree inferred from a given gene alignment. The Run1 and Run2 replicates were executed on two separate nodes (i.e., each analysis was run on a single node, but Run1 was executed on a different node than Run2) on a supercomputing cluster. Genes whose Run1 and Run2 generated topologically identical phylogenies were considered reproducible, while genes whose Run1 and Run2 generated topologically different phylogenies were considered irreproducible, but we also examined differences in the trees’ branch lengths and clade support values. We analyzed 19,414 gene alignments from 15 animal, plant, and fungal phylogenomic data sets with a wide range of taxon sampling (from 15 to 1178 taxa) that were constructed using diverse gene sampling approaches.